

Abstract
Leucyl/phenylalanyl-tRNA-protein transferase (L/F-transferase) is an N-end rule pathway enzyme, which catalyzes the transfer of Leu and Phe from aminoacyl-tRNAs to exposed N-terminal Arg or Lys residues of acceptor proteins. Here, we report the 1.6 Å resolution crystal structure of L/F-transferase (JW0868) from Escherichia coli, the first three-dimensional structure of an L/F-transferase. The L/F-transferase adopts a monomeric structure consisting of two domains that form a bilobate molecule. The N-terminal domain forms a small lobe with a novel fold. The large C-terminal domain has a highly conserved fold, which is observed in the GCN5-related N-acetyltransferase (GNAT) family. Most of the conserved residues of L/F-transferase reside in the central cavity, which exists at the interface between the N-terminal and C-terminal domains. A comparison of the structures of L/F-transferase and the bacterial peptidoglycan synthase FemX, indicated a structural homology in the C-terminal domain, and a similar domain interface region. Although the peptidyltransferase function is shared between the two proteins, the enzymatic mechanism would differ. The conserved residues in the central cavity of L/F-transferase suggest that this region is important for the enzyme catalysis.
Keywords: crystal structure, N-end rule pathway, proteolysis, GNAT superfamily fold
Leucyl/phenylalanyl-tRNA-protein transferase (EC: 2.3.2.6., L/F-transferase) (Kaji et al. 1965) is one of the essential enzymes controlling the half-lives of proteins in vivo in the N-end rule pathway (Tobias et al. 1991; Varshavsky 1992; Shrader et al. 1993). The two proteolytic systems, the Clp-dependent and the ubiquitin-dependent, are both referred to as the N-end rule pathways in prokaryotes and eukaryotes, respectively (Hwang et al. 1988; Katayama et al. 1988). In bacteria with the N-end rule pathway, the primary destabilizing N-terminal residues, Leu and Phe, are recognized by the ATP-dependent protease ClpAp (Tobias et al. 1991; Goldberg 1992). The analogous eukaryotic protein, arginyl-tRNA-protein transferase (ATE1, R-transferase), mediates arginyl transfer to Asp and Glu (Kaji et al. 1963). One of the primary destabilizing N-terminal residues, Arg, is recognized by the ubiquitin proteolytic system (Hochstrasser 1996; Haas and Siepmann 1997; Varshavsky 1997). Despite the enzymological similarities between the eukaryotic R-transferase and the prokaryotic L/F-transferase, there is no significant sequence similarity between them.
The L/F-transferase is encoded by the aat gene, which was first isolated from Escherichia coli (JW0868) (Leibowitz and Soffer 1970). It also exists in actinobacteria, cyanobacteria, proteobacteria, chlorobi, spirochaetes, and thermus-deinococcus (Fig. 1) and is widely distributed in eubacteria (Ichetovkin et al. 1997). L/F-transferase catalyzes the transfer of Leu and Phe from aminoacyl-tRNAs to exposed N-terminal Arg or Lys residues of acceptor proteins (Leibowitz and Soffer 1971).
Figure 1.
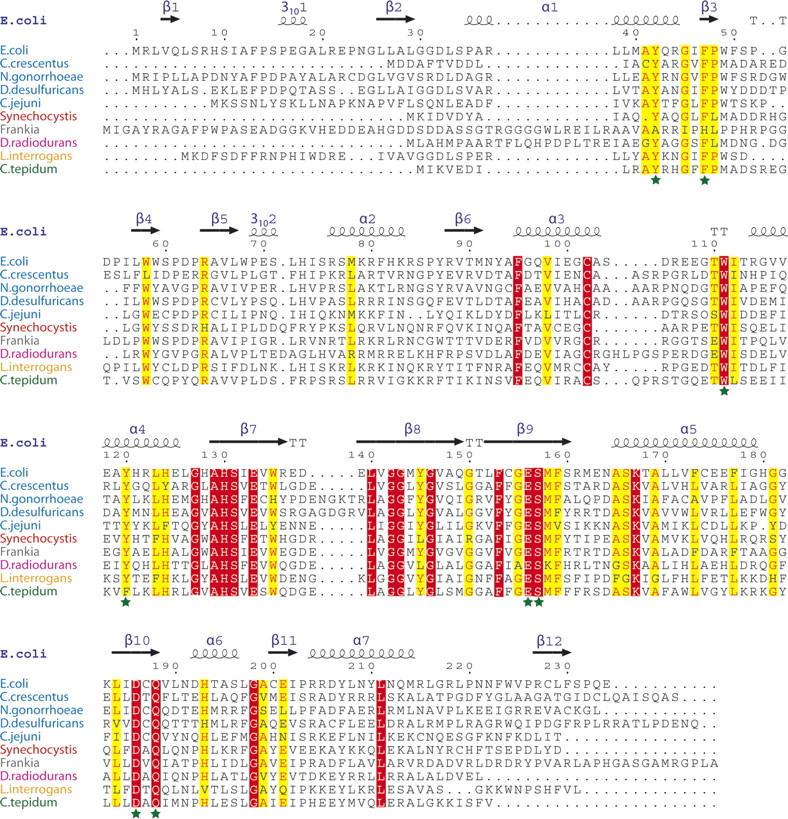
The secondary structure-based sequence alignment of L/F-transferase from E. coli. The classifications of proteobacteria, cyanobacteria, actinobacteria, thermus-deinococcus, spirochaetes, and chlorobi strains are shown with blue, red, gray, magenta, orange, and green letters, respectively. The highly conserved and completely conserved residues in all proteins are highlighted in yellow and red, respectively. The conserved residues in the cavity of L/F-transferase are indicated by dark green-colored stars.
Several previous studies have explored the biochemical and kinetic properties of L/F-transferase (Abramochkin and Shrader 1995, 1996). The L/F-transferase preferred Leu-, Phe-, and Met-tRNA as substrates, thus suggesting that an unbranched β-carbon and the side-chain hydrophobicity of the aminoacyl group of the aminoacyl-tRNA were recognized by the enzyme (Kaji et al. 1965; Scarpulla et al. 1976; Abramochkin and Shrader 1995). The research suggested that the recognition of the aminoacyl-tRNA by L/F-transferase started from the 5′ end to the single-stranded 3′-terminal CCA, where no base pairs were formed at all (Abramochkin and Shrader 1996).
The L/F-transferase enzymatic activity core domain encompasses ∼120 amino acids (Ichetovkin et al. 1997), but the critical residues that catalyze the peptidyltransferase reaction are still unclear. Here, we determined the crystal structure of E. coli L/F-transferase, the first three-dimensional structure of an L/F-transferase. The structure of L/F-transferase contains two domains and revealed the location of the enzyme catalytic region at the domain interface.
Results and Discussion
The L/F-transferase was crystallized by the hanging-drop vapor diffusion method. The L/F-transferase crystal belonged to the space group C2, with unit cell dimensions of a = 68.3 Å, b = 44.8 Å, and c = 76.4 Å. The structure of L/F-transferase was solved by the MAD method using the Se-Met crystal, and was refined to an R factor of 18.7% and an R-free factor of 21.7% to 1.6 Å resolution (Table 1). There was no observable electron density for the region encompassing residues 105–109, and the two C-terminal residues, and thus these residues were not included in the final structure. The L/F-transferase is a monomeric protein consisting of two domains that form a bilobate molecule, with dimensions of 54 × 32 × 34 Å (Fig. 2A). The smaller lobe of the molecule is formed by four β strands, with two parallel (β1, β2) and two anti-parallel (β3, β4), surrounded by an α helix (α1) and a 310-helix (3101) of the N-terminal domain (residues 1–63). The larger lobe of L/F-transferase comprises eight β strands with two parts of an anti-parallel β-sheet (β5, β10, β11; β6–β9, β12), and with six α helices (α2–α7) and a 310-helix (3102) (Fig. 2). An analysis of the molecular surface electrostatic potential indicated a long shallow cleft at the surface, running across the N-terminal and C-terminal domains. A large central cavity exists at the interface of the N-terminal and C-terminal domains, with the characteristic positive charge contributed by the corner of the cleft on the C-terminal domain surface (Fig. 3A), suggesting that they probably complement the negatively charged phosphate backbone of the substrate aminoacyl-tRNA. A structure database search was performed using the Dali server (Holm and Sander 1993). Three similar structures were found, including GCN5 histone acetyltransferase (Protein Data Bank [PDB] code 1PU9, Z-score = 10.3, sequence identity = 6%) (Clements et al. 2003), FemX transferase (PDB code 1XE4, Z-score = 9.8, sequence identity = 12%) (Maillard et al. 2005), and aminoglycoside 6′-N-acetyltransferase (PDB code 1S3Z, Z-score = 9.0, sequence identity = 12%) (Vetting et al. 2004). Although they lack significant sequence identity, the C-terminal domains of L/F-transferase and the others consist of the structurally conserved core region formed by four α helices and six β strands (α3–α6, β6–β11 in L/F-transferase), which is observed in the GCN5-related N-acetyltransferase (GNAT) family (Fig. 2B). In addition, the GNAT superfamily fold usually indicates the binding of acetyl-CoA, which donates the acetyl group that is transferred to a primary amine (Vetting et al. 2005).
Table 1.
X-ray crystallography statistics

Figure 2.

The structure of L/F transferase. (A) Stereo-view ribbon diagram of the overall structure of L/F-transferase. The N-terminal domain (1–63) is colored blue, and the C-terminal domain (64–232) is colored yellow. The part of the loop that is missing is marked by asterisks. (B) A cartoon demonstrating the folded topology of L/F-transferase. The orientation and the color scheme of the domains are the same as in the ribbon diagram. The conserved GNAT superfamily fold is colored pink.
Figure 3.

Molecular surface representation of L/F-transferase. The front view (A) and the back view (B) are colored by electrostatic potential (blue, +70KT; red, −70KT). The proposed substrate binding cleft is indicated by a gray line.
L/F-transferase and FemX catalyze similar peptidyltransferase reactions, involving the transfer of an uncharged amino acid from a donor aminoacyl-tRNA to an acceptor peptide. The FemX UDP-MurNAc-pentapeptide (UDP-MPP):L-alanine transferase from Weissella viridescens is a cell-wall peptidoglycan biosynthesis-related protein, which consists of two domains, and it has a long substrate binding cleft (Hegde and Shrader 2001; Biarrotte-Sorin et al. 2004). Based on their structural and peptidyltransferase reaction similarities, we superimposed the L/F-transferase and FemX structures. The N-terminal domains of the two structures indicated a very different structure, in which the N-terminal domain of L/F-transferase appeared to have a novel βαβαββ fold and to be smaller than that of FemX (Fig. 4). In contrast, the C-terminal domains of the two structures can be superimposed with a root-mean-square deviation (RMSD) of 2.7 Å for the structurally conserved GNAT superfamily fold (Fig. 4C; Dyda et al. 2000; Vetting et al. 2005). In addition, the two domains of the FemX structure share a similar fold structure, but the L/F-transferase did not exhibit the structural homology between the two domains. The FemX enzyme adds L-Ala to the ɛ-amino group of L-Lys (Fig. 4B, C) in the UDP-MPP. It is bound in the interface of the two domains, thus implying the existence of a catalytic site in this region. Although the L/F-transferase and FemX appear to utilize the different substrates, the similar structural features revealed that the two proteins may share the substrate binding and active regions.
Figure 4.

Structural comparison of the L/F-transferase and the FemX transferase. The conserved GNAT superfamily fold regions are shown in yellow and purple-blue color, respectively, in the structure of the C-terminal domain of L/F-transferase (A) and FemX transferase (B), belonging to the peptidyltransferase enzymes. The UDP-MPP bound in the FemX transferase structure is colored red and depicted as a ball-and-stick model. The L-Lys of UDP-MPP is shown in pale cyan. (C) Superposition of the overall backbone structures of L/F-transferase and FemX transferase (stereo view). The structures are indicated by a main-chain, with the N-terminal domains of L/F-transferase and FemX colored dark gray and gray, respectively. The C-terminal domains of L/F-transferase and FemX are colored as in A and B, respectively.
Thus, in the L/F-transferase structure, the domain interface region involves a broad space surrounded by α1 and β3 of the N-terminal domain, and α4, β5, β9, and β10 of the C-terminal domain (Fig. 2A). Most of the conserved residues are extensively distributed within the cleft of L/F-transferase. Especially, the conserved residues Tyr42, Phe47, Trp111, Tyr120, Glu156, Ser157, Asp186, and Gln188 are assembled in the central cavity of the long cleft (Figs. 1, 3A). Notably, the completely conserved residue Glu156 is located at the central position of the cavity, and its side-chain is directed toward Tyr42 and Tyr120, respectively belonging to α1 and α4, which are highly conserved in the L/F-transferase family (Fig. 1). Thus, Tyr42 is located within hydrogen-bonding distance to Tyr120 and Glu156. It seems that Tyr120 and Glu156 are structurally similar to the critical residues for UDP-MPP binding in the FemX complex structure (Hegde and Shrader 2001; Biarrotte-Sorin et al. 2004). The other completely conserved residues, Asp186 and Gln188, are located at the end of β5, and Asp186 is positioned within hydrogen-bonding distance to Glu156 via two water molecules (Fig. 5). Interestingly, two aromatic residues, Phe47 and Trp111, are located at the entrance of the cavity of the L/F-transferase, and both of their side-chains point toward the inside of the cavity, implying that the aromatic rings of the residues should be favorable for tRNA stacking. The domain interface region of L/F-transferase contains many key conserved residues that form a hydrogen-bond network, suggesting that the cavity region is important for enzyme catalysis.
Figure 5.

Detailed view of the domain interface region of L/F-transferase. The L/F-transferase structure is shown as a cartoon model, and the side-chains of the residues are shown as ball-and-stick models. The conserved residues of L/F-transferase are colored gray, and the two water molecules are shown by wheat-colored spheres. The hydrogen bonds are indicated by dashed black lines.
Materials and methods
Protein expression, purification, and crystallization
The selenomethionine-labeled L/F-transferase was expressed from the cloning vector pCA24N in E. coli strain B834(DE3) (Novagen). The cells were harvested and disrupted by sonication in 20 mM of Tris-HCl buffer (pH 8.0), containing 500 mM NaCl, 5 mM of 2-mercaptoethanol, and 1 mM of PMSF. The lysate was cleared by centrifugation for 30 min at 15,000 rpm. The supernatant was purified by two chromatography steps, on HiTrap Chelating HP5 and Hiload 16/60 Superdex 200 columns. The crystals of L/F-transferase were grown using the hanging-drop vapor diffusion method at 293 K. The protein solution was dialyzed against 20 mM of MES buffer (pH 6.0), containing 200 mM NaCl and 20 mM of DTT, and was concentrated to 1.1 mg/mL with an Amicon Ultra-15 Centrifugal Filter (Millipore). The best crystallization conditions employed a reservoir solution containing 100 mM of Tris-HCl buffer (pH 8.5), 200 mM NaCl, and 25% PEG 3350. The crystals were grown within 5 d to maximum dimensions of 0.1 × 0.1 × 0.2 mm.
Data collection, structure determination, and refinement
For data collection, all crystals were transferred to a cryoprotectant solution including 12% (v/v) of PEG 400, picked up in a 0.2-mm nylon loop, and then flash frozen in a cold nitrogen stream at 100 K. The MAD data sets were collected at three wavelengths, 0.9797 Å (edge), 0.9795 Å (peak), and 0.9680 Å (remote), to 1.6 Å resolution on the NW12A beamline at PF-AR (Tsukuba). Diffraction data were processed and scaled using the HKL2000 program (Otwinowski and Minor 1997). Data collection statistics are presented in Table 1.
For phase determination, eight selenium sites were located by using SOLVE (Terwilliger and Berendzen 1999). The resulting electron density map (figure of merit 0.58) was considerably improved by density modification with the program RESOLVE (Terwilliger 2002) (figure of merit 0.74). The model building was completed using the program O (Jones et al. 1991). Rigid-body, simulated annealing, energy minimization, and individual B-factor refinements were carried out using CNS (Brunger et al. 1998). The stereochemical quality of the final structural models was checked with PROCHECK (Laskowski et al. 1993). All figures were made with PyMOL (DeLano 2002), and superpositions of structures were prepared with LSQMAN (Kleywegt 1996).
The coordinates and structure factors have been deposited in the RCSB Protein Data Bank, with the accession code 2CXA.
Acknowledgments
We thank Noriko Ito-Matsuura and Rie Shibata for technical help during purification and crystallization. This work was supported by the RIKEN Structural Genomics/Proteomics Initiative (RSGI), the National Project on Protein Structural and Functional Analysis, and the Ministry of Education, Culture, Sports, Science and Technology of Japan.
Footnotes
Reprint requests to: Shigeyuki Yokoyama, Protein Research Group, RIKEN Genomic Sciences Center, 1-7-22 Suehiro-cho, Tsurumi, Yokohama 230-0045, Japan; e-mail: yokoyama@biochem.s.u-tokyo.ac.jp; fax: 81-45-503-9195.
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.062616107.
References
- Abramochkin G. and Shrader, T.E. 1995. The leucyl/phenylalanyl-tRNA-protein transferase. Overexpression and characterization of substrate recognition, domain structure, and secondary structure. J. Biol. Chem. 270: 20621–20628. [DOI] [PubMed] [Google Scholar]
- Abramochkin G. and Shrader, T.E. 1996. Aminoacyl-tRNA recognition by the leucyl/phenylalanyl-tRNA-protein transferase. J. Biol. Chem. 271: 22901–22907. [DOI] [PubMed] [Google Scholar]
- Biarrotte-Sorin S., Maillard, A.P., Delettre, J., Sougakoff, W., Arthur, M., and Mayer, C. 2004. Crystal structures of Weissella viridescens FemX and its complex with UDP-MurNAc-pentapeptide: Insights into FemABX family substrates recognition. Structure 12: 257–267. [DOI] [PubMed] [Google Scholar]
- Brunger A.T., Adams, P.D., Clore, G.M., DeLano, W.L., Gros, P., Grosse-Kunstleve, R.W., Jiang, J.S., Kuszewski, J., Nilges, M., and Pannu, N.S., et al. 1998. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 54: 905–921. [DOI] [PubMed] [Google Scholar]
- Clements A., Poux, A.N., Lo, W.S., Pillus, L., Berger, S.L., and Marmorstein, R. 2003. Structural basis for histone and phosphohistone binding by the GCN5 histone acetyltransferase. Mol. Cell 12: 461–473. [DOI] [PubMed] [Google Scholar]
- DeLano W.L.. 2002. The PyMOL molecular graphics system. DeLano Scientific, San Carlos, CA.
- Dyda F., Klein, D.C., and Hickman, A.B. 2000. GCN5-related N-acetyltransferases: A structural overview. Annu. Rev. Biophys. Biomol. Struct. 29: 81–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg A.L.. 1992. The mechanism and functions of ATP-dependent proteases in bacterial and animal cells. Eur. J. Biochem. 203: 9–23. [DOI] [PubMed] [Google Scholar]
- Haas A.L. and Siepmann, T.J. 1997. Pathways of ubiquitin conjugation. FASEB J. 11: 1257–1268. [DOI] [PubMed] [Google Scholar]
- Hegde S.S. and Shrader, T.E. 2001. FemABX family members are novel nonribosomal peptidyltransferases and important pathogen-specific drug targets. J. Biol. Chem. 276: 6998–7003. [DOI] [PubMed] [Google Scholar]
- Hochstrasser M.. 1996. Ubiquitin-dependent protein degradation. Annu. Rev. Genet. 30: 405–439. [DOI] [PubMed] [Google Scholar]
- Holm L. and Sander, C. 1993. Protein structure comparison by alignment of distance matrices. J. Mol. Biol. 233: 123–138. [DOI] [PubMed] [Google Scholar]
- Hwang B.J., Woo, K.M., Goldberg, A.L., and Chung, C.H. 1988. Protease Ti, a new ATP-dependent protease in Escherichia coli, contains protein-activated ATPase and proteolytic functions in distinct subunits. J. Biol. Chem. 263: 8727–8734. [PubMed] [Google Scholar]
- Ichetovkin I.E., Abramochkin, G., and Shrader, T.E. 1997. Substrate recognition by the leucyl/phenylalanyl-tRNA-protein transferase. Conservation within the enzyme family and localization to the trypsin-resistant domain. J. Biol. Chem. 272: 33009–33014. [DOI] [PubMed] [Google Scholar]
- Jones T.A., Zou, J.Y., Cowan, S.W., and Kjeldgaard, M. 1991. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A 47: 110–119. [DOI] [PubMed] [Google Scholar]
- Kaji H., Novelli, G.D., and Kaji, A. 1963. A soluble amino acid-incorporating system from rat liver. Biochim. Biophys. Acta 76: 474–477. [PubMed] [Google Scholar]
- Kaji A., Kaji, H., and Novelli, G.D. 1965. Soluble amino acid-incorporating system. I. Preparation of the system and nature of the reaction. J. Biol. Chem. 240: 1185–1191. [PubMed] [Google Scholar]
- Katayama Y., Gottesman, S., Pumphrey, J., Rudikoff, S., Clark, W.P., and Maurizi, M.R. 1988. The two-component, ATP-dependent Clp protease of Escherichia coli. Purification, cloning, and mutational analysis of the ATP-binding component. J. Biol. Chem. 263: 15226–15236. [PubMed] [Google Scholar]
- Kleywegt G.. 1996. Use of non-crystallographic symmetry in protein structure refinement. Acta Crystallogr. D Biol. Crystallogr. 52: 842–857. [DOI] [PubMed] [Google Scholar]
- Laskowski R.A., McArthur, M.W., Moss, D.S., and Thornton, J.M. 1993. PROCHECK—a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26: 283–291. [Google Scholar]
- Leibowitz M.J. and Soffer, R.L. 1970. Enzymatic modification of proteins. 3. Purification and properties of a leucyl, phenylalanyl transfer ribonucleic acid protein transferase from Escherichia coli. J. Biol. Chem. 245: 2066–2073. [PubMed] [Google Scholar]
- Leibowitz M.J. and Soffer, R.L. 1971. Enzymatic modification of proteins. VI. Site of acylation of bovine serum albumin in the leucine, phenylalanine-transfer reaction. J. Biol. Chem. 246: 4431–4438. [PubMed] [Google Scholar]
- Maillard A.P., Biarrotte-Sorin, S., Villet, R., Mesnage, S., Bouhss, A., Sougakoff, W., Mayer, C., and Arthur, M. 2005. Structure-based site-directed mutagenesis of the UDP-MurNAc-pentapeptide-binding cavity of the FemX alanyl transferase from Weissella viridescens . J. Bacteriol. 187: 3833–3838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski Z. and Minor, W. 1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276: 307–326. [DOI] [PubMed] [Google Scholar]
- Scarpulla R.C., Deutch, C.E., and Soffer, R.L. 1976. Transfer of methionyl residues by leucyl, phenylalanyl-tRNA-protein transferase. Biochem. Biophys. Res. Commun. 71: 584–589. [DOI] [PubMed] [Google Scholar]
- Shrader T.E., Tobias, J.W., and Varshavsky, A. 1993. The N-end rule in Escherichia coli: Cloning and analysis of the leucyl, phenylalanyl-tRNA-protein transferase gene aat. J. Bacteriol. 175: 4364–4374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terwilliger T.C.. 2002. Automated structure solution, density modification and model building. Acta Crystallogr. D Biol. Crystallogr. 58: 1937–1940. [DOI] [PubMed] [Google Scholar]
- Terwilliger T. and Berendzen, J. 1999. Automated MAD and MIR structure solution. Acta Crystallogr. D Biol. Crystallogr. 55: 849–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobias J.W., Shrader, T.E., Rocap, G., and Varshavsky, A. 1991. The N-end rule in bacteria. Science 254: 1374–1377. [DOI] [PubMed] [Google Scholar]
- Varshavsky A.. 1992. The N-end rule. Cell 69: 725–735. [DOI] [PubMed] [Google Scholar]
- Varshavsky A.. 1997. The ubiquitin system. Trends Biochem. Sci. 22: 383–387. [DOI] [PubMed] [Google Scholar]
- Vetting M.W., Magnet, S., Nieves, E., Roderick, S.L., and Blanchard, J.S. 2004. A bacterial acetyltransferase capable of regioselective N-acetylation of antibiotics and histones. Chem. Biol. 11: 565–573. [DOI] [PubMed] [Google Scholar]
- Vetting M.W., S. de Carvalho, L.P., Yu, M., Hegde, S.S., Magnet, S., Roderick, S.L., and Blanchard, J.S. 2005. Structure and functions of the GNAT superfamily of acetyltransferases. Arch. Biochem. Biophys. 433: 212–226. [DOI] [PubMed] [Google Scholar]