

Abstract
What is already known about this subject
The reviews already published on population pharmacokinetic/pharmacodynamic (PK/PD) analyses have focused on theory and have presented some clinical applications, evaluated validation practices in limited circumstances, defined the interest and sometimes the complexity of this approach in drug development or proposed a list of relevant articles.
None of them has exhaustively evaluated published analyses and more precisely the model-building steps.
In view of the statistical complexity of population PK/PD methodology, more attention is required to how models are built and how they are reported in the literature.
What this study adds
With a strict methodology and by establishing a standardized tool, this survey provides an exhaustive, objective and up-to-date review of model-building practices.
It reveals deficiencies in information reporting in most articles and the genuine need for guidance in publishing.
An initial, minimal list of items is suggested, which can be used by authors and reviewers in pharmacology journals.
The value of published peer-reviewed papers could be greatly improved if authors were to address the suggested list of items systematically.
Aims
A descriptive survey of published population pharmacokinetic and/or pharmacodynamic (PK/PD) analyses from 2002 to 2004 was conducted and an evaluation made of how model building was performed and reported.
Methods
We selected 324 articles in Pubmed using defined keywords. A data abstraction form (DAF) was then built comprising two parts: general characteristics including article identification, context of the analysis, description of clinical studies from which the data arose, and model building, including description of the processes of modelling. The papers were examined by two readers, who extracted the relevant information and transmitted it directly to a MySQL database, from which descriptive statistical analysis was performed.
Results
Most published papers concerned patients with severe pathology and therapeutic classes suffering from narrow therapeutic index and/or high PK/PD variability. Most of the time, modelling was performed for descriptive purposes, with rich rather than sparse data and using NONMEM software. PK and PD models were rarely complex (one or two compartments for PK; Emax for PD models). Covariate testing was frequently performed and essentially based on the likelihood ratio test. Based on a minimal list of items that should systematically be found in a population PK–PD analysis, it was found that only 39% and 8.5% of the PK and PD analyses, respectively, published from 2002 to 2004 provided sufficient detail to support the model-building methodology.
Conclusions
This survey allowed an efficient description of recent published population analyses, but also revealed deficiencies in reporting information on model building.
Keywords: bibliometry, model building, nonlinear mixed effect model, pharmacodynamic, pharmacokinetic, population model
Introduction
The earliest work in the field of population pharmacokinetics (PK) appeared in the late 1970s [1, 2]. During the 1990s, the number of papers describing the population approach applied to pharmacodynamics (PD) and dose–effect models increased dramatically [3]. Recently, the number of population PK/PD publications has escalated further. Success in this field can be attributed to the fact that this approach, which can be performed using different techniques (two-stage, nonlinear mixed effect model, Bayesian hierarchical model), has greatly improved the efficiency of PK analyses. It leads to quantification of variability and identification of relevant covariates by modelling the PK of a typical patient associated with different levels of variability. The population approach allows one to collect integrated information on PK from only a few measurements per subject and it can be applied easily in the clinic and in drug development [4–8]. In the clinic, the population approach can provide prior information for hospital therapeutic drug monitoring; it allows the study of PK and/or PD in specific populations of patients, understanding and anticipation of drug interactions or tolerances to treatment, and the study of compliance issues in oral and long-term therapy. In drug development, where it is promoted by the drug authorities, it is frequently used to combine data from different clinical trials and in the application process [9–12]. Coupled with Monte-Carlo simulations, it can be used to optimize future clinical trials according to the number of patients, inclusion and exclusion criteria, dosage regimen, study length, dates of visits, toxicity and efficacy.
The main drawback of population PK/PD methodology is its statistical complexity [4, 13]. It requires the use of many models (structural, interindividual, intraindividual and covariate), numerous statistical assumptions (random and fixed effects distribution) as enumerated by Karlsson et al. [14] and a variety of estimation methods (algorithms and approximations) [15, 16]. In this context, population analyses should be performed carefully and reported precisely [17–19]. Ideally, this description should include the purpose of the model and possible model applications, a relevant description of the data used for model building and description of their exploratory examination, listing of model-building steps, and a description of all submodels, of the final model and of the qualification process [11], the aim being to allow an independent modeller (with access to the data) to redo the analysis. Faced with the aforementioned complex issues, the quality and completeness of published analyses vary greatly. In the 1990s, a first exhaustive review of the literature from 1977 to 1996 was performed by Mentré and Ebelin to illustrate and comment on these different aspects [3].
The main objective of the present study was to perform an exhaustive survey of recently published population PK/PD analyses. A secondary objective was to assess whether model-building steps are correctly performed and reported, based on a list of minimal items that should always be documented. The methodology was as follows: first, papers cited in MEDLINE (Pubmed) from 2002 to 2004 were selected using specific keywords; second, a questionnaire was constructed in order to establish a data abstraction form (DAF); and third, all the selected papers were carefully read, the questions in the DAF were answered and information was entered into a database. This study includes a description of DAF building and paper selection, presents the survey results in terms of the data used in the modelling and in term of model building. Another paper, using the same database but a different DAF, addresses the question of population PK–PD model qualification [20]. These present results focus on model-building practices, on which no exhaustive and detailed review has ever been published.
Methods
Article selection
Initial article selection was performed in MEDLINE (Pubmed) using a list of keywords aimed at capturing papers concerning population PK, PD or PK–PD published from 1 January 2002 to 12 December 2004: ((population AND model *) OR (non AND linear AND mixed AND effect *) OR bayesian OR hierarchical OR NONMEM OR nlme OR NLMIXED OR P-PHARM OR WinNonMix OR *bugs OR NPLM OR NPEM OR Kinetica OR ADAPT OR ITRLS OR MP2) AND (PK-PD OR PK-PD OR PBPK OR pharmacokinetic * OR pharmacodynamic *). Additionally, the search was limited to ‘English language’ and ‘human data’. Based on the abstract or, if necessary, on the full article, papers were then further limited to those dealing with medications or analyses performed on original clinical data. Reviews and methodology articles were excluded.
Data abstraction form building
The methodology used for building the DAF was based on the work of Boutron et al. [21], who published several systematic reviews using a checklist of items. The DAF was constructed around a single statistical unit defined as a PK, PD or PK–PD model, rather than the paper itself. The rationale was based on the fact that our work focused mainly on describing the quality of published models and techniques used to qualify them, rather than on the quality of published papers. Consequently, when a paper described more than one model, it had as many entries in our database as the number of models described in the paper. A two-part grid was then created. The first part, devoted to general characteristics, included article identification, context of the analysis and description of clinical studies from which the data arose. Article identification (number of items = 4) contained generalities about the paper and the journal in which it was published. Context of the analysis (number of items = 7) collected information about authors and the treatment administered. It characterized the nature of the authors' laboratory (industry/academia), the therapeutic classes and names of administered drugs. Therapeutic classes were defined according to the Food and Drug Administration (FDA) National Drug Code Directory [22]. Clinical study(ies) (number of items = 49) described the clinical trial(s) from which the data arose, including phase of clinical development, objectives of clinical trial(s) (PK, dose finding, therapeutic drug monitoring, etc.), target population, treatment summary (routes of administration, treatment duration, number of doses, etc.) as well as experimental design. This section provided information about potential complexities of design (sparse data, cross-over study, etc.) and then potential difficulties for model building. The second part of the DAF (number of items = 214) aimed to provide precise information about the characteristics of the model-building process. Given that this last part was the most interesting with regard to modelling, a larger number of items were defined. Information was collected on the objectives of modelling (predictive or descriptive), the software used and the drugs included in the model. Added information was related to the PK, PD or PK–PD model, the amount of data actually included in the modelling process, the nature of the structural, inter- and intraindividual models and, when applicable, the nature of the covariate model. For each model, information was gathered about criteria used for final covariate model building and selection from an exhaustive list, including criteria such as graphs and statistical criteria such as the likelihood ratio test.
This DAF was built and finalized between July 2004 and January 2005 by nine independent PK–PD modellers (authors of this paper). The gamut of the author's backgrounds (Pharmaceutical Science, Veterinary Medicine, Medicine, Statistics, and Engineering), their different levels of experience and skill, positions (permanent or PhD student) and origins (industry and academia) improved the relevance of the questions and increased the understanding necessary to classify them according to the overall point of view of the modellers. The group ensured, especially, that the questions, whether single, multiple choice or open, were as simple and unambiguous as possible. Moreover, the group prioritized the use of different fields with defined checklists to provide for the efficient recording of information and subsequent automated analysis. Finally, a draft was tested by the two readers (C.D. and K.B.) using 20 additional articles, selected according to the criteria described above, but published either before 2002 or after 2004. The articles were examined by both readers, which assisted them in agreeing on the interpretation of different questions and resolving differences in answers. The final version of the DAF is available at: http://www.bichat.inserm.fr/equipes/Emi0357/download.html.
Data collection
The DAF was implemented in HTML and PHP languages. PhpMyAdmin (version 2.5.3) software was used, which is a tool written in PHP intended to handle the administration of a MySQL database server [23]. In this way, it was possible to enter interactively the items in the DAF while reading the papers through a clear and easily usable local web interface. At the end of reading, answers were then directly transmitted to the MySQL database. Variables were encoded as either numeric or character variables, including categorical or continuous data, depending on the type of answer. All coding was defined in the PHP script. The 324 articles retrieved were split between the two readers according to a randomization stratified by year. The reading finally selected required 4 months (from March to July 2005).
Statistical analysis
Once the reading was completed, the MySQL database was exported into SAS (version 8) and S-plus (version 6.2) statistical packages for statistical analysis. For each item within the different parts, descriptive statistics (mean, SD, minimum and maximum values) were used to report the results for continuous variables, whereas frequencies were used to describe categorical variables. New variables were generated and cross analyses were performed in order to highlight the results. For example, covariates were regrouped according to treatment characteristics, demographics, and clinical or biological characteristics of the patients. Journals were classified as statistical, pharmacological or clinical [24] and the time was split by year of publication. Differences across journals or years were tested with a χ2 test.
Results
Generalities
Among the 482 articles initially selected, some were excluded (see Figure 1). The remaining articles were distributed evenly across years: 108 for 2002, 100 for 2003 and 116 for 2004 (list available at: http://www.bichat.inserm.fr/equipes/.../). The articles came primarily from pharmacological (58%) and clinical journals (41%), with most coming from leading journals in the fields of cancer, anaesthesiology and antimicrobials (see Figure 2A). Only three articles came from statistical journals. Authors were primarily from academia (71%) or a mix of industry and academia (16%). The distribution of therapeutic classes is presented in Figure 2B. Consistent with Figure 2A, oncolytics, antimicrobials and pain relief drugs were among those where a population model has been most often published.
Figure 1.
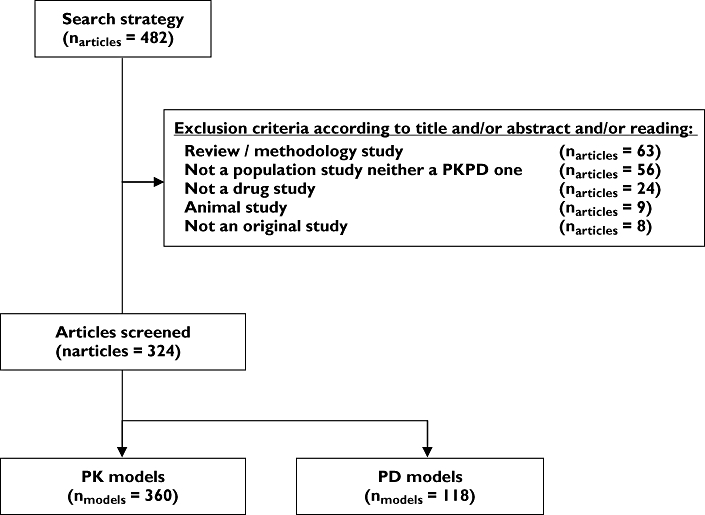
Screening of the articles selected in Pubmed using exclusion criteria (exclusion criteria were not mutually exclusive)
Figure 2.
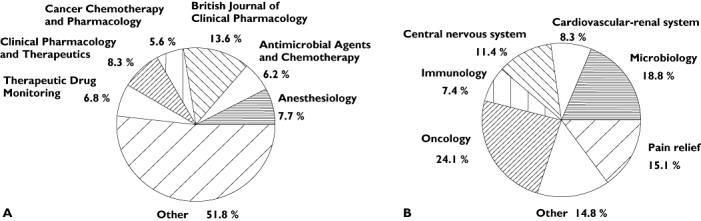
(A) Journals in which articles on population PK/PD (n = 324) were most frequently published; other: journals in which <5% of the papers were found. (B) Therapeutic classes most frequently studied in population PK/PD articles (n = 324); other: therapeutic classes found in <5% of the papers
Clinical studies
Generally, the data used for modelling came from clinical studies with a majority of PK objectives (68%), the rest being PK–PD (23%) or PD (9%). Clinical phases of these studies were reported for 66% of the models. The majority were in postmarketing authorization (34%) or in Phase I studies (20%). A typical model was characterized from the data of a single clinical study (77%), with single arm (57%), performed on patients (78%) and adults (68%). About 25% of the screened models dealt with paediatric patients, whereas only 2% concerned elderly patients. Drugs were administered as multiple doses (68%) or as a single dose (24%) (8% not reported). The routes of administration included i.v. infusion (52%), oral (39%) or i.v. bolus (7%). When studies included more than one arm, subjects were often randomized (65%). About 21% of clinical studies were typical dose-escalation studies and prospective design optimization (as described in [25]) was recovered for 9% of the models.
Modelling
Articles included a majority of models with a descriptive purpose (81%) or predictive and descriptive purposes (17%). Models with a descriptive purpose presented parameter estimates, as expected, for 99% of cases, then defined PK/PD variability (21%) and tested covariates (58%). The most frequent methodology was the nonlinear mixed-effect model (92%), rarely preceded (2%) or replaced (5%) by a two-stage approach. These models included one compound in 89% of cases and more than two compounds for only 5% of cases. The most frequently used software was NONMEM. Distribution of software and associated methods across models is presented in Table 1. Of note, for NONMEM analyses, algorithms were not reported in 32% of the cases. Among reported algorithms, First Order Conditional Estimation (FOCE) was the most often used (with or without interaction), closely followed by First Order (FO). Looking at developments over time, the use of ‘FOCE INTERACTION’ increased from 2002 to 2004 (from 7 to 24%, P = 0.006). The use of FOCE (INTERACTION) seemed less frequent for PD models, but the absence of estimation method reporting in this latter case was more frequent than for PK models.
Table 1.
Softwares and methods most used in model building among the PK (n = 360) and PD (n = 118) population models. For each method, we report the number of models and in brackets, the percentage referring to the corresponding category
| Softwares | PK models | PD models | ||
|---|---|---|---|---|
| NONMEM | 247 | (68.6) | 92 | (78.0) |
| FO | 58 | (23.5) | 22 | (23.9) |
| FOCE | 71 | (28.7) | 12 | (13.0) |
| FOCEI | 47 | (19.0) | 13 | (14.1) |
| FOCE centered | 2 | (0.8) | 1 | (1.1) |
| LAPLACE | 4 | (1.6) | 5 | (5.4) |
| NR* | 65 | (26.3) | 39 | (42.4) |
| NPEM | 20 | (5.6) | 2 | (1.7) |
| ADAPT | 17 | (4.7) | 8 | (6.8) |
| PPharm/Kinetica | 14 | (3.9) | 4 | (3.4) |
| MP2 | 13 | (3.6) | 0 | (0.0) |
| WinNonMix | 10 | (2.8) | 1 | (0.9) |
| Other | 32 | (8.9) | 8 | (6.8) |
| NR* | 7 | (1.9) | 3 | (2.5) |
NR: Not Reported.
PK models
Of the 324 articles selected, 360 PK models were retrieved. Nearly all studies (99%) reported the number of included subjects. The median was approximately 50 and only 26% of datasets included more than 100 subjects. The number of samples per subject (usually more than three) was defined on observed data in 16% of cases and on scheduled design in 3% of cases. Quantiles 10, 25, 50, 75 and 90% of number of points per subject were 2, 4, 9, 14 and 17, respectively. When the model included only one drug, the number of compartments was mostly limited to less than two (Figure 3). Conversely, when parent and metabolite compounds were modelled together (10% of the models), the number of compartments could reach a maximum of 13, as for irinotecan [26]. Linking the number of points per subject with the number of compartments, an increase was observed in the number of samples per subject when the number of compartments increased. For one compartment, the median was four, for two compartments it was 10 and for three compartments it was 12 points per subject. Parent and metabolite were modelled either simultaneously (51% of the published models) or sequentially (34%). With regard to random effect models, Inter-Individual Variability (IIV) model type was often reported, whereas the nature of the IIV matrix was not often reported (only 18% of the PK models). IIV models were either exponential (53%) or multiplicative (11%). Residual error was modelled mainly as additive (17%), combined (additive and multiplicative) (21%) or multiplicative (24%). Criteria used for selecting the final model were, when reported, graphics (43%), Akaike criterion, Bayesian information criteria, the likelihood, or objective function (38%), the likelihood ratio test (22%) or residuals distribution (15%). Finally, if each item is considered separately, the majority of analyses showed an adequate description (see data, interindividual variability, error model or building criteria in Figure 4, left plot).
Figure 3.
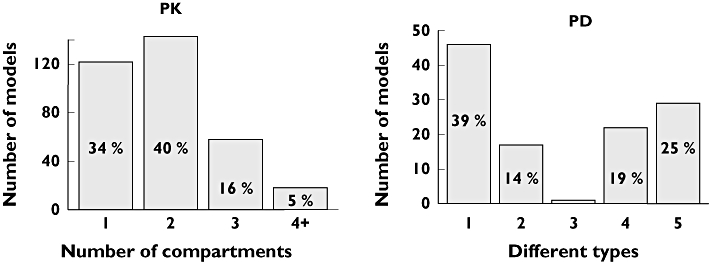
Number of compartments of each PK model (n = 360, left side) and PD model types (n = 118, right side). The different types of PD models are defined by: Emax models (1), indirect models (2), physiological models (3), other models for continuous data (including linear, exponential, power and log linear models) (4), or models for noncontinuous data (including Cox, logistic and time to event models, and models for count or ordered categorical data) (5). The percentage is indicated if only = 5%
Figure 4.
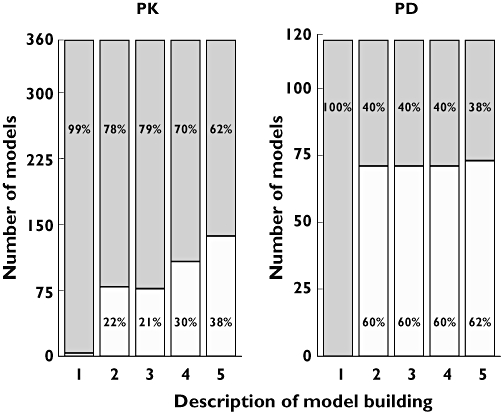
Description level of PK (n = 360) and PD (n = 118) model building. The grey part of each column represents the percentage of models with reported information; the white part, models with no reported information. Information is defined by the number of subjects and number of observations (total or per patient) included in the model, in respective columns (1) and (2), type of Inter-Individual Variability model in column (3), nature of error model in column (4), and description of criteria used in model building in column (5). The percentage is indicated only if = 5%. (Yes, ( ); No, (□))
); No, (□))
Covariates were considered for most PK models with a median number of six covariates tested. Although information was sometimes not reported, their selection was mostly performed based on graphs of posthoc estimates (see Table 2), followed by stepwise inclusion using only the likelihood ratio test (43% of cases). Other criteria independent of the likelihood, such as decrease of IIV or SE, or clinical relevance of the covariates were also used. A median of two covariates was kept in final models. Characteristics of the covariates tested finally retained in the model are reported in Table 3. For most models, demographic covariates or covariates on liver and kidney functions were the most often considered. Overall, very few papers (see, for example, Kerbusch et al. [27]), provided a complete and relevant description of covariates model building.
Table 2.
Building steps in the 251 (out of 360) PK models and in the 43 (out of 118) PD models when a covariate model is performed. For each method, we report the number of models and in brackets, the percentage referring to the corresponding category
| PK models | PD models | |||
|---|---|---|---|---|
| Covariate models | 251 | (69.7) | 43 | (36.4) |
| Selection based on post-hoc* | 144 | (57.4) | 25 | (58.1) |
| graphs | 102 | (70.8) | 12 | (48.0) |
| GAM (or bootstrap of GAM) | 32 | (22.2) | 5 | (20.0) |
| univariate test | 17 | (11.8) | 1 | (4.0) |
| tree | 1 | (0.7) | 0 | (0.0) |
| other | 30 | (20.8) | 6 | (24.0) |
| NR** | 31 | (21.5) | 9 | (36.0) |
| Building approach described | 138 | (55.0) | 15 | (34.9) |
| stepwise | 106 | (76.8) | 10 | (66.6) |
| forward | 28 | (20.3) | 5 | (33.3) |
| backward | 4 | (2.9) | 0 | (0.0) |
| Building criteria reported* | 208 | (82.9) | 31 | (72.1) |
| LRT | 168 | (80.8) | 26 | (83.9) |
| AIC, BIC (or SC), OF, L*** | 63 | (30.3) | 5 | (16.1) |
| IIV decrease | 56 | (26.9) | 4 | (12.9) |
| SE decrease | 26 | (12.5) | 2 | (6.5) |
| clinical relevance | 17 | (8.2) | 1 | (3.2) |
| Wald test | 0 | (0.0) | 0 | (0.0) |
| other | 30 | (14.4) | 6 | (19.4) |
Under each category, the percentages in one column sum up to more than 100% because several different methods could be used in the same model.
NR: Not reported.
AIC: Akaike criterion, BIC (or SC): Bayesian information criteria (or Schwarz criteria), OF: objective function, L: likelihood.
Table 3.
Type of covariate tested and selected in the 251 (out of 360) PK models and in the 43 (out of 118) PD models when a covariate model is performed. For each method, we report the number of models and in brackets, the percentage referring to the corresponding category
| PK models | PD models | |||||||
|---|---|---|---|---|---|---|---|---|
| Covariate models | 251 (69.7) | 43 (36.4) | ||||||
| Covariates | Tested* | Kept* | Tested* | Kept* | ||||
| Demographic | 233 | (92.8) | 186 | (74.1) | 31 | (72.1) | 10 | (23.3) |
| Liver/Kidney function | 122 | (48.6) | 105 | (41.8) | 12 | (27.9) | 8 | (18.6) |
| Biological parameter | 41 | (16.3) | 34 | (13.6) | 7 | (16.3) | 6 | (14.0) |
| Drug interaction | 58 | (23.1) | 47 | (18.7) | 9 | (20.9) | 3 | (7.0) |
| Dose | 33 | (13.1) | 26 | (10.4) | 4 | (9.3) | 3 | (7.0) |
| Metabolism induction/inhibition | 24 | (9.6) | 19 | (7.6) | 3 | (7.0) | 2 | (4.7) |
| Disease | 45 | (17.9) | 37 | (14.7) | 12 | (27.9) | 6 | (14.0) |
| Food | 14 | (5.6) | 13 | (5.2) | ||||
| Performance status | 21 | (8.4) | 18 | (7.2) | 2 | (4.7) | 1 | (2.3) |
| Time | 22 | (8.8) | 17 | (6.8) | 3 | (7.0) | 2 | (4.7) |
| Administration | 10 | (4.0) | 10 | (4.0) | 6 | (14.0) | 3 | (7.0) |
| Adverse events | 1 | (0.4) | 0 | (0.0) | ||||
| PK parameters | 4 | (9.3) | 3 | (7.0) | ||||
Under each category, the percentages in one column sum up to more than 100% because several different covariates could be tested and kept in the same model.
PD models
In the selected articles, only 118 PD models were reported, 77% of which concerned PK and PD models built simultaneously. For these models, 33% of the datasets included >100 subjects. For 60% of the cases, the number of observations was not reported. PD model types varied, but showed a majority of Emax models, followed by models for noncontinuous data (defined and presented in Figure, right side). In 20% of cases, the PD models included an effect compartment model. With regard to random effect models, the IIV model type was most frequently exponential (in 25% of the PD models), IIV matrix was rarely reported (in only 5%) and residual error was mainly modelled as additive (18%). Criteria used for selecting the final model were, when reported, graphics (22%), Akaike criterion, Bayesian information criteria, the likelihood, objective function (19%) or the likelihood ratio test (14%). Overall, the description of model building was less well reported than for PK ones (Figure 4, right plot). However, it is encouraging to note that for the residual error model, deficiencies in reporting information decreased from 2002 to 2004 (75 to 44% of the models, P = 0.02).
Covariates were tested for 36% of the PD models, with a median number of five covariates. Covariate selection, not always clearly described, was often performed with graphs of posthoc estimates and their inclusion, by either the stepwise or forward approach using only the likelihood ratio test (in 49% of cases, see Table 2). Most of the time, the number of covariates selected in the final model was very limited (i.e, 0 for 54% and one for 27% of the PD covariates models) and they included demographic, liver or kidney function, biological parameters or disease. Tested and retained covariates are reported in Table 3.
Discussion
The only method for obtaining an overview of a research area is through exhaustive reading of recent literature. For this purpose, a DAF has many advantages. It allows one to target all interesting questions and to discard information irrelevant to the study. Simple questions and a standardized DAF format ensure objectivity. Our interactive DAF has other advantages, in that it avoids errors in transferring data from paper to computer and allows for presentation of the data in a format compatible with statistical analysis. The number of experienced scientific modellers involved in the modelling of the DAF and in the interpretation of the results, as well as the diversity of their backgrounds, ensured the quality and reliability of this approach.
Overall, the models published in the screened papers were built primarily with clinical data from postmarketing studies. This can be explained partly by confidentiality issues with data from pharmaceutical industry. Academicians, whose activity is evaluated according to publishing frequency, not surprisingly, represented the majority of the authors. They often used data from hospital for therapeutic drug monitoring or data from public clinical trials. With regard to therapeutic classes, oncolytics, antimicrobial agents and anaesthetics were modelled most frequently. These classes can present a narrow therapeutic index as well as severe toxicity. Modelling in this context is very useful. One can optimize clinical trials, sampling times, dosing, rhythm of administration and number of patients in different arms. One can either perform therapeutic drug monitoring, limit toxicity or reduce to a certain extent tolerance and resistance. The data were often homogeneous (in the majority from one single clinical trial, one arm, including only one compound) and rich rather than sparse (number of points per patients equal to 4, 10 and 12 for 1, 2 and 3 compartments, respectively). This homogeneity probably explains why the majority of models are solely descriptive and are not used to predict data in other populations, since model usefulness is limited. With regard to methodology, the one-stage nonlinear mixed-effect approach was most frequently used over the 2002–2004 time period. Despite the fact that it has been demonstrated several times that the two-stage approach has many drawbacks in practical situations (including sparse data, data imbalance and subject-specific dosing history), 5% of models reviewed were built with it. Actually, the two-stage approach provides a poorly approximated asymptotic covariance matrix [17, 28] and is considered less efficient than the one-stage nonlinear mixed-effect modelling method [29]. In some cases, as in animal studies, the two-stage approach is still used due to the limited number of subjects (often n ≤ 6) and samples per subject (often n ≤ 3). In our case, the complexity of implementation of population analysis seems to be the main reason for the continuing popularity of the two-stage approach. Concerning the software, NONMEM remains the most widely used for analysis, both in academia and industry. It has been used for quite some time and is considered overall to be highly flexible with regard to the NM-TRAN scripting language and data input system, which allows one to define any type of dosage regimen, PK, PD or PK–PD models [30]. Surprisingly, there were still instances where this software was reported as misused, despite all the training and references which are available to users. For example, the FO method was used with multiplicative as well as exponential interindividual variability model, although it is known that it can induce bias on parameter estimates [31]. This may be linked to convergence problems method or excessive computation time for the more advanced FOCE method, even with fast, modern computers [32, 33]. Model complexity, however, did not seem to be an issue, as we did not notice that a PK model with a large number of compartments was systematically associated with the use of FO.
We found three times more PK than PD models, and most of the PD models were simultaneous PK–PD models. These were primarily Emax models, with effect compartments. Although such models can be useful in understanding drug–effect relationships and to quantify interindividual variability in clinical response, PD models are less frequent than PK ones. This frequency can be explained by several factors [34]. First, there is the difficulty of measuring and modelling drug effects compared with drug and metabolite concentrations, for which PK methodology is well established and standardized. A second reason is the absence of apparent causal relationships between intermediate effects quantified by biomarkers, most often utilized in PD models, and clinical end-points, which can be expressed as survival data and can often not be modelled with a methodology such as population PK–PD.
Covariate testing remains one of main objectives of population PK modelling (70%) and it is used to explain variability, but was involved in only 36% of the PD models. Indeed, among the 118 PD models, 80% were part of a PK–PD model and in these, testing covariates on the PK or PD model was performed in 67% of cases. It is understandable that after covariates have been included in the PK model, it was more difficult to find any potential covariates specific to PD models (tested in only 32%). PK often explains the main part of PD variability, except if there is substantial heterogeneity in the disease or in the response. It explains why only fewcovariates were found to be significant in the PD models (Table 3).
Given that the methodology of PK–PD population models is complex, involves many assumptions and can be used to make important decisions in drug development, it is crucial that it be reliable. As a first step, readers of published analyses should be able to understand and evaluate the numerous choices made in the analysis through detailed descriptions. The most recent FDA guidelines giving an overall description of what is expected in population PK study reports dates back to 1999 [17]. Recently, pharmacometricians from the Swedish medical product agency and from the European Agency for the Evaluation of Medicinal Products reported on what their agencies look for when assessing a population PK analysis in an article, and a draft guideline [18, 19]. Consulting also some reviews in the field [3, 4, 13, 30, 35], we established the DAF by defining the items we considered as interesting for a literature survey. However, through the review we performed, many deficiencies in the quality of reported information were observed. Due to the importance of this result, we considered it important to quantify and illustrate this observation. For this purpose, a limited list of items were defined that we considered essential to be addressed by authors in any type of journals. Those items extracted from the DAF were:
subject characteristics (healthy/patient)
dosage (single/multiple) and route of administration
total number of subjects and total number of observations or number of observations per patient
criteria used for model selection
nature of the structural, IIV and error models
method of estimation and software.
Our survey has revealed that only 39% of the published PK reports and only 8.5% of the PD models reported the aforementioned items (see, for example, the excellent paper of Van Kesteren [32]). Since this information appeared reasonable, these low percentages were surprising and demonstrate the need to establish guidelines when reporting population PK–PD analysis in the literature.
This minimal list of items should be found in all population PK/PD papers published in peer-reviewed journals and could be complemented by other items such as: description of raw data exploration, untested covariates, handling of missing data and data below the limit of quantification, different model-building steps and all aspects of model qualification. This last item is one of the most important and has been addressed in a separate paper which analyses the same database with similar methodology but with a different DAF [20]. However, the establishment of such a check list for population PK–PD publications would require consensual decisions between experts and editorial committees of journals publishing population analyses. Numerous meetings would probably be required before addressing this challenge and reaching a consensus. This was not the purpose of the present study, but its results naturally point to this conclusion.
Acknowledgments
This study was supported by Institut de Recherche International Servier. P.G. is funded by INSERM, Paris, France.
References
- 1.Sheiner LB, Rosenberg B, Melmon KL. Modelling of individual pharmacokinetics for computer-aided drug dosage. Comput Biomed Res. 1972;5:411–59. doi: 10.1016/0010-4809(72)90051-1. [DOI] [PubMed] [Google Scholar]
- 2.Sheiner LB, Rosenberg B, Marathe VV. Estimation of population characteristics of pharmacokinetic parameters from routine clinical data. J Pharmacokinet Biopharm. 1977;5:445–79. doi: 10.1007/BF01061728. [DOI] [PubMed] [Google Scholar]
- 3.Mentré F, Ebelin ME. Validation of population pharmacokinetic/pharmacodynamic analyses: review of proposed approaches. In: Aarons L, Balant LP, Danhof M, et al., editors. COST B1. European Cooperation in the Field of Scientific and Technical Research. The Population Approach. Measuring and Managing Variability in Response, Concentration and Dose. Brussels: Office for Official Publications of the European Communities; 1997. pp. 148–60. [Google Scholar]
- 4.Pillai GC, Mentré F, Steimer JL. Non-linear mixed effects modeling—from methodology and software development to driving implementation in drug development science. J Pharmacokinet Pharmacodyn. 2005;32:161–83. doi: 10.1007/s10928-005-0062-y. [DOI] [PubMed] [Google Scholar]
- 5.Sheiner LB, Steimer JL. Pharmacokinetic/pharmacodynamic modeling in drug development. Annu Rev Pharmacol Toxicol. 2000;40:67–95. doi: 10.1146/annurev.pharmtox.40.1.67. [DOI] [PubMed] [Google Scholar]
- 6.Steimer JL, Ebelin ME, van Bree J. Pharmacokinetic and pharmacodynamic data and models in clinical trials. Eur J Drug Metab Pharmacokinet. 1993;18:61–76. doi: 10.1007/BF03220009. [DOI] [PubMed] [Google Scholar]
- 7.Steimer JL, Vozeh S, Racine-Poon A, Holford N, O'Neill R. Pharmacokinetics of drugs. (Handbook of experimental pharmacology) In: Welling PG, Balant LP, editors. The Population Approach: Rationale, Methods, and Applications in Clinical Pharmacology and Drug Development. Berlin-Heidelberg: Springer-Verlag; 1994. pp. 404–51. [Google Scholar]
- 8.Whiting B, Kelman AW, Grevel J. Population pharmacokinetics. Theory and clinical application. Clin Pharmacokinet. 1986;11:387–401. doi: 10.2165/00003088-198611050-00004. [DOI] [PubMed] [Google Scholar]
- 9.Bhattaram VA, Booth BP, Ramchandani RP, Beasley BN, Wang Y, Tandon V, Duan JZ, Baweja RK, Marroum PJ, Uppoor RS, Rahman NA, Sahajwalla CG, Powell JR, Mehta MU, Gobburu JV. Impact of pharmacometrics on drug approval and labeling decisions: a survey of 42 new drug applications. AAPS J. 2005;7:E503–E512. doi: 10.1208/aapsj070351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chien JY, Friedrich S, Heathman MA, de Alwis DP, Sinha V. Pharmacokinetics/pharmacodynamics and the stages of drug development: role of modeling and simulation. AAPS J. 2005;7:E544–E559. doi: 10.1208/aapsj070355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ette EI, Williams PJ, Lane JR. Population pharmacokinetics III: design, analysis, and application of population pharmacokinetic studies. Ann Pharmacother. 2004;38:2136–44. doi: 10.1345/aph.1E260. [DOI] [PubMed] [Google Scholar]
- 12.Gobburu JV, Marroum PJ. Utilisation of pharmacokinetic–pharmacodynamic modelling and simulation in regulatory decision-making. Clin Pharmacokinet. 2001;40:883–92. doi: 10.2165/00003088-200140120-00001. [DOI] [PubMed] [Google Scholar]
- 13.Beal SL, Sheiner LB. Estimating population kinetics. Crit Rev Biomed Eng. 1982;8:195–222. [PubMed] [Google Scholar]
- 14.Karlsson MO, Jonsson EN, Wade JR. Assumption testing in population pharmacokinetic models: illustrated with an analysis of moxonidine data from congestive heart failure patients. J Pharmacokinet Biopharm. 1998;26:207–45. doi: 10.1023/a:1020561807903. [DOI] [PubMed] [Google Scholar]
- 15.Davidian M, Giltinan DM. Nonlinear Models for Repeated Measurement Data. London: Chapman & Hall; 1995. pp. 151–235. [Google Scholar]
- 16.Davidian M, Giltinan DM. Nonlinear models for repeated measurement data. An overview and update. J Agr Biol Envir St. 2003;8:387–419. [Google Scholar]
- 17.Food and Drug Administration. Guidance for Industry—Population Pharmacokinetics. [Google Scholar]
- 18.Wade JR, Edholm M, Salmonson T. A guide for reporting the results of population pharmacokinetic analyses: a Swedish perspective. AAPS Pharmsci. 2005;7:45. doi: 10.1208/aapsj070245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guideline on Reporting the Results of Population Pharmacokinetic Analyses. Available at http://www.emea.eu.int/pdfs/human/ewp/18599006en.pdf Last accessed 17 July 2006.
- 20.Brendel K, Dartois C, Comets E, Lemenuel-Diot A, Laveille C, Tranchand B, Girard P, Laffont CM, Mentre F. Are population pharmacokinetic and/or pharmacodynamic models adequately evaluated?: a survey of the literature from 2002 to 2004. Clin Pharmacokinet. 2007;46:221–34. doi: 10.2165/00003088-200746030-00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Boutron I, Tubach F, Giraudeau B, Ravaud P. Methodological differences in clinical trials evaluating nonpharmacological and pharmacological treatments of hip and knee osteoarthritis. J Am Stat Assoc. 2003;290:1062–70. doi: 10.1001/jama.290.8.1062. [DOI] [PubMed] [Google Scholar]
- 22.Food and Drug Administration. The National Drug Code Directory. 2005. Available at http://www.fda.gov/cder/ndc/tbldclas.txt Last accessed 3 May 2006.
- 23.Easyphp. 2003. Version 1.7. Available at http://www.easyphp.org/index.php3 Last accessed 3 May 2006.
- 24.The Thomson Corporation. Journal Citation Reports. Available at http://portal.isiknowledge.com/portal.cgi?DestAp=JCR&Func=Frame&Init=Yes&SID=Z2BCiK9enDagCACa4AH Last accessed 3 May 2006.
- 25.Retout S, Mentré F. Optimisation of individual and population designs using Splus. J Pharmacokinet Pharmacodyn. 2003;30:417–43. doi: 10.1023/b:jopa.0000013000.59346.9a. [DOI] [PubMed] [Google Scholar]
- 26.Xie R, Mathijssen RH, Sparreboom A, Verweij J, Karlsson MO. Clinical pharmacokinetics of irinotecan and its metabolites: a population analysis. J Clin Oncol. 2002;20:3293–301. doi: 10.1200/JCO.2002.11.073. [DOI] [PubMed] [Google Scholar]
- 27.Kerbusch T, Wahlby U, Milligan PA, Karlsson MO. Population pharmacokinetic modelling of darifenacin and its hydroxylated metabolite using pooled data, incorporating saturable first-pass metabolism, CYP2D6 genotype and formulation-dependent bioavailability. Br J Clin Pharmacol. 2003;56:639–52. doi: 10.1046/j.1365-2125.2003.01967.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ette EI, Williams PJ. Population pharmacokinetics II. Estimation methods. Ann Pharmacother. 2004;38:1907–15. doi: 10.1345/aph.1E259. [DOI] [PubMed] [Google Scholar]
- 29.Sheiner LB, Beal SL. Evaluation of methods for estimating population pharmacokinetic parameters. III. Monoexponential model: routine clinical pharmacokinetic data. J Pharmacokinet Biopharm. 1983;11:303–19. doi: 10.1007/BF01061870. [DOI] [PubMed] [Google Scholar]
- 30.Bonate PL. Recommended reading in population pharmacokinetic pharmacodynamics. AAPS Pharmsci. 2005;7:E363–E373. doi: 10.1208/aapsj070237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Beal SL, Sheiner L. NONMEM Users Guide—Part VIIConditional Estimation Methods. San Francisco: University of California; 1998. p. 11. [Google Scholar]
- 32.Van Kesteren C, Mathot RA, Raymond E, Armand JP, Dittrich C, Dumez H, Roche H, Droz JP, Punt C, Ravic M, Wanders J, Beijnen JH, Fumoleau P, Schellens JH. Population pharmacokinetics of the novel anticancer agent E7070 during four phase I studies: model building and validation. J Clin Oncol. 2002;20:4065–73. doi: 10.1200/JCO.2002.01.005. [DOI] [PubMed] [Google Scholar]
- 33.Dartois C, Lemenuel-Diot A, Laveille C, Tranchand B, Tod M, Girard P. Evaluation of uncertainty parameters estimated by different population PK software and methods. J Pharmacokinet Pharmacodyn. 2007;34:289–311. doi: 10.1007/s10928-006-9046-9. [DOI] [PubMed] [Google Scholar]
- 34.Girard P, Nony P, Boissel JP. The place of simultaneous pharmacokinetic pharmacodynamic modeling in new drug development: trends and perspectives. Fundam Clin Pharmacol. 1990;4(Suppl. 2):103s–115s. doi: 10.1111/j.1472-8206.1990.tb00068.x. [DOI] [PubMed] [Google Scholar]
- 35.Williams PJ, Ette EI. Determination of model appropriateness. In: Hui CK, Duffull S, editors. Simulation for Designing Clinical Trials. A Pharmacokinetic–Pharmacodynamic Modeling Perspective. New York: Marcel Dekker; 1997. pp. 73–104. [Google Scholar]