

Abstract
Protein libraries based on natural scaffolds enable the generation of novel molecular tools and potential therapeutics by directed evolution. Here, we report the design and construction of a high complexity library (30 × 1013 sequences) based on the 10th fibronectin type III domain of human fibronectin (10FnIII). We examined the bacterial expression characteristics and stability of this library using a green fluorescent protein (GFP)-reporter screen, SDS-PAGE analysis, and chemical denaturation, respectively. The high throughput GFP reporter screen demonstrates that a large fraction of our library expresses significant levels of soluble protein in bacteria. However, SDS-PAGE analysis of expression cultures indicates the ratio of soluble to insoluble protein expressed varies greatly for randomly chosen library members. We also tested the stabilities of several representative variants by guanidinium chloride denaturation. All variants tested displayed cooperative unfolding transitions similar to wild-type, and two exhibited free energies of unfolding equal to wild-type 10FnIII. This work demonstrates the utility of GFP-based screening as a tool for analysis of high-complexity protein libraries. Our results indicate that a vast amount of protein sequence space surrounding the 10FnIII scaffold is accessible for the generation of novel functions by directed as well as natural evolution.
Keywords: 10FnIII, combinatorial protein library, in vitro selection, GFP reporter screen
Novel, engineered proteins are important as therapeutics, diagnostics, and imaging agents in biological systems. Combining selection/directed evolution with scaffold-based protein libraries provides an excellent route to engineer new protein function. For example, protein libraries based on stable, well-expressed protein scaffolds have been implemented for in vitro selection techniques, such as phage display, ribosome display, and mRNA display (Koide et al. 1998; Beste et al. 1999; Xu et al. 2002; Ciarapica et al. 2003; Stumpp et al. 2003; Wahlberg et al. 2003; Binz et al. 2004; Cicortas Gunnarsson et al. 2004; Binz and Pluckthun 2005). The utility of such libraries depends on how tolerant the scaffolds are to randomization. In order for a library to generate molecules useful for in vivo applications or large-scale preparations, the selected variants must remain folded and soluble.
One of the most popular scaffolds for protein design has been a fragment of human fibronectin known as 10FnIII (Fig. 1). This small (94-amino-acid) domain was implemented originally by Koide et al. (1998) for the generation of a 108-member phage display library and selection of novel ubiquitin-binding proteins. The fibronectin type III fold is a β-sandwich with three loops, similar to the immunoglobulin VH structure. However, 10FnIII is particularly appealing relative to murine-derived monoclonal antibodies for three reasons: (1) The scaffold is entirely human in sequence, making it a potentially useful therapeutic protein; (2) it contains no disulfide bonds enabling it to be used inside cells as an intrabody; and (3) it expresses at high levels in bacteria. More recent work has demonstrated that 10FnIII is also compatible with mRNA display-based selections giving access to libraries that exceed immunologic diversity (1012–1013 sequences) (Xu et al. 2002; Getmanova et al. 2006). Indeed, these high-diversity libraries have resulted in new ligands with nanomolar to picomolar dissociation constants (Xu et al. 2002; Parker et al. 2005; Getmanova et al. 2006).
Figure 1.

Sequence and structure of 10FnIII. (A) Sequence of 10FnIII. Residues that are part of the β-strand framework are in bold. Residue positions that were selected for randomization are colored red. (B) Two structural views of 10FnIII. The crystal structure of the 10FnIII domain shown is part of the 7–10FnIII domains (left, PDB accession no. 1FNF). The randomized BC and FG loop regions are colored red. The first seven N-terminal residues are colored green. The solution structure of 10FnIII is illustrated in a side view without the unstructured N-terminal residues (right, PDB accession no. 1TTG).
Two problems related to 10FnIII libraries have been poorly addressed in the literature to date: (1) library construction and (2) the fraction of the library members that are actually expressed and folded. For example, the library design described originally by Koide et al. (1998) does not enable construction of trillion member libraries because it has steps that include bacterial transformation. On the other hand, the mRNA display library construction described by Xu et al. (2002) was laborious due to prescreening needed to eliminate stop codons in the 21 randomized positions, and may have altered structurally critical positions in the DE loop (Getmanova et al. 2006). Together, these issues place unwanted limitations on 10FnIII-based design experiments.
Optimally, library design should be balanced in minimizing the effort of construction and maximizing (1) structural and chemical diversity (the number of randomized positions), (2) the translation readability (the lack of stop codons or frame-shifts in the ORF), and (3) stability (the likelihood that a randomly chosen member will be well folded). Very little work has been done to characterize the behavior of the proteins present in display libraries generally (for an exception, see Binz et al. 2003), and 10FnIII libraries in particular. Here, we use the term “behavior” to encompass three aspects: (1) the total amount of soluble protein produced, (2) the ratio of folded to unfolded protein, and (3) the free energy of folding. Given the potential importance of the 10FnIII scaffold for design and therapy, we felt a deeper understanding of 10FnIII-based libraries was needed.
Our goal was to create a simple 10FnIII library strategy that led to very high complexity (∼30 trillion sequences) and enabled screening and analysis of both naïve and evolved clones.
To do this, we held the DE loop constant (randomized previously in Xu et al. 2002) to better preserve structure, but created larger and more diverse libraries compared with Koide et al. (1998). We also altered 10FnIII itself, removing the unstructured N-terminal seven residues (Main et al. 1992). Recent work supports this truncation indicating that the N terminus may interfere with binding in this scaffold (Getmanova et al. 2006).
We used several approaches to assay the quality of the naïve library. First, we explored using a green fluorescent protein (GFP)-based expression screen (Waldo et al. 1999) to examine the quality of library, the first time this approach has been applied to assay library quality. Waldo et al. (1999) demonstrated that the fluorescence of C-terminal GFP fusions in bacteria is highly dependent on the ability of the upstream protein to be folded. We then used this 10FnIII–GFP fusion construct to analyze the expression fitness of 94 randomly chosen 10FnIII variants. We also measured the amount of soluble and insoluble protein for 19 representative variants. Finally, we overexpressed four variants in Escherichia coli, purified them to homogeneity, and measured their chemical stability by guanidinium chloride denaturation. Our work provides an integrated, facile route to construct and analyze highly diverse 10FnIII-based libraries, giving general insight into the 10FnIII scaffold.
Results
10FnIII scaffold design
Our library design differs from previous 10FnIII-based scaffolds in loop randomization and scaffold length. 10FnIII has a structure similar to the immunoglobulin VH domain with three loops corresponding to antibody hypervariable regions. Figure 1 illustrates the sequence and structure of 10FnIII. The three loops, BC (residues 23–29), DE (residues 51–54), and the long FG loop (residues 77–86), are topologically analogous to CDR1, CDR2, and CDR3 of VH domains, respectively. We chose to randomize seven BC loop residues and 10 FG loop residues, while keeping the short DE loop constant. Although the DE loop length and sequence is variable among various FnIII domains, this loop forms a very rigid β-hairpin while the BC and FG loops have some conformational freedom that could adapt to sequence variations (Main et al. 1992; Dickinson et al. 1994b; Copie et al. 1998).
In order to minimize the size of the scaffold, we also truncated seven N-terminal residues which are unstructured in the solution structure of 10FnIII (Fig. 1B, right panel; Main et al. 1992). It would also be advantageous to eliminate these residues, as they may generally affect the binding surface generated by randomization of loops BC and FG as demonstrated by Getmanova et al. (2006) with selected variants that bind KDR. There, they truncated the first eight residues of the N terminus after selection and found binding improved by a factor of 3 on average. In the crystal structure, the N terminus extends between the two randomized loops to connect the 9th and 10th domains of Fibronectin (Fig. 1B, left panel; Leahy et al. 1996). The solution structure, lacking residues 1–7, illustrates the contiguous surface generated by the two randomized loops (Fig. 1B, right panel).
We tested the effect of truncating the seven N-terminal residues on 10FnIII stability by guanidinium chloride denaturation (Fig. 2). Wild-type 10FnIII and the truncation mutant 10FnIII(Δ1–7) were expressed in E. coli and purified by a combination of His6-tag nickel affinity chromatography and size-exclusion chromatography. Chemical denaturation was monitored by tryptophan fluorescence, and the wild-type and the truncation mutant displayed nearly identical denaturant concentrations at the midpoint of unfolding. Free energies of unfolding were determined from the denaturation data (Table 1) and were found to be identical within error, validating the rationale for eliminating residues 1–7 of 10FnIII in the design of our scaffold.
Figure 2.
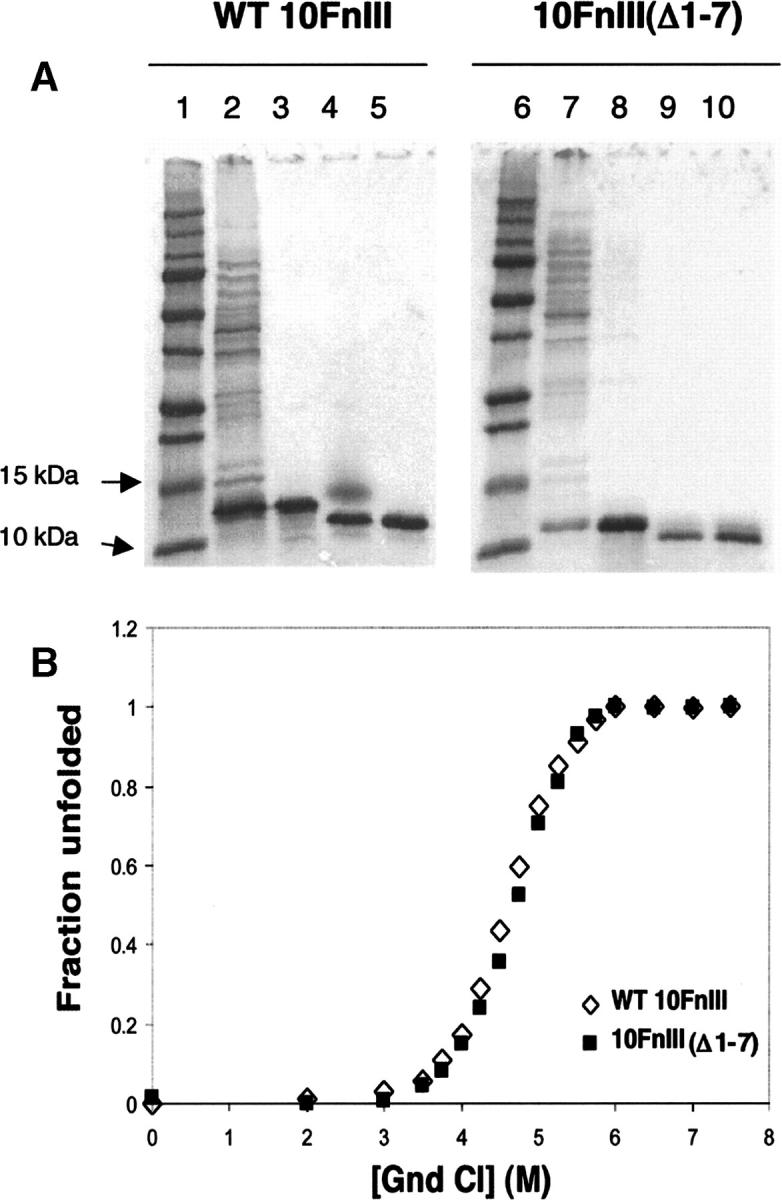
Purification and stability of WT 10FnIII compared to 10FnIII(Δ1–7). (A) Purification of WT 10FnIII (lanes 1–5) and 10FnIII(Δ1–7) (lanes 6–10). (Lanes 1,6) Protein standard. (Lanes 2,7) Cleared lysate. (Lanes 3,8) HisTrap column purification. (Lanes 4,9) Factor Xa cleavage of His6-tag. (Lanes 5,10) Gel filtration purification. (B) Guanidinium chloride denaturation of WT 10FnIII (open diamonds) and 10FnIII(Δ1–7) (closed squares) monitored by Trp fluorescence.
Table 1.
Expression characteristics and stability of 10FnIII variants

Library synthesis
Figure 3 illustrates the scheme for generating the library from eight synthetic oligonucleotides of about 60 nucleotides in length. The length versus number of oligos was chosen to minimize the number of steps in library assembly while minimizing errors resulting from limits on synthesis quality and purification fidelity. Constant regions were designed with codons optimized for translation in both mammalian and bacterial systems by avoiding unfavorable codons for either system. The random loop regions were coded by the degenerate NNS strategy (N = A, C, T, G; S = G, C). Two fragments were built up by Klenow polymerase extension of two oligonucleotides. The BC loop fragment was extended and amplified with two additional oligonucleotides by PCR. The two fragments were digested with BsaI, which left cohesive ends that joined the two fragments without creating mutations upon ligation. The ligation product was amplified by two additional oligonucleotides. The 5′ oligonucleotide contains a T7 promoter sequence for in vitro transcription and a portion of the tobacco mosaic virus translation enhancer sequence for translation in reticulocyte rich lysate (Roberts and Szostak 1997).
Figure 3.

Scheme for constructing 10FnIII library. Linear illustration of library DNA (A) generated from eight oligonucleotides (B).
The complexity of the library was ultimately determined by the total number of molecules generated by the ligation step assuming each product is unique. The goal was to generate at least 1013 molecules, so a 10-fold excess of each fragment was used. After purification, 3 × 1013 DNA molecules were recovered. The library was designed for in vitro selection experiments using mRNA display (Roberts and Szostak 1997; Liu et al. 2000; Takahashi et al. 2003). The efficiency of fusion formation of the 10FnIII library (∼5% of input template forms mRNA–protein fusions) enables the generation of >3 × 1013 protein–mRNA fusion molecules (data not shown).
Loop sequence composition
We next determined the amino acid composition of the randomized BC and FG loops in our DNA library (Fig. 4). This step is important because the observed ratio of A, G, T, and C at the random “N” positions (or G and C at the “S” positions) can differ substantially from the intended 1:1:1:1 due to mixing errors or differing reaction rates of the mononucleotides. To do this, we sequenced 29 10FnIII(Δ1–7) library variants, giving a total of 493 codons. The nucleotide composition for the codons shows that T was slightly overrepresented and G was slightly underrepresented in the synthetic oligos. Summing over both loops, these differences were evened out because the synthetic BC loop oligonucleotide coded the sense strand while the synthetic FG loop oligonucleotide coded the antisense strand.
Figure 4.

Random sequence composition. The frequencies of each amino acid (closed diamonds) are compared to the target NNS distribution (open circles) and the composition of natural protein interfaces involved in protein–protein binding (Chakrabarti and Janin 2002).
The loop amino acid composition is generally very close to the NNS target with only a few amino acids such as alanine and proline being overrepresented (Fig. 4). Since we wish to use these sequences to design binders, we also compared the amino acid composition in our library to that seen in typical protein–protein interfaces (Chakrabarti and Janin 2002). The NNS codons typically used for library design do not closely represent the natural protein interface composition, a consideration for future library designs. On the other hand, our pool contains a broad distribution of all amino acids with no residues having a very rare occurrence and no residues dominating the pool.
Fraction folded analyzed using a GFP reporter screen
Thus far we have discussed library complexity in terms of the number of independent DNA or protein sequences. For scaffold-based libraries, we must also consider another metric, the fraction of sequences that are both in-frame at the DNA level and fold correctly as proteins. The GFP reporter screen developed by Waldo et al. (1999) allows us to address both the frame and foldedness of our library. In this approach, an ORF is fused to the N terminus of GFP. ORFs that contain stop codons, frame shifts, or result in misfolded proteins will interfere with the expression or folding of GFP, resulting in nonfluorescent bacterial colonies. Intact ORFs that produce folded proteins should result in fluorescence that is proportional to the amount of protein expressed.
We obtained the Waldo et al. (1999) vector and modified it to increase the number of transformants that could be obtained (see Materials and Methods). We cloned our 10FnIII(Δ1–7) library into the modified vector (denoted pAO3) and screened ∼2000 colonies. Approximately 45% of these colonies gave visible fluorescence when illuminated with near UV light. We picked 10 of the nonfluorescent colonies and found that all contained either stop codons or frame shifts predicted to eliminate GFP expression. We also picked 94 colonies with varying fluorescence, confirmed that they contained inserts, and sequenced 29 of these as mentioned previously. All of these sequenced 10FnIII(Δ1–7) variants were in-frame and contained no stop codons. From this work we conclude that the vast majority of nonfluorescent colonies contain errors in the 10FnIII(Δ1–7) library ORF and that ∼45% of our library sequences are both in-frame and full-length.
The value of 45% in-frame sequences is in good agreement with predictions made using our library design. We used the observed loop nucleotide composition from the 29 sequenced clones to calculate the fraction of library variants that would lack stop codons. This analysis predicted that 58% of the library would contain no stop codons. The fact that we observe a slightly decreased fraction (45%) of good sequences is in agreement with fact that our calculations did not account for insertion and deletion errors in DNA synthesis. The effect of frame shifts and truncations on library diversity is minimal. Including only full-length, in-frame sequences our library contains >13 trillion unique sequences.
Library expression fitness profile
The GFP screen revealed that for intact library sequences, the fluorescence intensity of GFP fusions varied a great deal from colony to colony. This observation is in line with the original intention of the screen as a tool to quantify the amount of folded soluble protein expressed inside the cell (Waldo et al. 1999). We next sought to quantify the range of fluorescence using the 94 variants we had cloned previously. To do this, bacteria from each colony were cultured and subjected to quantitative fluorescence analysis as previously described (Waldo et al. 1999). Figure 5A shows the fluorescence obtained for each of the 94 variants (labeled Fn01–94) using the wild-type 10FnIII(Δ1–7) as a standard (fluorescence defined as 100%).
Figure 5.
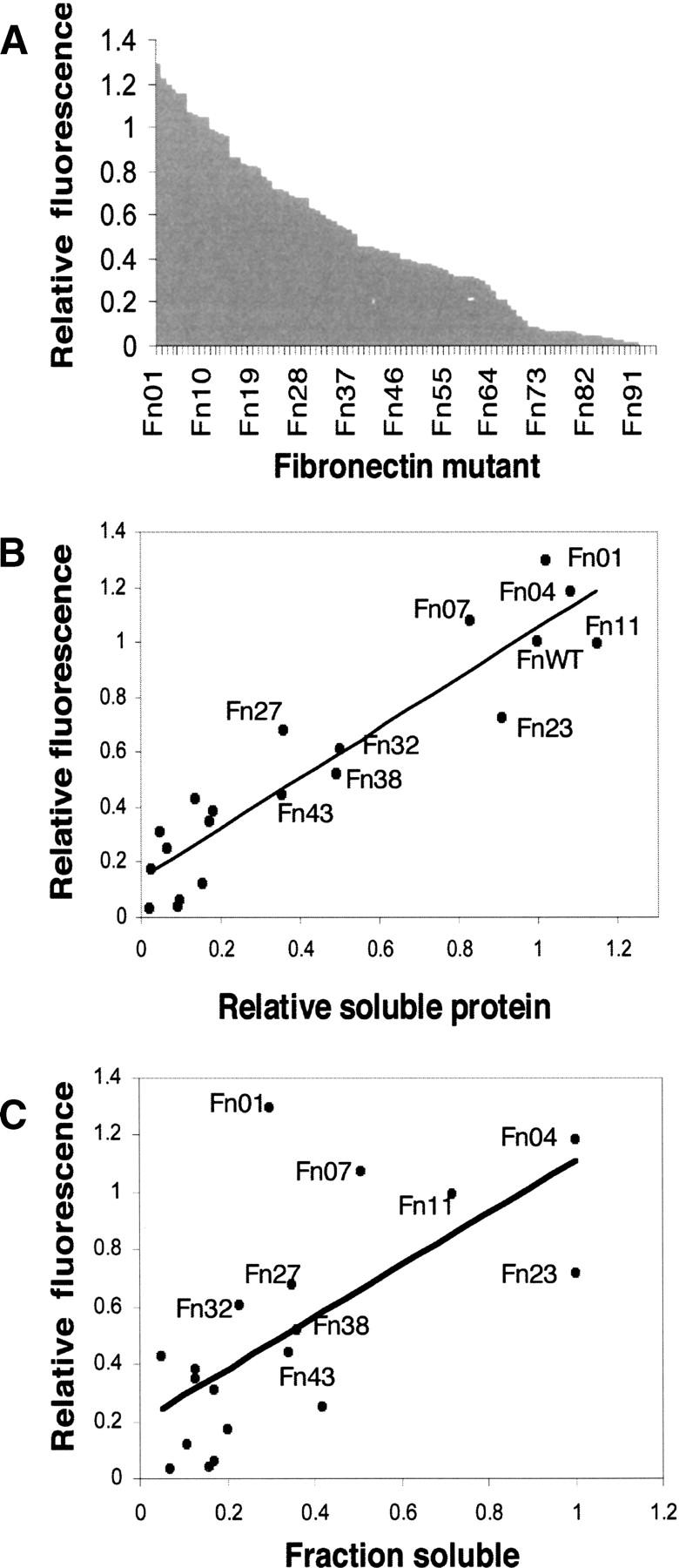
Expression analysis of 10FnIII library. (A) Expression fitness landscape of the 10FnIII library. Fluorescence intensity values of 94 Fn variant–GFP fusions were obtained from cell suspensions and normalized to WT 10FnIII(Δ1–7)-GFP. Each variant is marked by a dash on the X axis. (B) Relation between relative fluorescence and Fn variant soluble expression. Nineteen variants plus WT 10FnIII(Δ1–7) were subcloned into an expression vector without GFP. The relative expression values represent the amount of protein expressed in the soluble fraction relative to wild type measured by band densitometry. (C) The relation between relative fluorescence and Fn variant solubility, determined by band densitometry of soluble and insoluble fractions.
Taken together, the quantitative fluorescence information provides a two-dimensional folding fitness landscape for the library. Several features of this landscape are worth noting. Overall, the fluorescence intensities range from <1%–130% of the standard. However, ∼20% of the library gives fluorescence that is quantitatively similar (80%–130%) to the standard, consistent with the idea that these molecules are well folded. Finally, more than half of the library (62/94 clones) gives fluorescence that is within a factor of 3 of the standard, indicating that the majority of the library variants express at usable levels.
The quantitative GFP analysis should be proportional to the amount of soluble, folded protein expressed. The GFP analysis does not reveal either the amount of each variant that is misfolded or the total amount of expressed protein. To address these two issues, we removed the GFP tags and determined the fraction of soluble and insoluble protein for 19 representative variants and the WT 10FnIII(Δ1–7) using SDS PAGE analysis (see Materials and Methods) (Fig. 5C, X axis). This analysis revealed that the amount of soluble 10FnIII variant measured by gel correlates well with the GFP fluorescence measured in whole cells (Fig. 5B). However, there are large variations in the fraction of total protein that is soluble for the variants we tested. We note that the fraction of soluble protein does not correlate well with the quantitative GFP fluorescence (Fig. 5C).
In the context of 10FnIII, the GFP screen best reflects the amount of soluble protein present, rather than the fraction of the total protein that is soluble. Thus, the GFP screen gives very high scores to proteins such as Fn01, a relatively poorly behaved protein (∼30% of total protein is folded) as well as Fn04 and Fn23, two very well-behaved proteins (∼100% of total protein folded). This apparent paradox is resolved by the fact that Fn01 scores highly in the GFP screen because this clone expresses a truly prodigious amount of both folded and unfolded protein.
Stability of 10FnIII variants
The final question we wished to address was the free energy change caused by introducing 17 nonwild type residues into the 87 residue 10FnIII(Δ1–7) scaffold. Four representative variants, Fn04, Fn23, Fn32, and Fn38 were overexpressed and purified to homogeneity similar to 10FnIII and 10FnIII(Δ1–7) described previously. For each protein, the yield was proportional to the quantitative fluorescence measurements (Table 1). Fn32 expressed the least amount of protein (4 mg/L) and Fn04 expressed the same amount as WT 10FnIII(Δ1–7) (20 mg/L).
All four protein variants displayed cooperative unfolding transitions with m values similar to WT 10FnIII and WT 10FnIII(Δ1–7) (Fig. 6; Table 1). These observations indicated that the proteins were compact, folded, and well behaved at room temperature. The midpoints of unfolding varied from 2 M to 4.5 M guanidinium chloride concentrations, and all the denaturation data could be fit to a two-state folding free energy model (Santoro and Bolen 1988). This analysis revealed that Fn23, Fn32, WT 10FnIII(Δ1–7), and WT 10FnIII have the same unfolding free energy within error (7.1–7.7 kcal/mol). Fn04 and Fn38 were less stable than WT 10FnIII by 2.2 kcal mol−1 and 3.3 kcal mol−1, respectively. Interestingly, protein stability does not correlate with expression. Fn32 is the most stable, but expressed the least protein, whereas the less stable Fn04 expressed the most protein.
Figure 6.

Guanidinium chloride denaturation of 10FnIII variants compared to WT10 FnIII(Δ1–7). (Closed triangles) Fn04; (open circles) Fn23; (open squares) Fn32; (closed circles) Fn38; (crosses) WT 10FnIII(Δ1–7).
The four variants illustrate the sequence differences between the library and the starting scaffold design, WT 10FnIII(Δ1–7) (Table 2). Each protein has only one randomized residue in common with the WT 10FnIII(Δ1–7) loops. None of these identity positions is repeated and all sequences are unique among the four variants. Remarkably, these data suggest that the 10FnIII(Δ1–7) structure can tolerate random mutations accounting for ∼20% of the domain.
Table 2.
Sequences of Fn variants tested for stability

Discussion
We have demonstrated a simple and efficient method of creating a large diversity protein library. Our synthesis scheme required no in vivo steps or in vitro preselections. Our goal was to assemble a high complexity library while retaining high stability and solubility.
Our approach used a shortened version of 10FnIII that we denote 10FnIII(Δ1–7). Our rationale for truncation of the scaffold was based on information from structures of the domain as well as mutation analysis. The seven N-terminal residues are structured in the crystal structure of the 7th–10th fibronectin III domains of fibronectin (Leahy et al. 1996), but not in the solution structure of 10FnIII alone (Main et al. 1992). Crystals of the 10th domain alone could be obtained only after elimination of these residues (Dickinson et al. 1994a). Pro 5 plays an important structural role contributing to the hydrophobic core in the structure of the 7th–10th FnIII domains. However, Cota et al. (2000) demonstrated that mutation of Pro5 to alanine does not effect stability, as would be expected if it does not contribute to the hydrophobic core of 10FnIII alone. Also, Asp7 makes unfavorable electrostatic interactions with other negatively charged residues on the surface of 10FnIII (Koide et al. 2001), further validating the removal of the unstructured residues.
These unstructured residues could bias the outcome of selections or affect binding affinity due to the proximity of the unstructured N terminus to the randomized loops. Recent work on in vitro selected 10FnIII variants indicates that removing the N-terminal eight residues actually improves binding by threefold to the target protein, the extracellular domain of KDR (Getmanova et al. 2006). Our chemical denaturation analysis here provides the first demonstration that 10FnIII(Δ1–7) has equivalent thermodynamic stability to the wild-type domain, and is therefore suitable as a protein scaffold.
We chose to randomize two loops, BC and FG, which are tolerant to mutations. The long FG loop is very flexible, indicated by lack of long-range NOEs observed in NMR experiments (Main et al. 1992; Copie et al. 1998). The DE loop, however, is very rigid and intolerant to mutation compared with the BC and FG loops. For example, previous work showed that inserting four glycine residues into the DE loop substantially destabilized 10FnIII (Batori et al. 2002). In line with this view, Parker et al. (2005) found that an unstable KDR binding variant selected from a three-loop 10FnIII library could be stabilized by replacing the selected DE loop residues with the wild-type sequence.
As long as 17 residues over two loops is sufficient for generating a ligand binding surface, further randomization is not necessary. In order to minimize the likelihood of selecting poorly expressing, unstable variants we chose a more conservative approach in our library design compared to the one used previously for mRNA display selections (Xu et al. 2002; Getmanova et al. 2006). However, the extent of our library randomization is greater than the randomization by Koide et al. (1998) in order to create a larger ligand binding surface (17 vs. 10 randomized residues). The fibronectin type III domain is structurally similar to the camelid antibody VH domain. This domain is known to bind one protein antigen (RNase A) via CDR1 and CDR3 alone (analogous to loops BC and FG, respectively) with a binding surface area typical of protein–protein interactions (Decanniere et al. 1999). Binding surfaces of natural proteins have on average 23 residues that lose accessibility to solvent (Chakrabarti and Janin 2002). On average, 12 of these residues are buried. All 17 residues randomized in our library will not necessarily contribute to a potential binding interface. However, as our library contains more potential binding residues than the average number of buried interface residues in natural proteins, our library will likely be able to generate protein recognition molecules with surface areas similar to those of natural proteins.
We chose a novel approach to analyze the integrity of our libraries, implementing a GFP-based screen to determine expression and folding statistics. Although this screen had previously been used to screen for improved expression of natural protein point mutants (Waldo et al. 1999), we found that it was also very useful for assaying our engineered library (Fig. 5). In our library, the GFP signal best correlates with the absolute amount of soluble protein present, not with the fraction of a given protein that is soluble. Thus, some of the brightly fluorescing colonies contain protein that is poorly behaved (<50% soluble), while others contain variants that are very well behaved (100% soluble). The difference we observe from Waldo et al. (1999) may be due to either the relatively small size of the 10FnIII(Δ1–7), the wide range of expression possible among the variants, or a combination of the two. Despite these minor differences, we believe the screen is very valuable both as a tool to assay the library, and as a future strategy to evolve proteins with increased expression of soluble protein.
We observe a wide range of solubility for the proteins in our library (X axis, Fig. 5C). Our work is the first time that such properties have been measured for a random loop library, and the range is probably endemic to any diverse library based 10FnIII. Our analysis reveals that the majority of the library (66%) expresses a good amount of soluble protein, within a factor of 3 of wt 10FnIII. It is worth noting that the expression characteristics of 10FnIII-based libraries are likely far superior to the most commonly used antibody-based scaffolds (Maynard and Georgiou 2000). It is also clear that very well behaved proteins can be derived from both naïve and selected 10FnIII libraries. Indeed, the KDR binder isolated previously is currently in preclinical development as an anticancer therapeutic. Finally, preliminary work indicates that one or more point mutations to the 10FnIII constant region may greatly enhance both the fraction and total soluble protein expressed, at least on a case-by-case basis (C.A. Olson and R.W. Roberts, unpubl.).
Finally, we find it remarkable that the 29 naïve variants revealed no obvious protein sequence bias at any position in either the BC or FG loops. Indeed, cursory analysis reveals that even clone Fn23, which contains a stretch of three leucines in the FG loop, is still highly expressed and soluble (Fig. 5; Tables 1, 2). The fact that all four variants differ from WT 10FnIII at 16 positions and all four differ from each other at all 17 positions suggests a large amount of diversity is tolerated. If our screen is representative, then over half of all possible sequences surrounding the 10FnIII(Δ1–7) library are accessible for function. This represents an enormous amount of sequence space that can be sampled using directed or natural evolution. Overall, our results demonstrate the importance of combining a balanced library design and the utility of GFP-based library screening.
Materials and methods
Library synthesis
The library was constructed from eight oligonucleotides synthesized at the Yale Keck oligonucleotide synthesis facility. All oligonucleotides were purified by denaturing urea PAGE, recovered by Elutrap electroelution, and ethanol precipitated. Nucleotide positions labeled “1” were mixed with 20% T, 30% C, 30% A, and 20% G. Nucleotide positions labeled “2” were mixed with 60% C and 40% G. Sixteen picamoles of Fnoligo3 (5′-CCAGCCTCCTGATCAGCTGG112112112112112112112CGCTACTACCGCATCACCTACG-3′) containing the randomized BC loop sequence were annealed to 16 pmol Fnoligo4 (5′-GCACGGTGAATTCCTGGACAGGGCTATTGCCACCAGTTTCACCGTAGGTGATGCGGTAGTAGCG-3′) and extended by Klenow DNA polymerase (New England Biolabs) at room temperature for 20 min. After agarose gel electrophoresis the product was amplified and extended by Fnoligo2 (5′-CAATTACAATGCTCGAGGTCGTCGCTGCGACTCCGACCAGCCTCCTGATCAGCTGG-3′) and Fnoligo5 (5′-CCTACCGGTCTCAGCTGATGGTAGCAGTGGACTTGCTGCCAGGCACGGTGAATTCCTGGACAGG-3′) in an 8-mL PCR reaction using Taq DNA polymerase; 1500 pmol of product was recovered after native PAGE (1× TBE, 50 mM NaCl) and electroelution (0.5× TBE); 1600 pmol each of Fnoligo6 (5′-CCTACCGGTCTCACAGCGGCCTGAAACCTGGTGTCGACTATACCATCACGGTGTACGCCGTCACG-3′) and Fnoligo7 (5′-CGGTAGTTGATGGAGATCGG211211211211211211211211211211CGTGACGGCGTACACCGTGA-3′) containing the randomized FG loop positions were extended by Klenow DNA polymerase in a 4-mL reaction. After native PAGE purification, electroelution, and ethanol precipitation, 1000 pmol of product were recovered. The fragments were digested with BsaI at 50°C for 3 h in 1.6 mL reactions using 10 U of enzyme per 10 μg of DNA. Digested fragments were purified with Qiagen spin columns. Of each fragment, 167 pmol were ligated together in a 1-mL reaction using T4 DNA ligase (New England Biolabs). After native PAGE purification, electroelution, and ethanol precipitation, 50 pmol of product were recovered, resulting in a final complexity of 3 × 1013. The library was extended by two additional primers, Fnoligo1 (5′-TTCTAATACGACTCACTATAGGGACAATTACTATTTACAATTACAATGCTCGAGGTCGTCG-3′) and Fnoligo8 (5′ GGAGCCGCTACCGGATCCGGTGCGGTAGTTGATGGAGATCGG), and amplified in a 10-mL PCR to generate 20 copies of the 3 × 1013 independent sequences.
GFP reporter cloning and library expression analysis
The GFP sequence, linker, and stop stuffer was taken from the vector described by Waldo et al. (1999) and placed in pET 16b (creating plasmid pAO3). This was done to increase ligation and transformation efficiencies as a result from cloning into NcoI rather than NdeI, thereby facilitating the screening of a larger number of library members. The internal NcoI restriction site within the GFP sequence was eliminated, and the GFP construct was PCR amplified and digested for ligation into the NdeI and BamHI sites in pET 16b. The GFP construct contains a stop stuffer sequence for library cloning with NdeI and BamHI. In order to preserve the BamHI site in the construct, the 3′ primer contained a BclI restriction site, which forms compatible cohesive ends with the BamHI sequence. Upon ligation of the GFP construct into the NdeI–BamHI sites in pET 16b, the original BamHI site is eliminated while the internal BamHI site is preserved for library cloning. The 10FnIII library was cloned into the NcoI–BamHI sites of the new GFP reporter construct. Ligation products were transformed into E. coli. DH5-α. Purified supercoiled plasmid library was then used for transformation of E. coli. BL21(DE3). Transformants were plated directly on nitrocellulose membranes (Schleicher and Schuell) on LB-Agar plates and grown 10 h at 37°C. The membranes were then transferred to LB-agar plates containing 1 mM IPTG. All solid and liquid media was supplemented with 100 μg/mL ampicillin. The library was illuminated by a handheld UV lamp and GFP expressing fusions were counted and picked for frozen glycerol solution stocks.
Fluorescence measurements of Fn variant cultures were carried out identically to the protocol developed by Waldo et al. (1999). For analysis of expression characteristics in the non-GFP construct, selected variant inserts were digested out of the reporter construct using NcoI and BamHI and ligated directly into a modified pET 16b vector, which was constructed so that a stop codon immediately follows the BamHI sequence. Note that this leaves a C-terminal Gly-Ser extension, which is not predicted to affect stability and is representative of the Fn library, which contains a (GlySer)3 linker to space the mRNA away from the protein upon fusion formation. All variants assayed for expression characteristics were sequenced. Each variant was expressed in a 10-mL culture for 4 h at 37°C and then pelleted. The cell pellets were washed with buffer once (20 mM Tris–HCl, pH 8.0, 150 mM NaCl) and then resuspended with 1 mL of buffer and incubated on ice with 1 mg/mL lysozyme (Sigma) for 45 min. The cells were further lysed by sonicating with three 10-sec pulse sequences at 50% duty cycle. The lysate was cleared by centrifugation at 20,000g for 20 min twice. The cell pellet was washed three times with buffer and then resuspended with 1 mL of buffer. Standard volumes were run on 4%–20% SDS PAGE gels. The fraction of protein for each variant that was soluble was determined by densitometric scanning of Coomassie-stained gel bands. To relate the amount of protein expressed in the soluble fraction among Fn variants, densitometric intensities of gel bands were related to WT 10FnIII(Δ1–7). Two variants plus wild type were expressed and analyzed in triplicate to test variations in expression levels. Levels of soluble protein expressed for Fn01, Fn23, and WT 10FnIII(Δ1–7) had standard deviations of 19%, 14%, and 13%, respectively.
Cloning, expression, and purification of Fn variants
Wild-type and truncated 10FnIII were amplified by PCR with Herculase DNA Polymerase (Stratagene) from the FN7–10 plasmid from Leahy et al. (1996) generously provided by Dr. Harold P. Erickson. The 5′ primer extended the fragments, coding for a His6-tag and factor Xa protease recognition sequence. These fragments were ligated into the pET 16b expression vector (Novagen) with NcoI–BamHI restriction sites (New England Biolabs). The resulting plasmid is similar to the original pET 16b vector except the NdeI restriction sequence immediately following the fXa recognition sequence, which codes for His-Met, is removed. The four Fn variants prepared were cloned and purified identical to the wild-type proteins. One liter of LB culture was inoculated with 10 mL of overnight preculture and grown to mid-log phase (OD = 0.4–0.5) at 37°C. Protein expression was induced by addition of IPTG (Sigma) to 1 mM final concentration. Cultures were pelleted and frozen at −79°C. The cell pellets were thawed on ice and resuspended in 20 mL nickel binding buffer (20 mM Tris-HCl, 500 mM NaCl, 10 mM imidazole at pH 8.0) plus 0.5 mg/mL lysozyme (Sigma), and incubated on ice for 45 min. PMSF (Sigma) was added to a 1 mM final concentration. The cell lysis was aided by sonicating on ice with five pulses at 50% duty cycle lasting 30 sec and separated by 1 min. The lysate was pelleted 2 × 20 min at 13,000 rpm. The cleared lysate was loaded onto a His-Trap Nickel affinity column (Amersham) and washed with 10 column volumes of binding buffer, followed by buffer containing 20 mM and then 30 mM imidazole. All proteins were eluted with 4 column volumes of buffer containing 500 mM imidazole. Buffer was exchanged to factor Xa cleavage buffer (20 mM Tris-HCl at pH 8.0, 100 mM NaCl, 2 mM CaCl2) either using Millipore centriprep centrifugal filtration devices (3000 kDa MWC) or dialysis membranes from Spectrum. Factor Xa (Novagen) cleavage was carried out with 10 U of enzyme per 1 mg of protein overnight at room temperature. Cleavage removed the His6-tag leaving no extra N-terminal residues, except for a Methionine, which was included to represent the in vitro expressed library. The cleavage reaction was directly loaded onto a sephracryl S-100 size exclusion column (Amersham) which was equilibrated with 50 mM Acetate (pH 5.5) and 100 mM NaCl.
Chemical denaturation
Guanidinium chloride (Fluka) induced unfolding transitions were monitored by Trp fluorescence (Shimadzu RF-5301, excitation = 280 nm, emission = 350 nm, slit widths = 5 nm). Measurements were carried out in sodium acetate buffer (pH 5.5), 100 mM NaCl at 30°C. Protein sample concentrations were 3 μM. Free energies of unfolding were obtained using a linear extrapolation method (Santoro and Bolen 1988). Fits were obtained with KaleidaGraph (Synergy software).
Acknowledgments
We thank Dr. Harold P. Erickson for donation of the vector containing the fibronectin 7th–10th FnIII domains, and Dr. Geoffrey S. Waldo for donation of the GFP reporter vector. Thanks to Dr. Premal Shah for technical assistance. C.A.O. was supported by an NSF graduate fellowship. This work was supported by the American Foundation for AIDS Research (RWR, 106573-36-RGNT) and the National Institutes of Health (RWR, RO1 GM60416).
Footnotes
Reprint requests to: Richard W. Roberts, 3710 McClintock Avenue, RTH-507, Los Angeles, CA 90089-1211, USA; e-mail: richard.roberts@usc.edu; fax: (213) 821-4207.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.062498407.
References
- Batori V., Koide, A., and Koide, S. 2002. Exploring the potential of the monobody scaffold: Effects of loop elongation on the stability of a fibronectin type III domain. Protein Eng. 15: 1015–1020. [DOI] [PubMed] [Google Scholar]
- Beste G., Schmidt, F.S., Stibora, T., and Skerra, A. 1999. Small antibody-like proteins with prescribed ligand specificities derived from the lipocalin fold. Proc. Natl. Acad. Sci. 96: 1898–1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binz H.K. and Pluckthun, A. 2005. Engineered proteins as specific binding reagents. Curr. Opin. Biotechnol. 16: 459–469. [DOI] [PubMed] [Google Scholar]
- Binz H.K., Stumpp, M.T., Forrer, P., Amstutz, P., and Pluckthun, A. 2003. Designing repeat proteins: Well-expressed, soluble and stable proteins from combinatorial libraries of consensus ankyrin repeat proteins. J. Mol. Biol. 332: 489–503. [DOI] [PubMed] [Google Scholar]
- Binz H.K., Amstutz, P., Kohl, A., Stumpp, M.T., Briand, C., Forrer, P., Grutter, M.G., and Pluckthun, A. 2004. High-affinity binders selected from designed ankyrin repeat protein libraries. Nat. Biotechnol. 22: 575–582. [DOI] [PubMed] [Google Scholar]
- Chakrabarti P. and Janin, J. 2002. Dissecting protein–protein recognition sites. Proteins 47: 334–343. [DOI] [PubMed] [Google Scholar]
- Ciarapica R., Rosati, J., Cesareni, G., and Nasi, S. 2003. Molecular recognition in helix–loop–helix and helix–loop–helix-leucine zipper domains. Design of repertoires and selection of high affinity ligands for natural proteins. J. Biol. Chem. 278: 12182–12190. [DOI] [PubMed] [Google Scholar]
- Cicortas Gunnarsson L., Nordberg Karlsson, E., Albrekt, A.S., Andersson, M., Holst, O., and Ohlin, M. 2004. A carbohydrate binding module as a diversity-carrying scaffold. Protein Eng. Des. Sel. 17: 213–221. [DOI] [PubMed] [Google Scholar]
- Copie V., Tomita, Y., Akiyama, S.K., Aota, S., Yamada, K.M., Venable, R.M., Pastor, R.W., Krueger, S., and Torchia, D.A. 1998. Solution structure and dynamics of linked cell attachment modules of mouse fibronectin containing the RGD and synergy regions: Comparison with the human fibronectin crystal structure. J. Mol. Biol. 277: 663–682. [DOI] [PubMed] [Google Scholar]
- Cota E., Hamill, S.J., Fowler, S.B., and Clarke, J. 2000. Two proteins with the same structure respond very differently to mutation: The role of plasticity in protein stability. J. Mol. Biol. 302: 713–725. [DOI] [PubMed] [Google Scholar]
- Decanniere K., Desmyter, A., Lauwereys, M., Ghahroudi, M.A., Muyldermans, S., and Wyns, L. 1999. A single-domain antibody fragment in complex with RNase A: Non-canonical loop structures and nanomolar affinity using two CDR loops. Structure Fold. Des. 8: 361–370. [DOI] [PubMed] [Google Scholar]
- Dickinson C.D., Gay, D.A., Parello, J., Ruoslahti, E., and Ely, K.R. 1994a. Crystals of the cell-binding module of fibronectin obtained from a series of recombinant fragments differing in length. J. Mol. Biol. 238: 123–127. [DOI] [PubMed] [Google Scholar]
- Dickinson C.D., Veerapandian, B., Dai, X.P., Hamlin, R.C., Xuong, N.H., Ruoslahti, E., and Ely, K.R. 1994b. Crystal structure of the tenth type III cell adhesion module of human fibronectin. J. Mol. Biol. 236: 1079–1092. [DOI] [PubMed] [Google Scholar]
- Getmanova E.V., Chen, Y., Bloom, L., Gokemeijer, J., Shamah, S., Warikoo, V., Wang, J., Ling, V., and Sun, L. 2006. Antagonists to human and mouse vascular endothelial growth factor receptor 2 generated by directed protein evolution in vitro. Chem. Biol. 13: 549–556. [DOI] [PubMed] [Google Scholar]
- Koide A., Bailey, C.W., Huang, X., and Koide, S. 1998. The fibronectin type III domain as a scaffold for novel binding proteins. J. Mol. Biol. 284: 1141–1151. [DOI] [PubMed] [Google Scholar]
- Koide A., Jordan, M.R., Horner, S.R., Batori, V., and Koide, S. 2001. Stabilization of a fibronectin type III domain by the removal of unfavorable electrostatic interactions on the protein surface. Biochemistry 40: 10326–10333. [DOI] [PubMed] [Google Scholar]
- Leahy D.J., Aukhil, I., and Erickson, H.P. 1996. 2.0 A crystal structure of a four-domain segment of human fibronectin encompassing the RGD loop and synergy region. Cell 84: 155–164. [DOI] [PubMed] [Google Scholar]
- Liu R., Barrick, J.E., Szostak, J.W., and Roberts, R.W. 2000. Optimized synthesis of RNA–protein fusions for in vitro protein selection. Methods Enzymol. 318: 268–293. [DOI] [PubMed] [Google Scholar]
- Main A.L., Harvey, T.S., Baron, M., Boyd, J., and Campbell, I.D. 1992. The three-dimensional structure of the tenth type III module of fibronectin: An insight into RGD-mediated interactions. Cell 71: 671–678. [DOI] [PubMed] [Google Scholar]
- Maynard J. and Georgiou, G. 2000. Antibody engineering. Annu. Rev. Biomed Eng. 2: 339–376. [DOI] [PubMed] [Google Scholar]
- Parker M.H., Chen, Y., Danehy, F., Dufu, K., Ekstrom, J., Getmanova, E., Gokemeijer, J., Xu, L., and Lipovsek, D. 2005. Antibody mimics based on human fibronectin type three domain engineered for thermostability and high-affinity binding to vascular endothelial growth factor receptor two. Protein Eng. Des. Sel. 18: 435–444. [DOI] [PubMed] [Google Scholar]
- Roberts R.W. and Szostak, J.W. 1997. RNA–peptide fusions for the in vitro selection of peptides and proteins. Proc. Natl. Acad. Sci. 94: 12297–12302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoro M.M. and Bolen, D.W. 1988. Unfolding free energy changes determined by the linear extrapolation method. 1. Unfolding of phenylmethanesulfonyl α-chymotrypsin using different denaturants. Biochemistry 27: 8063–8068. [DOI] [PubMed] [Google Scholar]
- Stumpp M.T., Forrer, P., Binz, H.K., and Pluckthun, A. 2003. Designing repeat proteins: Modular leucine-rich repeat protein libraries based on the mammalian ribonuclease inhibitor family. J. Mol. Biol. 332: 471–487. [DOI] [PubMed] [Google Scholar]
- Takahashi T.T., Austin, R.J., and Roberts, R.W. 2003. mRNA display: Ligand discovery, interaction analysis and beyond. Trends Biochem. Sci. 28: 159–165. [DOI] [PubMed] [Google Scholar]
- Wahlberg E., Lendel, C., Helgstrand, M., Allard, P., Dincbas-Renqvist, V., Hedqvist, A., Berglund, H., Nygren, P.A., and Hard, T. 2003. An affibody in complex with a target protein: Structure and coupled folding. Proc. Natl. Acad. Sci. 100: 3185–3190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldo G.S., Standish, B.M., Berendzen, J., and Terwilliger, T.C. 1999. Rapid protein-folding assay using green fluorescent protein. Nat. Biotechnol. 17: 691–695. [DOI] [PubMed] [Google Scholar]
- Xu L., Aha, P., Gu, K., Kuimelis, R.G., Kurz, M., Lam, T., Lim, A.C., Liu, H., Lohse, P.A., and Sun, L., et al. 2002. Directed evolution of high-affinity antibody mimics using mRNA display. Chem. Biol. 9: 933–942. [DOI] [PubMed] [Google Scholar]