

Abstract
Thr93, Ser94, Thr140, and Ser306 are conserved in all adenylosuccinate lyases (ASL) and are close to other amino acids previously identified by mutagenesis as being in the active site. To test their involvement in the enzyme's function, each of these amino acids was replaced by alanine. All the mutants exhibit circular dichroism spectra which are similar to that of wild-type enzyme, indicating there is no appreciable change in secondary structure. T93A exhibits 0.5% of the Vmax of wild-type ASL with a 10-fold increase in Km for adenylosuccinate. S94A has 65% of the Vmax of wild-type ASL with little change in Km. T140A exhibits 0.03% of the activity of wild-type enzyme with an 11-fold increase in Km. S306A has 0.4% of the Vmax of wild-type ASL with a sevenfold increase in Km. Measurements of the pH-Vmax profile reveal a pK2 value for S94A of 7.83 and S306A of 7.65, in contrast to 8.24 for the wild-type enzyme and 8.42 for T93A. Thr93 may orient adenylosuccinate optimally for catalysis, while Ser94 stabilizes protonated His89, a determinant of pK2. Thr140 may, through hydrogen bonding, interact with Asn270, an amino acid essential for catalysis. Ser306 may be involved in a hydrogen bond network that ultimately stabilizes protonated His68, which is probably the general acid in the reaction of enzyme with substrate. The results of this paper demonstrate the importance in the catalytic function of ASL of hydrogen bonds and hydrogen bonding networks involving serine and threonine.
Keywords: adenylosuccinate lyase, purine biosynthesis, site-directed mutagenesis, hydrogen bonding
Adenylosuccinate lyase (ASL) catalyzes the conversion of adenylosuccinate (SAMP) to AMP and fumarate, a key step in purine biosynthesis (Ratner 1972). Point mutations at various positions within the human form of this enzyme result in the disorder Adenylosuccinate Lyase Deficiency, which is characterized by psychomotor retardation, autistic features, epilepsy, and muscle wasting (Jaeken and Van den Berghe 1984; Jaeken et al. 1988; Marie et al. 1999; Race et al. 2000; Van den Berghe and Jaeken 2001; Spiegel et al. 2006).
The crystal structure of Thermatoga maritima ASL indicates that the enzyme is a highly helical homotetramer, with each of its four active sites composed of different portions of three submits (Toth and Yeates 2000). Biochemical evidence in support of the multisubunit active site has been provided by experiments in which two inactive variants of Bacillus subtilis ASL, with mutations in regions contributed to an active site by different subunits, functionally complement each other with substantial recovery of enzymatic activity (Lee et al. 1999; Brosius and Colman 2000, 2002; Segall and Colman 2004). Although a high-resolution structure has not been determined for B. subtilis ASL, a homology model has been constructed using the structure of T. maritima ASL as a template (Brosius and Colman 2002; Segall and Colman 2004). Since the ASLs from these two bacteria share 49% identity plus an additional 23% strong similarity in amino acid sequence, this homology model should provide an excellent indication of the structure of B. subtilis ASL.
Affinity labeling and mutagenesis studies of B. subtilis ASL have implicated His141 and His68 as the general base/acid in the catalytic reaction (Lee et al. 1997, 1998, 1999). Mutagenesis studies have revealed additional amino acid residues, including His89, Lys268, and Asn270, that assist in the catalytic reaction, most likely by contributing to substrate binding and orientation in the active site (Brosius and Colman 2000, 2002; Segall and Colman 2004). It appears that a network of amino acid residues is required to coordinate the multiple negatively charged groups on SAMP through electrostatic and hydrogen bonding interactions, and that each of these residues is indispensable for high catalytic activity.
The importance of hydrogen bonding networks in enzymes has been well documented in terms of maintaining active site integrity and optimal substrate orientation for catalysis (Radisky et al. 2005; Jao et al. 2006; Zimmerman and Ferry 2006). Thr93, Ser94, Thr140, and Ser306 are conserved in all ASLs, as illustrated in Figure 1 for representative species from bacteria (e.g., B. subtilis and T. maritima) to humans. Among these, Thr93, Ser94, and Thr140 are located close to other amino acids that have previously been identified as within the active site, including His68, His89, His141, and Asn270 (Figs. 5, 6, see below). Ser306 is further from the active site but is positioned to hydrogen bond to Arg310, a residue recently shown to influence the catalytic rate, probably through a chain of electrostatic interactions involving His68 (Sivendran et al. 2005). Thus, these four amino acids were selected as targets for site-directed mutagenesis. Alanine was substituted for each of these four hydroxylic amino acids, one at a time, to determine whether their –OH functionalities were involved in the ASL-catalyzed conversion of SAMP to AMP and fumarate. A preliminary version of some of this work has been presented (Cashman et al. 2005).
Figure 1.

Amino acid sequence alignment of ASLs from two bacterial species, as well as from human, mouse, and chicken in the region of Thr93, Ser94, Thr140, and Ser306 (shown in bold). The amino acid numbers are those of the B. subtilis ASL.
Figure 5.

Structural model of B. subtilis ASL in the vicinity of Thr93 and Ser94. The distance between the –OH of Thr93 and a carboxyl oxygen of adenylosuccinate (SAMP) as well as that between the –OH of Ser94 and the δN of His89 are shown.
Figure 6.

Structural model of B. subtilis ASL in the region of Thr140. The model shows distances between Thr140, Asn270, and the substrate adenylosuccinate (SAMP).
Results
Activity and purity of wild-type and variant adenylosuccinate lyases
The mutant ASLs (T93A, S94A, T140A, and S306A) were all expressed well. Each yielded a single protein band on polyacrylamide gel electrophoresis in the presence of SDS, with a migration consistent with the known subunit molecular mass (∼50 kDa). Each ASL sample gave a single N-terminal sequence on Edman degradation for the first 15 amino acids, confirming the purity of these ASL preparations.
All four mutant enzymes retained sufficient catalytic activity to determine steady-state kinetic parameters at pH 7.0, as shown in Table 1. S94A ASL exhibited values for Km and Vmax that were very close to those observed for the wild-type enzyme. In contrast, the replacement of Thr93 by Ala resulted in a 200-fold decrease in Vmax and a 10-fold increase in Km, while the mutation at S306 caused a 250-fold decrease of Vmax and sevenfold increase of Km. However, replacement of T140 had the most profound effect on catalysis, causing a 3600-fold decrease of Vmax and 11-fold increase of Km.
Table 1.
Kinetic parameters for wild-type and mutant enzymes

pH-Vmax profiles of wild-type and mutant ASLs
The pH dependence of Vmax for wild-type ASL is bell-shaped (Palenchar et al. 2003), indicating that Vmax is influenced by two ionization events. The pH-Vmax profiles of the T93A, S94A, and S306A mutant enzymes are also bell-shaped. Figure 2 shows a representative graph of Vmax versus pH for the S94A mutant. Table 2 summarizes the values of pK1 and pK2 for wild-type and the three mutant enzymes. The pH-Vmax profile for T93A ASL is minimally perturbed, suggesting that this mutation does not influence the local environment of the amino acid residues that determine the effects of pH on enzyme activity. In contrast, the S94A mutant displays a marked decrease in pK2, as compared to wild-type ASL (Table 2). A large decrease in pK2 is also apparent in the pH-Vmax profile for the S306A mutant. (Because of the very low specific activity of the T140A enzyme, the pH dependence of Vmax could not be obtained.)
Figure 2.

pH dependence of Vmax for the S94A mutant ASL.
Table 2.
pK values determined from pH-Vmax profiles of wild-type and mutant ASLs

Circular dichroism of wild-type and mutant enzymes
To determine whether the global secondary structure of the enzyme was affected by the mutations to Ala at positions 93, 94, 140, and 306, far-UV circular dichroism (CD) spectra were obtained for wild-type ASL and each of these mutants. As shown in Figure 3, the spectra from the wild-type and mutant enzymes exhibited pronounced minima at both 208 and 222 nm, indicating a predominance of α-helical structure, as earlier studies of ASL have shown (Palenchar et al. 2003; Segall and Colman 2004). The CD spectra of T140A and S306A are similar to that of wild-type enzyme (Fig. 3). CD spectra of T93A and S94A are superimposable on that of wild-type enzyme (data not shown). We conclude that the mutations at positions 93, 94, 140, and 306 did not have an appreciable effect on the secondary structure of the enzyme.
Figure 3.

CD spectra of wild-type, T140A, and S306A enzymes.
Molecular masses of wild-type and mutant ASLs
ASL is a tetramer of identical subunits (Palenchar and Colman 2003). Although wild-type ASL has been shown to exist as an equilibrium mixture of dimeric (∼100,000 Da) and tetrameric (∼200,000 Da) forms, only the tetrameric form is catalytically functional (Palenchar and Colman 2003). In the present study, native polyacrylamide gel electrophoresis and analytical ultracentrifugation were used to evaluate whether the mutations affected the oligomeric state of the enzyme at pH 7.0. The results are shown in Table 3. All the enzymes exhibit molecular weights of about 200 kDa, showing that the wild-type and mutant enzymes exist predominantly in a tetrameric state. Thus, no major changes in the oligomeric state of ASL resulted from the four amino acid replacements made in this study.
Table 3.
Molecular weight of wild-type and mutant ASLs at pH 7.0

Intersubunit complementation
Although ASL contains both four identical subunits and four active sites, each of these active sites is formed from contributions of different regions from three subunits (Brosius and Colman 2002). As a result of this structural arrangement, we have previously shown that two different ASL mutant enzymes, in which each harbors a mutation in one of these separate regions, can complement each other forming some functionally intact active sites in a process referred to as intersubunit complementation (Lee et al. 1999; Brosius and Colman 2000; Segall and Colman 2004). We have also demonstrated that mixtures of mutants with changes at amino acids provided to the active site by the same subunit do not show an increase in activity (Segall and Colman 2004). For a tetramer with random dissociation and random reassociation of subunits, recovery of activity up to 25% that of wild-type enzyme is expected (Lee et al. 1999). Representative results of the complementation experiments are illustrated in Figure 4, which shows the time-dependent reactivation of the inactive T140A and S306A mutant enzymes when paired with variant enzymes whose point mutations come from different subunits.
Figure 4.

Complementation studies by reactivation of inactive mutant enzymes. The following mutant enzymes were mixed: T140A + K268Q (open circles); S306A + K268Q (closed upright triangles); T140A + H68Q (open squares); S306A + H141Q (upside-down open triangles); T140A + H141Q (closed circles); and S306A + H68Q (closed squares).
Table 4 summarizes the maximum regain of activity of the inactive T93A, T140A, and S306A when mixed with each of three other mutant enzymes. (Because of the high specific activity of the S94A mutant enzyme under standard assay conditions, intersubunit complementation studies of this type could not be conducted for this mutant.) When the T93A mutant was combined with K268A ASL (a completely inactive mutant enzyme described in an earlier study; Brosius and Colman 2002), the observed ASL activity reached 10% of that of the wild-type enzyme, while T93A plus completely inactive H141Q mutant (characterized previously; Lee et al. 1999) yielded 3% of wild-type activity. In contrast, no activity was gained when T93A was paired with H68Q, indicating that Thr93 and His68 are contributed by the same subunit to the active site. (H68Q alone has a specific activity of only 0.016 μmol min−1 mg−1; Lee et al. 1998.)
Table 4.
Complementation of inactive mutant ASLs

T140A achieved maximum activity that is 15.4% that of the wild-type enzyme when paired with K268Q, and 11.5% when incubated with H68Q, but gained no activity when paired with H141Q. S306A when combined with K268Q reached 15.4% of the activity of wild-type enzyme, and 4.6% of the wild-type enzyme activity when paired with H141Q. In contrast, no reactivation was observed when S306A and H68Q were mixed. These results demonstrate that Thr140 and His141 are contributed to the active site by the same subunit, and that Ser306 and His68 come from the same subunit.
Discussion
The intersubunit region of ASL previously identified as the active site (Lee et al. 1999; Toth and Yeates 2000; Brosius and Colman 2002; Segall and Colman 2004) also contains serine and threonine residues that are conserved in ASLs of many species. The present study sought to examine, by site-directed mutagenesis, the roles of the hydroxyl groups of these conserved serines and threonines. Alanine was substituted for each of the amino acids presently studied, since this replacement results in the elimination of a side-chain that can participate in hydrogen bonding without appreciably changing residue volume. In fact, each of these four mutations has minimal effect on the secondary or quaternary structure of ASL as indicated by their circular dichroism spectra and molecular weights.
As shown in Figure 5, the hydroxyl group of Thr93 is positioned within hydrogen bonding distance (2.6 Å) of one of the carboxylate oxygens on the succinyl portion of adenylosuccinate. The large decrease in Vmax and increase in Km for the T93A variant, along with the absence of a significant effect on the pH-rate profile for this mutant, are consistent with a role for this threonine in orienting adenylosuccinate within the active site. Since the T93A mutant enzyme regains activity by complementation, the global structure is largely unchanged and the loss of activity is strictly due to the point mutation at position 93. Other amino acid residues have also been shown to contribute to the positioning of SAMP within the active site by hydrogen bonding or electrostatic interactions: Asn270, Gln212, and Arg301 (Segall and Colman 2004). The succinyl moiety of SAMP is the location of the chemical transformation. Therefore, precise orientation is necessary for the enzymic general acid and base to function properly.
Ser94 does not have a major influence on the catalytic function of the enzyme at pH 7.0, but it helps to establish the pH range over which the enzyme is active, since replacement of this residue with alanine (in the S94A enzyme) lowers the pK2 for the enzyme. Evidence has previously been presented that the alkaline side of the pH-Vmax curve is due to the deprotonation of histidine, based on the temperature dependence of pK (Brosius and Colman 2000). The pK2 value appears to reflect the deprotonation of both His68 and His89 (Lee et al. 1999; Brosius and Colman 2000). Figure 5 shows that the hydroxyl group on the side-chain of Ser94 is nearly close enough to the side-chain δN of His89 to hydrogen bond to this residue. The X-ray crystal structure of the enzyme from T. maritima includes a water molecule that is well positioned to mediate interaction between the corresponding Ser and His in this enzyme, and presumably also in the B. subtilus ASL. Typically the pK of the imidazole of a protein histidine is 6–6.5 (Tipton and Dixon 1979); however, a water-mediated interaction between Ser94 and His89 could raise the pK of the histidine by favoring the protonated form of the imidazole. It has been proposed that the protonated His89 contributes to the catalytic function of ASL by coordination of a ribose hydroxyl of SAMP (Brosius and Colman 2000, 2002). In the H94A mutant, the absence of a hydroxyl group at position 94 would disrupt the hydrogen bonding network. This structural model (Fig. 5) can thus explain the decrease in pK2 observed for the S94A variant.
The importance of Thr140 for the catalytic function of ASL is indicated by the 3600-fold decrease in Vmax along with an 11-fold increase in Km for adenylosuccinate in the T140A mutant. Thr140 is located immediately adjacent in sequence to His141 (the histidine that has been postulated to be the general base in ASL catalysis) and it is conserved in at least 46 species. The structural model shown in Figure 6 suggests that Thr140 functions indirectly in catalysis by maintaining the correct orientation of amino acid residues that interact directly with the substrate SAMP. The hydroxyl group of Thr140 is only 3.12 Å from the side-chain carbonyl oxygen of N270, which is close to hydrogen bonding distance of one of the carboxyls of the substrate, SAMP. The backbone carbonyl oxygen of Thr140 is only 2.88 Å from the side-chain carbonyl oxygen of Asn270, also sufficiently close for hydrogen bonding. A recent report suggested that Asn270 functions in a critical role in ASL catalysis through binding to one of the carboxylate oxygens on the succinyl moiety of SAMP (Segall and Colman 2004). Therefore, the importance of maintaining Asn270 in a catalytically productive orientation provides a reasonable explanation for the severe decrease in Vmax and modest increase in Km at pH 7.0 for the T140A mutant. (Additional amino acids in the region of Thr93, Ser94, and Thr140 are indicated by the amino acid sequences shown in Fig. 1.)
Ser306 also makes a contribution to the function of ASL since the S306A mutant enzyme has a Vmax that is decreased 250-fold and a Km that is increased sevenfold, as compared to wild-type enzyme. Futhermore, the S306A enzyme exhibits a markedly decreased pK2. While Thr140 likely mediates ASL catalysis through one intervening amino acid residue, Ser306, which is completely conserved in at least 46 species, probably affects the catalytic reaction through a chain of three other amino acids: Arg310, Asp69, and His68. There is evidence that Asp69 interacts electrostatically with His68, stabilizing the functionally important protonated state of this histidine (Sivendran et al. 2005). The positively charged guanidinium group of Arg310, in turn, is sufficiently close to the negatively charged carboxylate of Asp69 to promote the optimal orientation of this residue by electrostatic interaction. These interactions are illustrated in the ASL structure shown in Figure 7. The hydroxyl group of Ser306 is only 3.1 Å from a N of the guanidinium group of Arg310, allowing for hydrogen bonding between these two amino acids, and there is a comparable distance between the backbone oxygen of Ser306 and the backbone nitrogen of Arg310. Thus, Ser306 may constitute an additional amino acid in this functional chain of amino acid residues extending to His68. The characteristics of the S306A mutant enzymes suggest a role for this serine in the optimal positioning of Arg310 and imply that perturbation of this orientation has a similar effect on catalysis as loss of the positive charge at position 310, as in R310Q, the drastic consequences of which have recently been reported (Sivendran et al. 2005).
Figure 7.
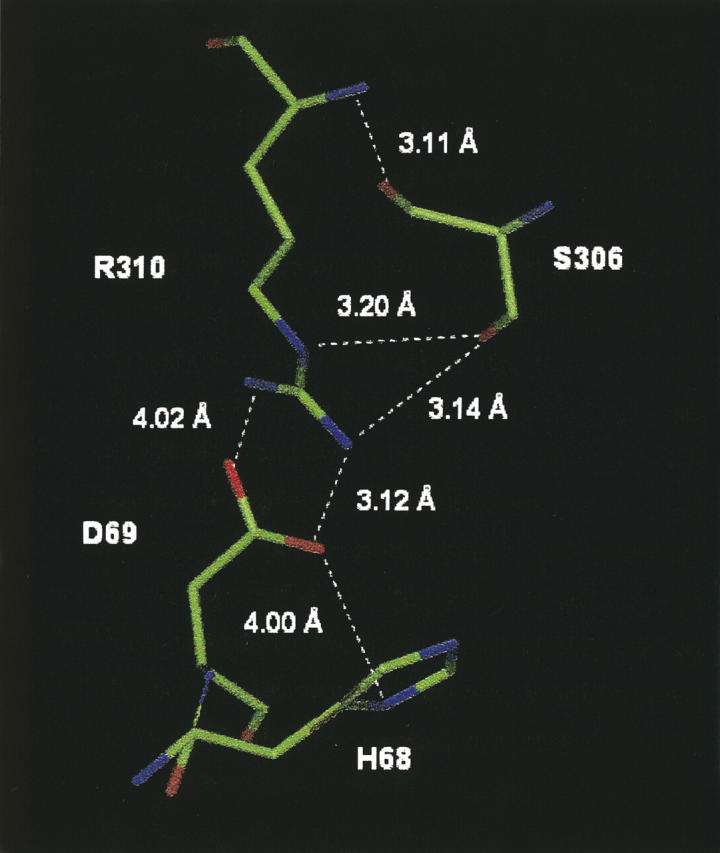
Structural model of B. subtilis ASL in the region of Ser306. The chain of hydrogen bonding and electrostatic interactions between Ser306, Arg310, Asp69, and His68 are shown.
Each of the mutant enzymes also exhibits a small increase in the pK1 value of its pH-Vmax profile as compared to wild-type ASL. Although the identity of the functional group responsible for this ionization remains uncertain, a previous study postulated that pK1 reflects the deprotonation of carboxylic acid groups on the succinyl portion of enzyme-bound SAMP. We thus interpret the increases observed in this value in terms of changes in the active site orientation of SAMP for each of the mutants; small differences in the environment of these carboxylic acid groups may cause a shift in their pK values.
Intersubunit complementation
Since each of the four active sites of the ASL homotetramer is composed of different regions of three identical subunits, pairs of ASL variants with amino acid substitutions at sites contributed to an active site by different subunits can functionally complement one another when mixed at equal concentrations (Lee et al. 1999; Brosius and Colman 2002; Segall and Colman 2004). As no activation occurs when T93A is paired with H68Q, His68 and Thr93 are contributed to the active site by the same subunit. The results of the present study also demonstrate that Ser306 is contributed to an intersubunit active site of ASL by the same subunit that provides His68. Thr140 and His141 originate from a second, separate subunit and Lys268 is contributed by a third. In addition, the abilities of the variant enzymes in the present study to complement ASL mutants bearing substitutions at different sites indicate that the mutations do not substantially affect the capabilities of the enzymes to form functional intersubunit contacts, and confirm the evidence that shows no major structural perturbations as a result of the amino acid substitutions.
The maximum extent of reactivation varies considerably among the different pairs of mutant enzymes (from 3.0% to 15.5% of wild-type activity, as shown in Table 4). For a tetramer with random dissociation of subunits, and random reassociation of monomers, the maximum extent of reactivation is expected to be 25% (Lee et al. 1999). The lower-than-theoretical degree of reactivation indicates that these processes are not random. There are three types of subunit interface in the tetramer, which differ in the contact area between the subunits (Segall and Colman 2004). The variation in the extent of reactivation has been attributed to the differences in the particular subunit interface involved for each pair of mutants tested for complementation, the exact location of the mutated amino acid within the subunit interface, and whether subunit dissociation or reassociation is rate-limiting in the overall complemention process (Lee et al. 1999; Brosius and Colman 2000, 2002; Segall and Colman 2004).
In conclusion, Thr93, Ser94, Thr140, and Ser306 perform very important roles in the function of ASL through hydrogen bonding interactions directly with adenylosuccinate, as well as indirectly through hydrogen bonds to active site amino acid residues. This is not surprising in light of earlier work that has demonstrated the remarkable sensitivity of ASL catalysis to the integrity of the network of amino acid residues in proximity to an active site (Lee et al. 1999; Brosius and Colman 2002; Segall and Colman 2004; Sivendran et al. 2005; Spiegel et al. 2006). The results of this study provide a clear example of the importance and variety of roles that the hydroxylic amino acid residues serine and threonine can perform in an enzyme and also emphasize the importance to catalysis by ASL of amino acid residues that interact with bound substrate through other residues, rather than directly.
Materials and methods
Materials
BioSynthesis Inc. provided the oligonucleotide primers used for site-directed mutagenesis and sequencing. Adenylosuccinate, as well as imidazole, and HEPES were supplied by Sigma Chemical Co. Concentrated protein assay reagent was purchased from BioRad. All other chemicals were of reagent grade.
Site-directed mutagenesis
The pBHis plasmid containing B. subtilis ASL was provided to us as a generous gift from Dr. Jack E. Dixon (UCSD Sch. Med., La Jolla, CA) The Stratagene QuikChange Mutagenesis kit, along with the plasmid, was used to construct mutations to this gene. The oligonucleotide primers used to produce the mutants were the following (and their respective complementary sequences): CATTACGGCTTAGCGTCAACTGACGTTG (T93A), CGGCTTAACGGCGACTGACGTTGTTG (S94A), GATGGGGCGCGCACACGGCGTAC (T140A), and GCGATATTTCTCATGCTTCAGCAGAACG (S306A). DNA sequencing to ensure the presence of the engineered mutations was conducted by the University of Delaware Center for Agricultural Biotechnology with an ABI Prism model 377 DNA sequencer (PE systems). The B. subtilis ASLs were expressed in Escherichia coli strain BL21(DE3) as His-tag fusion proteins, as described previously (Lee et al. 1997).
Wild-type and mutant enzymes were purified in a single step procedure using nickel affinity chromatography, as reported in an earlier study (Segall and Colman 2004; Lee et al. 1997); and protein purity was assessed electrophoretically using a 12% polyacrylamide gel in the presence of 0.1% sodium dodecyl sulfate. N-terminal amino acid sequencing in the gas phase, utilizing an Applied Biosystems Procise sequence analyzer, was also used to confirm protein purity. Except where indicated differently, absorbance at 280 nm (E1% = 10.6) was used to determine protein concentration (Lee et al. 1997). Purified protein was stored at −80°C in separate aliquots.
It should be noted that the mutant as well as wild-type B. subtilis ASLs studied here all have six additional histidines at the N terminus to facilitate the purification. Thus, each mutant differs from the wild type by only one amino acid. While the properties of the His-tagged and non-His-tagged B. subtilis ASLs have not been compared explicitly, the corresponding forms of human ASL have been studied. The His-tagged and non-His-tagged human ASLs were shown to exhibit very similar values of Vmax and Km for adenylosuccinate (Lee and Colman 2007).
Kinetics of wild-type and mutant adenylosuccinate lyase
Catalytic activity toward SAMP was measured by following the decrease in absorbance at 282 nm that accompanies the conversion of SAMP to AMP plus fumarate. (The difference in extinction coefficient between SAMP and AMP = 10,000 M−1cm −1; Tornheim and Lowenstein 1972.) For assays in the presence of SAMP concentrations >120 μM, the decrease in absorbance at 290 nm (Δɛ = 4050 M−1 cm−1) was monitored instead. Assays under standard conditions were performed at 25°C and pH 7.0, using about 8 μg/mL wild-type ASL (and higher concentrations of mutant enzymes) in the presence of 60 μM SAMP and 50 mM HEPES buffer. Specific activity was defined as micromoles of SAMP reacted per minute per milligram of enzyme under these conditions. Km and Vmax values toward SAMP were obtained for wild-type and mutant enzymes under standard assay conditions, except for the variation in the concentration of SAMP (between 2 and 300 μM), and the data were calculated via SigmaPlot using the Michaelis-Menten equation. All enzymes were preincubated at 25°C for at least 30 min prior to the assay.
pH-Vmax profiles for wild-type and mutant ASL enzymes
Vmax determinations were performed for wild-type, T93A, S94A, and S306A enzymes as a function of pH using MES (pH 6.0–6.9), HEPES (pH 6.8–8.0), and TAPS (pH 7.9–9.0) buffers. (These experiments were not done for the T140A ASL because of its extremely low specific activity.) In all of these assays, the SAMP concentration was held constant at 300 μM, which is high relative to the wild-type enzyme's Km for adenylosuccinate (Brosius and Colman 2000), and the reaction rates were monitored by following the change in absorbance at 290 nm. For the mutant enzymes, Km values were determined at several of the pH conditions tested and used to extrapolate to Vmax using the Michaelis-Menten equation. The pH profile data were fit to the equation: Vmax = V0/(1 + 10(pK 1 − pH) + 10(pH − pK 2 )) where Vmax is the maximum velocity at a given pH, V0 is the intrinsic, pH-independent value of Vmax, and pK1 and pK2 are the pK values obtained for the left and right sides of the curve (Dixon and Webb 1964).
Circular dichroism of adenylosuccinate lyase mutants
Far-UV CD spectra were obtained for wild-type ASL and each of these mutants. For the T93A and S94A mutants, spectra were acquired (at 0.2-nm intervals) on a Jasco J710 spectropolarimeter and averaged over five scans. For the T140A and S306A enzymes, an Aviv Quick Start 215 Circular Dichroism Spectrometer was used and spectra (with points at 1-nm intervals) were averaged over sets of three acquisitions. Wild-type ASL was measured for comparison, using both instruments. Final spectra were expressed in terms of molar ellipticity values ([θ], deg cm2/dmol) as a function of wavelength, using the equation [θ] = θ/10nCl. In this expression, θ is the experimentally determined ellipticity, n is the amount of residues in each enzyme subunit (437 including the His6-tag), C is the molar concentration of enzyme subunits, and l is the cuvette pathlength (0.1 cm).
Protein samples at concentrations of 0.2–0.5 mg/mL were prepared in 20 mM potassium phosphate buffer, pH 7.0, containing 20 mM potassium chloride. Enzymes were preincubated for at least 30 min at 25°C before obtaining measurements. Scans of buffer alone were performed over the same wavelength range and used as a background correction. Protein concentrations were measured using an assay based on that of Bradford, in which wild-type ASL at a known concentration served as the standard (Bradford 1976).
Molecular mass determination of mutant enzymes
Molecular masses were obtained for wild-type and mutant ASLs using analytical ultracentrifugation or native polyacrylamide gel electrophoresis. Analytical ultracentrifugation was performed using a Beckman Optima XL-A analytical ultracentrifuge. The instrument was run at 8000 rpm, 10,000 rpm, and 14,000 rpm, using an An-60 Ti rotor for sedimentation equilibrium studies and UV absorbance was monitored at 280 nm. All measurements were obtained at 25°C with enzyme samples adjusted to 0.4 mg/mL in 20 mM potassium phosphate buffer with 20 mM potassium chloride, pH 7.0. Three determinations of molecular weight were made for each type of enzyme used.
For native polyacrylamide gel electrophoresis, a series of gels was run at pH 7.0, 25°C ranging from 5% to 10% (acrylamide); the ratio of acrylamide:bisacrylamide was maintained constant at 37.5:1, as previously described (Palenchar et al. 2003). Molecular weight standards consisted of ferritin (450 kDa) alcohol dehydrogenase (140 kDa), bovine serum albumin (67 kDa), and ovalbumin (45 kDa). Each protein sample (8 μL at a concentration of 2 mg/mL) was applied to each type of gel. Prior to measurement, enzyme samples were preincubated at 25°C for 30 min.
Intersubunit complementation of mutant enzymes
Equal-volume (100 μL) mixtures of mutant enzymes in 0.1 M sodium phosphate buffer, pH 7.0, to give a final concentration of 0.7 mg/mL for each enzyme, were slowly frozen at −20°C. Upon slowly thawing at room temperature, each sample was gently mixed and incubated at 25°C. Assays under standard conditions were then performed at various timepoints with the enzyme remaining at this temperature, until a maximum level of activity was reached. Previous studies have suggested that slow freezing of multisubunit proteins in sodium phosphate buffer results in a decrease in pH that promotes subunit dissociation (Chilson et al. 1965), and this approach has been used successfully with ASL (Segall and Colman 2004). Each individual enzyme used in the experiment was also separately assayed (as a control) under the same conditions and at the same total concentration of enzyme following at least 30 min of preincubation at 25°C.
Acknowledgments
This work has been supported by NIH 1-RO1-DK60504 and by the University of Delaware HHMI Undergraduate Biological Sciences Education Program (for M.A.C.). We thank Dr. Yu Chu Huang (Univ. of Delaware) for performing the N-terminal amino acid sequencing and Lushanti Ariyananda (Univ. of Delaware) for conducting the analytical ultracentrifugation experiments.
Footnotes
Reprint requests to: Roberta F. Colman, Department of Chemistry and Biochemistry, University of Delaware, Newark, DE 19716, USA; e-mail: rfcolman@udel.edu; fax: (302) 831-6335.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.062650007.
Abbreviations: ASL, adenylosuccinate lyase; SAMP, adenylosuccinate; B. subtilis, Bacillus subtilis; T. maritima, Thermatoga maritima.
References
- Bradford M.M.. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72: 248–254. [DOI] [PubMed] [Google Scholar]
- Brosius J.L. and Colman, R.F. 2000. A key role in catalysis for His89 of adenylosuccinate lyase of Bacillus subtilis . Biochemistry 39: 13336–13343. [DOI] [PubMed] [Google Scholar]
- Brosius J.L. and Colman, R.F. 2002. Three subunits contribute amino acids to the active site of tetrameric adenylosuccinate lyase: Lys268 and Glu275 are required. Biochemistry 41: 2217–2226. [DOI] [PubMed] [Google Scholar]
- Cashman M.A., Segall, M.L., and Colman, R.F. 2005. Evaluation of the role of Thr93 and Ser94 in the function of adenylosuccinate lyase. FASEB J. 19: A306. [Google Scholar]
- Chilson O.P., Costello, L.A., and Kaplan, N.O. 1965. Effects of freezing on enzymes. Fed. Proc. 24: 555–565. [PubMed] [Google Scholar]
- Dixon M. and Webb, E.C. 1964. The enzymes, 2d ed. Academic Press, New York.
- Jaeken J. and Van den Berghe, G. 1984. An infantile autistic syndrome characterized by the presence of succinylpurines in body fluids. Lancet 2: 1058–1061. [PubMed] [Google Scholar]
- Jaeken J., Wadman, S.K., Duran, M., van Sprang, F.J., Holl, R.A., Theunissen, P.M., de Cock, P., Van den Bergh, F., Vincent, M.F., and Van den Berghe, G. 1988. Adenylosuccinase deficiency: An inborn error of purine nucleotide synthesis. Eur. J. Pediatr. 148: 126–131. [DOI] [PubMed] [Google Scholar]
- Jao S.C., Huang, L.F., Hwang, S.M., and Li, W.S. 2006. Tyrosine387 and Arginine404 are critical in the hydrolytic mechanism of E. coli Aminopeptidase P. Biochemistry 45: 1547–1553. [DOI] [PubMed] [Google Scholar]
- Lee P. and Colman, R.F. 2007. Expression, purification and characterization of stable, recombinant human adenylosuccinate lyase. Protein Expr. Purif. 51: 227–234. [DOI] [PubMed] [Google Scholar]
- Lee T.T., Worby, C., Dixon, J.E., and Colman, R.F. 1997. Identification of His141 in the active site of Bacillus subtilis adenylosuccinate lyase by affinity labeling with 6-(4-bromo-2,3-dioxobutyl)thiadenosine 5′-monophosphate. J. Biol. Chem. 272: 458–465. [PubMed] [Google Scholar]
- Lee T.T., Worby, C., Bao, Z., Dixon, J.E., and Colman, R.F. 1998. Implication of His68 in the substrate site of Bacillus subtilis adenylosuccinate lyase by mutagenesis and affinity labeling with 2-[(4-bromo-2,3-dioxobutyl)thio]adenosine 5-monophosphate. Biochemistry 37: 8481–8489. [DOI] [PubMed] [Google Scholar]
- Lee T.T., Worby, C., Bao, Z., Dixon, J.E., and Colman, R.F. 1999. His68 and His141 are critical contributors to the intersubunit catalytic site of adenylosuccinate lyase of Bacillus subtilis . Biochemistry 38: 22–32. [DOI] [PubMed] [Google Scholar]
- Marie S., Cuppens, H., Heuterspreute, M., Jaspers, M., Tola, E.Z., Gu, X.X., Legius, E., Vincent, M., Jaeken, J., and Cassiman, J., et al. 1999. Mutation analysis in adenylosuccinate lyase deficiency: Eight novel mutations in the reevaluated full ADSL coding sequence. Hum. Mutat. 13: 197–202. [DOI] [PubMed] [Google Scholar]
- Palenchar J.B. and Colman, R.F. 2003. Characterization of mutant enzyme found in human adenylosuccinate lyase deficiency: Asn276 plays and important structural role. Biochemistry 42: 1831–1841. [DOI] [PubMed] [Google Scholar]
- Palenchar J.B., Crocco, J.M., and Colman, R.F. 2003. The characterization of mutant Bacillus subtilis adenylosuccinate lyases corresponding to severe human adenylosuccinate lyase deficiencies. Protein Sci. 12: 1694–1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Race V., Marie, S., Vincent, M., and Van den Berghe, G. 2000. Clinical, biochemical, and molecular genetic correlations in adenylosuccinate lyase deficiency. Hum. Mol. Genet. 9: 2159–2165. [DOI] [PubMed] [Google Scholar]
- Radisky E.S., Lu, C.-J.K., Kwan, G., and Koshland Jr, D.E. 2005. Role of the intramolecular hydrogen bond network in the inhibitory power of chymotrypsin inhibitor 2. Biochemistry 44: 6823–6830. [DOI] [PubMed] [Google Scholar]
- Ratner S.. 1972. Argininosuccinases and adenylosuccinate lyases. In The enzymes, 3d ed, (ed. P.D. Boyer). Vol. Vol. 7, pp. 167–197. Academic Press, New York. [Google Scholar]
- Segall M.L. and Colman, R.F. 2004. Gln212, Asn270, and Arg301 are critical for catalysis by adenylosuccinate lyase from Bacillus subtilis . Biochemistry 43: 7391–7402. [DOI] [PubMed] [Google Scholar]
- Sivendran S., Segall, M.L., Rancy, P.C., and Colman, R.F. 2005. Effect of Asp69 and Arg310 on the pK of His68, a key catalytic residue of adenylosuccinate lyase. FASEB J. 19: A307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiegel E.K., Colman, R.F., and Patterson, D. 2006. Adenylosuccinate lyase deficiency (minireview). Mol. Genet. Metab. 89: 19–31. [DOI] [PubMed] [Google Scholar]
- Tipton K.F. and Dixon, H.B.F. 1979. Effects of pH on enzymes. In Methods in Enzymology (ed. D.L. Purich). Vol. Vol. 63, pp. 183–234. Academic Press, New York. [DOI] [PubMed] [Google Scholar]
- Tornheim K. and Lowenstein, J.M. 1972. The purine nucleotide cycle: The production of ammonia from aspartate by extracts of rat skeletal muscle. J. Biol. Chem. 247: 162–169. [PubMed] [Google Scholar]
- Toth E.A. and Yeates, T.O. 2000. The structure of adenylosuccinate lyase, an enzyme with dual activity in the de novo purine biosynthetic pathway. Structure 8: 163–174. [DOI] [PubMed] [Google Scholar]
- Van den Berghe G. and Jaeken, J. 2001. Adenylosuccinate lyase deficiency. In The metabolic and molecular basis of inherited diseases, 8th ed, (eds. C.R. Scriver et al.). Vol. Vol. 2, pp. 2653–2662. McGraw-Hill, New York. [Google Scholar]
- Zimmerman S.A. and Ferry, J.G. 2006. Proposal for a hydrogen bond network in the active site of the prototypic γ-class carbonic anhydrase. Biochemistry 45: 5149–5157. [DOI] [PubMed] [Google Scholar]