

Abstract
Coproporphyrinogen oxidase (CPO) is the sixth enzyme in the heme biosynthetic pathway, catalyzing two sequential oxidative decarboxylations of propionate moieties on coproporphyrinogen-III forming protoporphyrinogen-IX through a monovinyl intermediate, harderoporphyrinogen. Site-directed mutagenesis studies were carried out on three invariant amino acids, aspartate 400, arginine 262, and arginine 401, to determine residue contribution to substrate binding and/or catalysis by human recombinant CPO. Kinetic analyses were performed on mutant enzymes incubated with three substrates, coproporphyrinogen-III, harderoporphyrinogen, or mesoporphyrinogen-VI, in order to determine catalytic ability to perform the first and/or second oxidative decarboxylation. When Asp400 was mutated to alanine no divinyl product was detected, but the production of a small amount of monovinyl product suggested the Km value for coproporphyrinogen-III did not change significantly compared to the wild-type enzyme. Upon mutation of Arg262 to alanine, CPO was again a poor catalyst for the production of a divinyl product, with a catalytic efficiency <0.01% compared to wild-type, including a 15-fold higher Km for coproporphyrinogen-III. The efficiency of divinyl product formation for mutant enzyme Arg401Ala was ∼3% compared to wild-type CPO, with a threefold increase in the Km value for coproporphyrinogen-III. These data suggest Asp400, Arg262, and Arg401 are active site amino acids critical for substrate binding and/or catalysis. Possible roles for arginine 262 and 401 include coordination of carboxylate groups of coproporphyrinogen-III, while aspartate 400 may initiate deprotonation of substrate, resulting in an oxidative decarboxylation.
Keywords: coproporphyrinogen oxidase, enzyme kinetics, catalytic aspartate
The critical roles of heme and other related metalloporphyrins are seen throughout nature as prosthetic groups crucial to the function of many proteins (Jordan 1991). In hemoglobin and myoglobin, heme is critical for their function of oxygen transport and storage, respectively. In addition, the heme-containing cytochromes function in electron transport in the mitochondrial membrane of eukaryotic cells. The biosynthesis of heme in mammals occurs through a pathway involving eight enzyme-catalyzed reactions (Jordan 1991). The heme biosynthetic pathway starts with glycine and succinyl coenzyme A, ending with a molecule of heme containing a coordinated ferrous (Fe2+) ion bound to the four pyrrolic nitrogens (Jordan et al. 1972). Coproporphyrinogen oxidase (CPO, E.C. 1.3.3.3) is the sixth enzyme in this heme biosynthetic pathway and catalyzes the conversion of coproporphyrinogen-III (copro'gen-III) to protoporphyrinogen-IX (proto'gen-IX), a divinyl product, via two sequential oxidative decarboxylations of the A and B ring propionates (Fig. 1) (Jordan et al. 1972). The conversion of copro'gen-III to proto'gen-IX occurs through the well-established monovinyl intermediate harderoporphyrinogen (hardero'gen), and molecular oxygen is required for the catalysis in aerobes (Kennedy et al. 1970; Jackson et al. 1980). Mesoporphyrinogen-VI (meso'gen-VI) has been shown to be a substrate for this enzyme and undergoes only the first oxidative decarboxylation (Fig. 1) (Lash et al. 1994, 1999; Jones et al. 2002). The mechanism by which CPO performs catalysis is poorly understood, in part, due to the lack of knowledge about the amino acid residues involved in active site binding and catalysis. It has been shown, however, that recombinant human CPO does not require a metal cofactor for catalysis (Medlock and Dailey 1996). CPO exists predominately as a homodimer of ∼70–75 kDa with a monomeric size of ∼35–37.5 kDa, and mammalian CPO is located in the mitochondria (Medlock and Dailey 1996).
Figure 1.

Metabolism of porphyrinogens by CPO. Both copro'gen-III and meso'gen-VI are converted to monovinyl product. If R = P, hardero'gen is converted to divinyl product proto'gen-IX, if R = Et, a second oxidative decarboxylation is not performed.
The elucidation of the three-dimensional crystal structure of coproporphyrinogen oxidase from Saccharomyces cerevisiae (Phillips et al. 2004) and Homo sapiens (Lee et al. 2005) has allowed for analysis of structure–function relationships. The human and yeast forms of oxygen-dependent coproporphyrinogen oxidase each crystallized as homodimers and exhibit comparable tertiary topology. From the crystallographic data, each monomer of coproporphyrinogen oxidase contains a seven-stranded β-sheet surrounded by 10 α-helices. Upon dimerization, the β8 moieties from each monomer interact to form a two-stranded antiparallel sheet at the middle of the dimer interface, forming key contacts for dimer stabilization. The contact surface of each monomer is comprised mainly of nonpolar amino acids, providing the foundation for a hydrophobic interface. Several conserved amino acids are located in a deep cleft, forming the putative active site of CPO, with each monomer housing its own active site (Phillips et al. 2004; Lee et al. 2005). Using the yeast enzyme crystal structure, the substrate copro'gen-III was modeled into the cleft to locate critical amino acids (Phillips et al. 2004). The human enzyme crystallized with a molecule of citrate in the cleft, which helped probe surrounding residues in order to propose an active site (Lee et al. 2005). The cleft is formed by one side of the β-sheet and helices α7–α9, with α2 located above the cleft, suggesting its role as a lid that can open and close, varying the accessibility of the active site to bulk solvent.
The conserved residues aspartate 400, arginine 401, and arginine 262 (Fig. 2) of human CPO are all thought to be contained within the proposed active site of CPO based on three-dimensional structure analysis (Fig. 3). To further the understanding of catalysis performed by coproporphyrinogen oxidase, it is important to determine if these amino acids are involved in either binding of substrate, maintenance of active site architecture, or the catalytic mechanism. Therefore, we conducted site-directed mutagenesis studies to replace each amino acid with alanine. Upon protein isolation, kinetic analyses were performed on mutated recombinant enzyme in order to determine the kinetic parameters Km, k cat and k cat/Km. Kinetic analysis of substrates with each mutated enzyme will give a more thorough understanding of the necessity of each amino acid for substrate binding or catalysis in the first, second, or both oxidative decarboxylations catalyzed by coproporphyrinogen oxidase. The ability to determine which amino acids are required for binding and catalysis will give insights into the mechanism of CPO.
Figure 2.

Alignment of amino acid sequences of coproporphyrinogen oxidase from representative organisms. Boxed in black are identical amino acids and boxed in gray are similar amino acids. The conserved arginine 262, aspartate 400, and arginine 401 residues are marked with an asterisk. The amino acid sequence from amino acids 182–441 of human CPO (Homo sapiens; accession BAA04033) is compared to CPO from Arabidopsis thaliana (accession AAF86536), Chlamydomonas reinhardtii (accession AAD28475), Drosophila melanogaster (accession AAD46837), Saccharomyces cerevisiae (accession CAA89966), Escherichia coli (accession BAA16325), and Synechocystis sp. (accession BAA16863).
Figure 3.

Three-dimensional representation of the proposed active site of human CPO. Shown is one monomer of the dimeric enzyme. The distance from the guanidinyl nitrogen of Arg262 to the carboxyl carbon of Asp400 (A), the guanidinyl nitrogen of Arg262 to the guanidinyl carbon of Arg401 (B), and the carboxyl carbon of Asp400 to the guanidinyl carbon of Arg401 (C) are shown. Molecular graphics images were produced using the UCSF Chimera package from the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (supported by NIH P41 RR-01081) (Pettersen et al. 2004). Coordinates, PDB accession code 2AEX, were obtained from the Protein Data Bank (Lee et al. 2005).
Results
Wild-type recombinant human CPO and each of the purified mutant enzymes were assayed in the presence of increasing concentrations of three substrates, copro'gen-III, the natural substrate which undergoes both the first and second oxidative decarboxylations, meso'gen-VI, which undergoes only the first oxidative decarboxylation, and hardero'gen, which undergoes only the second decarboxylation. The dependence of catalytic rate on substrate concentration is illustrated in Figures 4 and 5, and the resulting kinetic parameters are summarized in Table 1.
Figure 4.

Initial velocity as a function of [S] plot for wild-type incubated with copro'gen-III showing kinetic parameters for total product accumulation (A), divinyl product accumulation (B), and monovinyl product after incubation with meso'gen-VI (C). Data were collected and analyzed as described in Materials and Methods. Kinetic data were analyzed by nonlinear regression analysis using SigmaPlot (Systat Software, Inc.) and are averages of triplicate determinations.
Figure 5.

Initial velocity as a function of [S] plot for mutant enzymes Asp400Ala incubated with copro'gen-III (A). Only the monovinyl product was detected; thus, monovinyl product kinetic parameters are equal to that of total product parameters for Asp400Ala. Arg262Ala incubated with copro'gen-III showing kinetic parameters for total product accumulation (B) and divinyl product accumulation (C). Arg401Ala incubated with copro'gen-III for total product accumulation (D) and divinyl product accumulation (E), or monovinyl product accumulation from incubation with meso'gen-VI (F). Data were collected and analyzed as described in Materials and Methods. Kinetic data were analyzed by nonlinear regression analysis using SigmaPlot and are averages of triplicate determinations.
Table 1.
Kinetic parameters of purified mutant and wild-type CPO enzymes

Aspartate 400 is important for efficient catalysis
Mutation of the conserved aspartate 400 to alanine severely impaired the catalytic ability of recombinant human CPO relative to wild-type enzyme. Incubation of Asp400Ala with copro'gen-III yielded small amounts of the monovinyl product, hardero'gen, with no detectable conversion to the divinyl product proto'gen-IX. Taking into account the sensitivity of product detection (∼2 nM), the amount of purified enzyme used in the assays, and the time of incubation (up to 2 h), the k cat for divinyl product formation for Asp400Ala is <0.0005% compared to wild-type CPO. As a result, the total product accumulated is equal to that of monovinyl production. There was also no detectable product formation when Asp400Ala was incubated with either meso'gen-VI, which undergoes just the first oxidative decarboxylation, or hardero'gen, which undergoes just the second oxidative decarboxylation. When considering total product formation, the Km value for Asp400Ala of 0.78 μM is essentially identical to the 0.79 μM value for wild-type enzyme (Table 1). The catalytic efficiency (k cat/Km) of Asp400Ala is just 0.047% of wild-type total product production, due to a 2250-fold decrease in k cat. When taking into account that the molar amount of hardero'gen formed is less then the molar amount of CPO present within the assay, and that no divinyl product is being formed, Asp400Ala is severely catalytically compromised, and is performing, on average, less than a single turnover.
Catalysis by arginine 262 and arginine 401 mutant enzymes
Mutation of arginine 262 and arginine 401 to alanine resulted in both decreased k cat values and increased Km values when compared to wild-type enzyme values using copro'gen-III as a substrate (Table 1). Replacement of Arg401 by Ala resulted in an increase in the apparent Km value for copro'gen-III of approximately threefold and a decrease in the k cat value of 16-fold for production of proto'gen-IX. Mutant enzyme Arg401Ala was able to convert meso'gen-VI to monovinyl product, although over 10-fold less efficiently than wild-type CPO, due to a fourfold higher Km and a threefold lower k cat (Table 1). Mutation of arginine 262 to alanine, however, had even more severe consequences for catalysis by CPO. The catalytic efficiency of Arg262Ala was drastically decreased, with a k cat/Km value of 0.0036%, for divinyl product formation, relative to wild-type CPO. This decrease in efficiency is attributed to a 15-fold increase in the Km for copro'gen-III and k cat decrease over 2000-fold. In contrast to Arg401Ala, no product formation was detected when mutant enzyme Arg262Ala was incubated with meso'gen-VI (Table 1). Whereas wild-type CPO is able to catalyze the conversion of hardero'gen to proto'gen-IX (J.R. Stephenson, C.L. Cooper, A-S I. M. Keck, T.D. Lash, and M.A. Jones, in prep.), we were unable to attain saturating conditions of hardero'gen with either Arg262Ala or Arg401Ala, suggesting increased Km values in each case (data not shown). When comparing total product formation using copro'gen-III, which accounts for the sum of monovinyl and divinyl products, Arg262Ala has an 11-fold higher Km value relative to that of wild-type CPO and a turnover number ∼1% of the wild-type value. Arg401Ala exhibited an 11-fold increase in Km and a k cat value ∼44% of the wild-type value (Table 2).
Table 2.
Ratio of monovinyl to divinyl products for purified mutant and wild-type CPO enzymes using copro'gen-III

Discussion
Conservation of primary structure in CPO
In a multiple sequence alignment comparing CPO sequences from mammals, plants, and bacteria multiple regions of conservation are apparent, making it difficult to predict individual amino acids that are critical for catalysis. The recently reported three-dimensional structures of CPO from yeast (Phillips et al. 2004) and humans (Lee et al. 2005) have provided information to help focus on a handful of potentially critical active site amino acids. Among these amino acids are arginine 262, aspartate 400, and arginine 401, amino acids that are conserved across species (Fig. 2). In Figure 2 as well as in a more extensive amino acid sequence alignment of ∼20 putative oxygen-dependent CPO sequences from the protein database, arginine 262, aspartate 400, and arginine 401 are absolutely conserved.
Catalysis by uroporphyrinogen decarboxylase and porphobilinogen deaminase
Phillips et al. (2003) reported that uroporphyrinogen decarboxylase (URO-D), the enzyme that precedes CPO in the heme biosynthetic pathway, contains a catalytic aspartate residue which coordinates the four pyrrole NH groups of copro'gen-III in the three-dimensional structure. This coordination gives rise to a bowl-shaped conformation in which the pyrrolic NH groups are pointing down toward the invariant aspartate and the pyrrole rings are angled upwards. Porphobilinogen deaminase, the third enzyme in the heme biosynthetic pathway, has also been shown to contain a catalytic aspartate residue (Woodcock and Jordan 1994). Thus, the conserved aspartate 400 in CPO was selected as an important amino acid for site-directed mutagenesis studies. Likewise, the positively charged functional group of arginine makes this residue an ideal candidate for interaction with the anionic propionate moieties of copro'gen-III. It is well established that arginine residues can serve as positively charged recognition sites for anionic substrates (Riordan et al. 1977), namely the carboxylate ion moieties of copro'gen-III.
Properties of porphyrinogen molecules
It should be noted that porphyrinogens are conformationally mobile molecules. Using octamethylporphyrinogen, von Maltzan (1982) has shown that these molecules contain four nonplanar strain-free conformations, including chair, saddle, chaise longe, and the bowl forms. Although the saddle, chair, and chaise longe conformations are the most favored in solution, crystal structures of related calix[4]pyrroles show the bowl conformation was most favored in the presence of anionic binding interactions (Gale et al. 1996). This, along with the URO-D crystal structure, is consistent with a model in which aspartate 400 interacts with the pyrrolic NH groups to stabilize the bowl conformation within the active site of CPO. These data also correlate to the crystallographic data of the yeast enzyme in which the authors proposed that aspartate 274, which aligns with aspartate 400 of human CPO, was coordinating to the four pyrrolic hydrogens.
Proposed catalytic mechanism for CPO
Recently, a mechanism was proposed by Lash (2005) in which initial deprotonation of the pyrrole NH produces a species with azacyclopentadienyl anion character. The electron-rich α-carbon can then react with molecular oxygen to form a transient peroxide anion that triggers the decarboxylation of the propionate moiety. This study sought to identify active site amino acids that could be implicated in the catalytic mechanism of CPO.
Catalytic contribution of aspartate 400
Wild-type human CPO performs the second oxidative decarboxylation more readily than the first with the k cat value for hardero'gen about 30 times higher than that using meso'gen-VI (J.B. Morgenthaler, R.L. Barto, T.D. Lash, and M.A. Jones, in prep.). However, the Asp400Ala mutant enzyme was unable to produce divinyl product. In addition, this mutant enzyme was an extremely poor catalyst for the first oxidative decarboxylation, producing a terminal monovinyl product at a rate <0.04% of the rate at which wild-type CPO catalyzes divinyl production. Thus, due to the large decrease in catalytic efficiency with negligible changes in the Km for copro'gen-III, it is highly likely aspartate 400 is a central component in the catalytic mechanism. Such a role is supported by the crystal structure of uroporphyrinogen decarboxylase (URO-D), the enzyme proceeding CPO in the heme pathway, which was cocrystallized with its product (substrate for CPO) copro'gen-III (Phillips et al. 2003). In the URO-D structure, the pyrrole units are folded upward from the plane forming a bowl shape, and thus the central pyrrolic NH groups point at the invariant aspartate 86 residue. The positive charge of the protonated pyrrole is stabilized by the negative charge of the aspartate residue, thus leading to the protonation of a pyrrole Cα atom (Phillips et al. 2003). In the case of CPO, following the mechanism proposed by Lash (2005), it can be postulated that the anionic aspartate 400 is also coordinating the pyrrolic nitrogens at the bottom of the bowl, thus leading to the deprotonation of the pyrrole NH and subsequent base-catalyzed oxidative decarboxylation of copro'gen-III.
Possible roles of arginine 262 and arginine 401 in the active site of CPO
The positive charge of an arginine side chain makes this amino acid a likely candidate for interaction with the negative propionate side chains of copro'gen-III in the enzyme active site. Analysis of the three-dimensional structure of the proposed active site of coproporphyrinogen oxidase (Phillips et al. 2004) implicated arginine 262 and arginine 401 as prospective binding candidates, prompting our site-directed mutagenesis studies. Upon mutation of Arg262 and Arg401, copro'gen-III Km values increased 15-fold and threefold, respectively. Arg401Ala retained significant catalytic function, with k cat values for proto'gen-IX production at 6% and total product formation at 44% of the wild-type rate. In addition, the arginine 401 mutation did not attenuate usage of meso'gen-VI. However, not only could Arg262Ala not catalyze a divinyl product from meso'gen-VI, it had a catalytic efficiency for divinyl production from copro'gen-III of <0.01% of the wild-type k cat/Km value. These data suggest the side chain of Arg262 may be at a location within the active site to participate more directly in oxidative decarboxylation than Arg401. Also, due to the difference in catalytic ability to perform an oxidative decarboxylation on meso'gen-VI, it can be postulated that the two arginines are binding substrate at separate, yet fundamentally necessary, propionate moieties. The absence of propionate groups on the C and D rings of meso'gen-VI only allows the formation of a monovinyl product (Lash et al. 1994, 1999; Jones et al. 2002). The ability of Arg401Ala to perform an oxidative decarboxylation on meso'gen-VI suggests that a mutation to alanine was not detrimental to binding. Thus, arginine 401 is most likely coordinating to one or both of these C or D ring propionates, which are less important in substrate positioning in the active site, since the mutation still allowed for catalysis of the oxidative decarboxylation, although not as efficiently as wild-type CPO. However, Arg262Ala was unable to perform an oxidative decarboxylation on meso'gen-VI, indicating that arginine 262 is very important to the binding of one or both of the propionate moieties on the A or B ring of meso'gen-VI and mutation to alanine leads to insufficient binding.
Refinement of the active site model for CPO
Through studies with porphyrinogen analogs using chicken red cell hemolysates as the enzyme source, it has been shown that modifications of B and C ring propionates reduce catalytic ability of CPO (Elder et al. 1978; Jackson et al. 1978, Yoshinaga and Sano 1980; Lash et al. 1999; Jones et al. 2002). This led to a model in which a binding site was necessary for the B ring propionate, which positioned the A ring propionate correctly for the first oxidative decarboxylation. Upon formation of monovinyl product, a binding site for the C ring propionate is necessary for the second oxidative decarboxylation to take place. Both oxidative decarboxylations are proposed to occur while the substrate is within the active site. This sequestering of the intermediate within the active site could help keep the intermediate stabilized in the reduced form to protect from oxidizing species produced during the reaction. Therefore, it can be speculated that within the dimeric enzyme, each monomer's active site performs independent catalysis of copro'gen-III to proto'gen-IX.
These studies, coupled with the kinetic parameters presented in this study, have allowed for the development of a more detailed model in which arginine 262 is involved in a binding site for the B ring propionate, and arginine 401 is the binding partner for the propionate on the C ring. The binding of copro'gen-III to these arginines puts the substrate in the correct position for the central pyrrole NH groups to coordinate with aspartate 400, allowing for the stabilization of the bowl conformation for the porphyrinogen. This interaction leads to deprotonation of the A ring NH and subsequent oxidative decarboxylation (Fig. 6).
Figure 6.
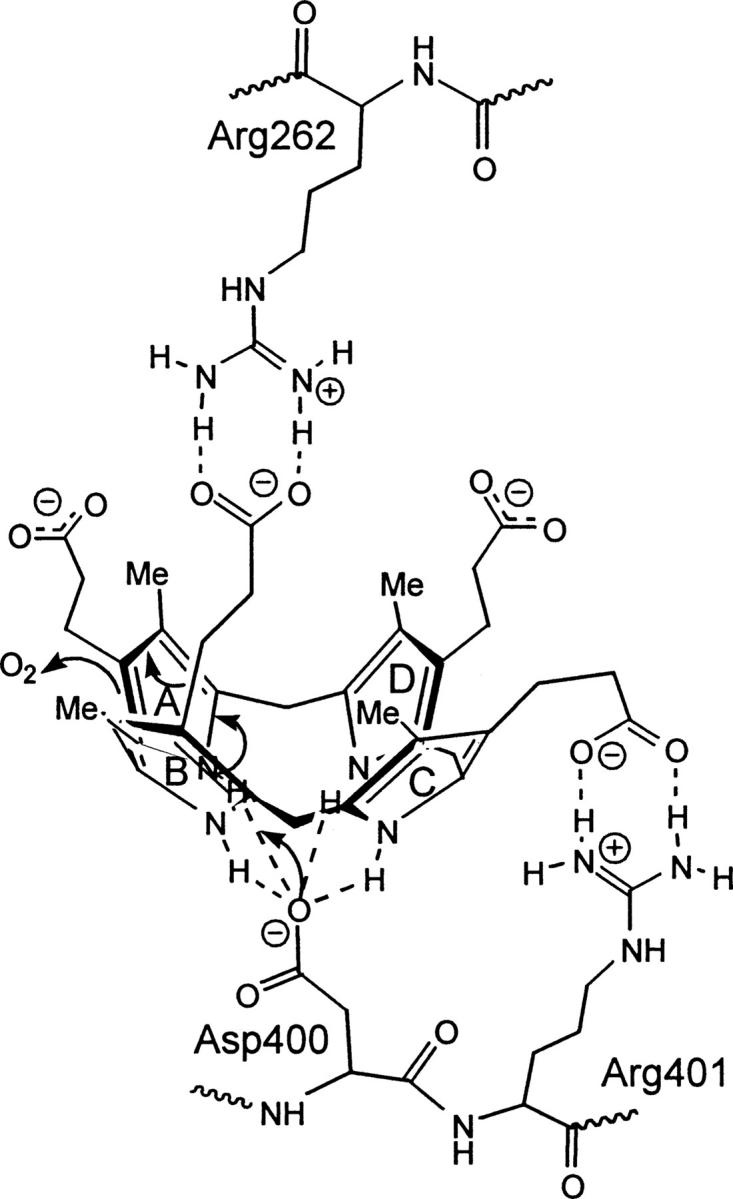
Proposed CPO active site interactions of copro'gen-III with arginine 262, aspartate 400, and arginine 401. Arginine 262 forms key interactions with the propionate moiety on ring B and arginine 401 forms key interactions with the propionate on ring C of copro'gen-III. The anionic residue, aspartate 400, is proposed to play a role in catalysis by coordinating to the central pyrrolic NH groups to stabilize a bowl conformation of copro'gen-III, thus leading to initial deprotonation of the A ring and subsequent oxidative decarboxylation. A speculated binding partner or role for ring D has not yet been determined.
Effect of mutation on relative rates of the first and second oxidative decarboxylations
During catalysis utilizing copro'gen-III, a certain amount of the intermediate hardero'gen accumulates and is detected by HPLC. For the wild-type enzyme under initial velocity and saturating concentrations of copro'gen-III, the ratio of monovinyl product, hardero'gen, to divinyl product, proto'gen-IX, is ∼0.50 (Table 2). Upon mutation of the putative active site amino acids Asp400, Arg262, and Arg401, however, the ratio of monovinyl to divinyl product changes drastically. In the case of mutant enzyme Asp400Ala, this ratio cannot be calculated since no detectable divinyl product was produced. For Arg262Ala and Arg401Ala mutant enzymes the monovinyl intermediate accumulates to a much higher concentration, with monovinyl/divinyl ratios of 32 and 12, respectively (Table 2). These data suggest that upon mutation the rate of the second oxidative decarboxylation is decreasing relative to the first oxidative decarboxylation.
CPO mutations associated with porphyria
Hereditary coproporphyria (HCP) is a genetic disorder of heme biosynthesis characterized by the excretion of coproporphyrin-III in the feces of afflicted patients (Nordmann et al. 1983). The primary gene defect causing HCP encodes the enzyme CPO in which the activity of this enzyme is greatly reduced (Nordmann et al. 1983). Symptoms associated with HCP include photosensitivity, psychological disturbances, and abdominal pain (Elder et al. 1997). Chromosome 9 contains the gene encoding human coproporphyrinogen oxidase (Cacheux et al. 1994), and >20 different mutations in this gene causing hereditary coproporphyria have so far been identified (Martasek 1998), including mutation of two arginines (Martasek et al. 1997; Lamoril et al. 2001), one of which was investigated further in this study. It is of interest that of the three amino acids selected for mutagenesis studies, only arginine 401 has been reported as mutated in porphyric patients (Lamoril et al. 2001). An arginine 401 to tryptophan mutation in CPO was identified in a patient and was shown to possess similar functionality as a Lys304Glu mutation involved in harderoporphyria (Lamoril et al. 1995), which is characterized by a buildup of harderoporphyrin in the feces. This report is consistent with the data presented here in that the mutation of Arg401Ala resulted in accumulation of the monovinyl intermediate. We have thus proposed a role of arginine 401 in binding of substrate. The ability of the Arg401Ala mutant to perform catalysis to a greater extent than Asp400Ala and Arg262Ala supports the finding of this mutation in the population, as it is not as detrimental to overall catalysis, whereas mutation of aspartate 400 or arginine 262 may be considered highly lethal, supporting their importance in the enzyme mechanism.
Summary
The data presented here have allowed for the extension of a model for the unusual oxidative decarboxylations carried out by the essential enzyme coproporphyrinogen oxidase in the synthesis of heme. In addition, the data provide a biochemical perspective to help define a role for a documented mutation in porphyric patients. Due to the complexity of the large porphyrinogen substrates utilized by CPO, it is likely there are additional amino acids critical for binding and catalysis. Further mutagenesis studies are currently in progress on additional potential active site residues to determine their contribution to catalysis and/or binding of substrate in CPO.
Materials and methods
Construction of expression vector pET21d–CPO
Large-scale production of recombinant CPO was performed using the pET-inducible protein expression system. The NcoI/HindIII fragment from the expression vector pHHCPO (a generous gift from Dr. Harry Dailey, University of Georgia) was subcloned into the multiple cloning site of pET21d (Novagen), resulting in expression of the cDNA encoding CPO under the control of the lac operon. Overexpression of recombinant CPO was induced with isopropyl-β-D-thiogalactopyranoside (IPTG).
Site-directed mutagenesis
Site-directed mutagenesis was performed using the PCR-mediated overlap extension method (Horton et al. 1989) utilizing Platinum Pfx DNA Polymerase (Invitrogen). Three universal oligonucleotides and each mutagenic nucleotide (Integrated DNA Technologies) were used, resulting in recombinant human coproporphyrinogen oxidase mutants (Table 3). In each case, the conserved amino acid was changed to alanine. Oligonucleotide #1 has the sequence 5′-CGCGGATCCATGGCTCACCATCACCATCACC-3′ and contained an NcoI restriction enzyme site (underlined). Oligonucleotide #2 had the sequence 5′-TTGCAATACGGAACTTCCAGAATTTCAGCTTCTTTGG-3′ and contained a 5′ noncomplementary overhang of 10 nucleotides. Oligonucleotide #3 had the sequence 5′-GGGGTACCAAGCTTATTCTGCCTGCATCAACGC-3′ and contained a HindIII restriction enzyme site (underlined). The encoded wild-type CPO lacked the mitochondrial-targeting signal located on 100 amino acids at the amino terminus. The sequence Met-Ala-His-His-His-His-His-His was encoded at the amino terminus prior to proline 34 to serve as a 6x-His-Tag for affinity purification using a metal affinity column. Restriction endonucleases NcoI and HindIII were used to digest each PCR product, which were then ligated with NcoI and HindIII digested pET21d. The cDNA of each mutant was sequenced to verify the mutation using automated fluorescent sequencing on a 3130 Genetic Analyzer using a BigDye terminator DNA sequencing kit from Applied Biosystems.
Table 3.
Wild-type CPO cDNA sequence aligned with mutagenic oligonucleotides for Arg262, Asp400, and Arg401

Expression and purification of His-tagged wild-type and mutant CPO proteins
Escherichia coli strain BL21(DE3)RIL (Stratagene) was used as the expression host. One liter cultures containing pET21d–CPO were grown in a shaking incubator at 37°C and 300 rpm to an optical density of 0.8–1.0 at 600 nm in Luria Broth medium with antibiotics (ampicillin and chloramphenicol at 100 μg/mL and 34 μg/mL, respectively). Overexpression of protein was induced by the addition of 1 mM IPTG followed by incubation at 37°C with 300 rpm shaking for ∼3 h. E. coli cells expressing either wild-type or mutant forms of CPO were harvested by centrifugation at 4620g for 5 min. Bacterial cell pellets were resuspended in 40 mL of CPO resuspension buffer (50 mM NaH2PO4, 300 mM NaCl, 50 mM Tris at pH 8) and lysed using a French Pressure Cell at 20, 000 psi with the addition of phenylmethylsulfonylfluoride as a protease inhibitor. The lysed cells were centrifuged at 27,200g for 30 min and the supernatant was passed through a 4-mL column of Ni-NTA agarose metal affinity resin (Qiagen). The column was washed with resuspension buffer containing 20 mM imidazole and desired protein was eluted with resuspension buffer containing 250 mM imidazole at a final pH of 7.0. The protein concentration of each fraction was determined by the method of Bradford (1976) using a Bio-Rad Protein Assay kit and BSA as a standard, and purification was evaluated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (Fig. 7).
Figure 7.

SDS-PAGE (12% polyacrylamide) of purified wild-type CPO, Arg262Ala, Asp400Ala, and Arg401Ala. Approximately 2–3 μg of protein were added per lane.
Substrate preparation
Three different substrates were used for kinetic analysis of recombinant enzymes. The use of these three substrates allowed for the relative comparison between the ability of wild-type and mutant enzymes to perform the first and second oxidative decarboxylations. Each substrate was prepared from the corresponding porphyrin methyl esters by incubation overnight with 8.3 M HCl and reduction with 3% sodium amalgam as previously reported (Lash et al. 1999; Jones et al. 2002). Coproporphyrin-III tetramethyl ester was purchased from Aldrich Chemical. Harderoporphyrin trimethyl ester and mesoporphyrin-VI dimethyl ester were previously synthesized (Lash et al. 1999, 2001, 2002). The reduced porphyrinogens were used directly after reduction and concentrations were quantitated spectrophotometrically at 406 nm in 8.3 M HCl.
Enzyme assay
Recombinant wild-type and mutant CPO were assayed at 37°C using the microprocedure previously reported (Jones et al. 2003). Following incubation and extraction, the porphyrins were converted to their respective methyl esters and separated by HPLC using a normal phase silica column and a mobile phase of ethyl acetate:cyclohexane (35:65 [v/v] for coproporphyrin-III and harderoporphyrin and 25:75 [v/v] for mesoporphyrin-VI). Porphyrins were detected at 404 nm, and amount of product was calculated from the integrated peak areas. Initial velocity (Vo) was evaluated for each mutant with each substrate using incubation times ranging from 15 sec to 20 min. For kinetic analysis, both wild-type and mutant enzymes were assayed under apparent initial velocity conditions with the three substrates at various concentrations from 0.05 to 25 μM. Kinetic data were performed in triplicate and analyzed by nonlinear regression analysis using SigmaPlot. Data are reported for formation of monovinyl intermediate, divinyl product, and the sum of these (indicated as total product) for both wild-type and mutant enzymes and kinetic parameters are given in Table 1.
Acknowledgments
We thank Dr. Harry Dailey, University of Georgia, for the generous gift of plasmid pHHCPO. This work was supported by Sigma Xi Grant-In-Aid of Research and Abbott Laboratories Graduate Fellowship to J.R.S. and Illinois State University Research Grant to M.A.J.
Footnotes
Reprint requests to: Marjorie A. Jones, Department of Chemistry, Illinois State University, Normal, IL 61790-4160, USA; e-mail: majone3@ilstu.edu; fax: (309) 438-5538.
Abbreviations: CPO, coproporphyrinogen oxidase; copro'gen-III, coproporphyrinogen-III; proto'gen-IX, protoporphyrinogen-IX; hardero'gen, harderoporphyrinogen; meso'gen-VI, mesoporphyrinogen-VI.
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.062636907.
References
- Bradford M.M.. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72: 248–254. [DOI] [PubMed] [Google Scholar]
- Cacheux V., Martasek, P., Fougerousse, F., Delfau, M., Druart, L., Tachdjian, G., and Grandchamp, B. 1994. Localization of the human coproporphyrinogen oxidase gene to chromosome band 3q12. Hum. Genet. 94: 557–559. [DOI] [PubMed] [Google Scholar]
- Elder G.H., Evans, J.O., Jackson, J.R., and Jackson, A.H. 1978. Factors determining the sequence of oxidative decarboxylation of the 2- and 4-propionate substituents of coproporphyrinogen III by coproporphyrinogen oxidase in rat liver. Biochem. J. 169: 215–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elder G.H., Hift, R.J., and Meissner, P.N. 1997. The acute porphyrias. Lancet 349: 1613–1617. [DOI] [PubMed] [Google Scholar]
- Gale P.A., Sessler, J.L., Kral, V., and Lynch, V. 1996. Calix[4]pyrroles: Old yet new anion-binding agents. J. Am. Chem. Soc. 118: 5140–5141. [Google Scholar]
- Horton R.M., Hunt, H.D., Ho, S.N., Pullen, J.K., and Pease, L.R. 1989. Engineering hybrid genes without the use of restriction enzymes; Gene splicing by overlap extension. Gene 77: 51–59. [DOI] [PubMed] [Google Scholar]
- Jackson A.H., Elder, G.H., and Smith, S.G. 1978. The metabolism of coproporphyrinogen-III into protoporphyrin-IX. Int. J. Biochem. 12: 877–882. [DOI] [PubMed] [Google Scholar]
- Jackson A.H., Jones, D.M., Philip, G., Lash, T.D., Battle, A.M., del, C., and Smith, S.G. 1980. Synthetic and biosynthetic studies of porphyrins, part IV. Further studies of the conversion of coproporphyrinogen-III to protoporphyrin-IX: Mass spectrometric investigations of the incubation of specifically deuteriated coproporphyrinogen-III with chicken red cell haemolysates. Int. J. Biochem. 12: 681–688. [DOI] [PubMed] [Google Scholar]
- Jones M.A., He, J., and Lash, T.D. 2002. Kinetic studies of novel di- and tri-propionate substrates for the chicken red blood cell enzyme coproporphyrinogen oxidase. J. Biochem. 131: 201–205. [DOI] [PubMed] [Google Scholar]
- Jones M.A., Thientanavanich, P., Anderson, M.D., and Lash, T.D. 2003. Comparison of two assay methods for activities of uroporphyrinogen decarboxylase and coproporphyrinogen oxidase. J. Biochem. Biophys. Methods 55: 241–249. [DOI] [PubMed] [Google Scholar]
- Jordan P.M., ed. 1991. Biosynthesis of tetrapyrroles. Elsevier, London.
- Jordan P.M., Shemin, D., and Boyer, P.D. 1972. The enzymes, Vol. vol. 7. Academic Press, New York.
- Kennedy G.Y., Jackson, A.H., Kenner, G.W., and Suckling, C.J. 1970. Isolation, structure and synthesis of a tricarboxylic porphyrin from harderian glands of rat. FEBS Lett. 6: 205–206. [DOI] [PubMed] [Google Scholar]
- Lamoril J., Martasek, P., Deybach, J.-C., De Silva, V., Grandchamp, B., and Nordmann, Y. 1995. A molecular defect in coproporphyria oxidase gene causing harderoporphyria, a variant form of hereditary coproporphyria. Hum. Mol. Genet. 4: 275–278. [DOI] [PubMed] [Google Scholar]
- Lamoril J., Puy, H., Whatley, S.D., Martin, C., Woolf, J.R., Da Silve, V., Deybach, J.-C., and Elder, G.H. 2001. Characterization of mutations in the CPO gene in British patients demonstrates absence of genotype-phenotype correlation and identifies relationship between hereditary coproporphyria and harderoporphyria. Am. J. Hum. Genet. 68: 1130–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lash T.D.. 2005. The enigma of coproporphyrinogen oxidase: How does this unusual enzyme carry out oxidative decarboxylations to afford vinyl groups? Bioorg. Med. Chem. Lett. 15: 4506–4509. [DOI] [PubMed] [Google Scholar]
- Lash T.D., Drinan, M.A., Zhen, C., Mani, U.N., and Jones, M.A. 1994. Synthetic substrates for coproporphyrinogen oxidase: Mesoporphyrinogen-VI revisited. Bioorg. Med. Chem. Lett. 4: 1607–1612. [Google Scholar]
- Lash T.D., Mani, U.N., Drinan, M.A., Zhen, C., Hall, T., and Jones, M.A. 1999. Normal and abnormal heme biosynthesis. 1. Synthesis and metabolism of di- and monocarboxylic porphyrinogens related to coproporphyrinogen-III and harderoporphyrinogen: A model for the active site of coproporphyrinogen oxidase. J. Org. Chem. 64: 464–477. [Google Scholar]
- Lash T.D., Hall, T., Mani, U.N., and Jones, M.A. 2001. Normal and abnormal heme biosynthesis. 3. Synthesis and metabolism of tripropionate analogues of coproporphyrinogen-III: Novel probes for the active site of coproporphyrinogen oxidase. J. Org. Chem. 66: 3753–3759. [DOI] [PubMed] [Google Scholar]
- Lash T.D., Keck, A.-S.I.M., Mani, U.N., and Jones, M.A. 2002. Unprecedented overmetabolism of a porphyrinogen substrate by coproporphyrinogen oxidase. Bioorg. Med. Chem. Lett. 12: 1079–1082. [DOI] [PubMed] [Google Scholar]
- Lee D.S., Flacksova, E., Bodnarova, M., Demeler, B., Martasek, P., and Raman, C.S. 2005. Structural basis of hereditary coproporphyria. Proc. Natl. Acad. Sci. 102: 14232–14237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martasek P.. 1998. Hereditary coproporphyria. Semin. Liver Dis. 18: 25–32. [DOI] [PubMed] [Google Scholar]
- Martasek P., Camadro, J.-M., Raman, C.S., Lecomte, M.C., Le Caer, J.P., Demeler, B., Grandchamp, B., and Labbe, P. 1997. Human coproporphyrinogen oxidase. Biochemical characterization of recombinant normal and R231W mutant enzymes expressed in E. coli as soluble, catalytically active homodimers. Cell. Mol. Biol. 43: 47–58. [PubMed] [Google Scholar]
- Medlock A.E. and Dailey, H.A. 1996. Human coproporphyrinogen oxidase is not a metalloprotein. J. Biol. Chem. 271: 32507–32510. [DOI] [PubMed] [Google Scholar]
- Nordmann Y., Grandchamp, B., De Verneuil, H., Phung, L., Cartigny, B., and Fontaine, G. 1983. Harderoporphyria: A varient hereditary coproporphyria. J. Clin. Invest. 72: 1139–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen E.F., Goddard, T.D., Huang, C.C., Couch, G.S., Greenblatt, D.M., Meng, E.C., and Ferrin, T.E. 2004. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 25: 1605–1612. [DOI] [PubMed] [Google Scholar]
- Phillips J.D., Whitby, F.G., Kushner, J.P., and Hill, C.P. 2003. Structural basis for tetrapyrrole coordination by uroporphyrinogen oxidase. EMBO J. 22: 6225–6233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips J.D., Whitby, F.G., Warby, C.A., Labbe, P., Yang, C., Pflugrath, J.W., Ferrara, J.D., Robinson, H.A., Kushner, J.P., and Hill, C.P. 2004. Crystal structure of the oxygen-dependant coproporphyrinogen oxidase (Hem13p) of Saccharomyces cerevisiae . J. Biol. Chem. 279: 38960–38968. [DOI] [PubMed] [Google Scholar]
- Riordan J.F., McElvany, K.D., and Borders, C.L. 1977. Arginyl residues: Anion recognition sites in enzymes. Science 195: 884–886. [DOI] [PubMed] [Google Scholar]
- von Maltzan B.. 1982. Synthesis of 2, 3, 7, 8, 12, 13, 17, 18-octamethylporphyrinogen in almost quantitative yield. Angew. Chem. Int. Ed. Engl. 21: 785–788. [Google Scholar]
- Woodcock S.C. and Jordan, P.M. 1994. Evidence for participation of aspartate-84 as a catalytic group at the active site of porphobilinogen deaminase obtained by site-directed mutagenesis of hemC gene from Escherichia coli . Biochemistry 33: 2688–2695. [DOI] [PubMed] [Google Scholar]
- Yoshinaga T. and Sano, S. 1980. Coproporphyrinogen oxidase. II. Reaction mechanism and role of tyrosine residues on the activity. J. Biol. Chem. 225: 4727–4731. [PubMed] [Google Scholar]