

Abstract
The Nostoc punctiforme genes Np275 and Np276 are two adjacently encoded proteins of 98 and 75 amino acids in length and exhibit sequences composed of tandem pentapeptide repeats. The structures of Np275 and a fusion of Np275 and Np276 were determined to 2.1 and 1.5 Å, respectively. The two Nostoc proteins fold as highly symmetric right-handed quadrilateral β-helices similar to the mycobacterial protein MfpA implicated in fluoroquinolone resistance and DNA gyrase inhibition. The sequence composition of the intervening coding region and the ability to express a fused protein by removing the stop codon for Np275 suggests Np275 and Np276 were recently part of a larger ancestral pentapeptide repeat protein.
Keywords: structure determination, pentapeptide repeat protein, right-handed quadrilateral β-helix, cyanobacterium, fusion protein
Members of the pentapeptide repeat protein (PRP) family consist of tandemly encoded repeats with the consensus sequence [S,T,A,V][D,N][L,F][S,T,R][G] (Bateman et al. 1998; Vetting et al. 2006). Sequence analysis suggests there are few excursions (loops, etc.) from the main structural element encoded by pentapeptide repeats. The first structure of a PRP family member was the mycobacterial fluoroquinolone resistance protein, MfpA (Hegde et al. 2005). MfpA folds as a right-handed quadrilateral β-helix (RHQBH) with shape and charge properties reminiscent of B-form DNA. MfpA, and its functional homolog Qnr, were found to bind to and inhibit DNA gyrase (Hegde et al. 2005; Tran et al. 2005), providing a mechanism by which these proteins could protect cells from the cytotoxic effect of fluoroquinolones. Similarly, the McbG protein provides self-resistance to Microcin B17, a DNA supercoiling inhibitor, although it is predicted that the mode of binding to DNA gyrase would be different (Kolter and Moreno 1992; Vetting et al. 2006). The function and mode of action of most PRP proteins are unknown. The HglK protein has been associated with transport or localization of glycolipids in cyanobacterium (Black et al. 1995), and the RfrA protein of Synechocystis sp. Strain 6803 was shown to be associated with magnesium uptake (Chandler et al. 2003); however, the method by which these proteins exert their effects is unknown. Proteins with PRP domains have been found in the genomes of almost all organisms, with the exception of yeast, although each typically has only one or two occurrences. Cyanobacteria appear to be unique in the multiplicity of proteins containing PRP domains. Nostoc punctiforme, a nitrogen-fixing cyanobacterium that can occupy a wide variety of ecological niches and can exist in several vegetative states, has 40 proteins with PRP domains (Meeks et al. 2001; Vetting et al. 2006). Npun02000275 (Np275) and Npun02000276 (Np276) are two adjoining, chromosomally encoded, Nostoc punctiforme proteins and are constructed almost entirely from pentapeptide repeats. They are the smallest proteins with a PRP domain in the Nostoc punctiforme genome with 98 and 75 residues, respectively. In contrast to MfpA, which has a mixed phenylalanine/leucine composition for the central i residue, and often has a serine or threonine in the i−2 position, Np275 and Np276 have predominantly leucine and alanine at the i and i−2 positions, respectively. In this respect, the repeats of Np275 and Np276 are more representative of the typical pentapeptide repeat domains that have been sequenced to date (Pfam-00805). Interestingly, the intervening sequence between the stop codon for Np275 and the start codon for Np276 not only follows the pentapeptide consensus sequence, but does so in the correct frame, suggesting that Np275 and Np276 were recently a single longer protein. In order to further characterize the PRP fold, the structure of Np275 and its fusion with Np276 were determined by X-ray crystallography.
Results and Discussion
Molecular replacement solution
The highly regular nature of the PRP fold suggested that molecular replacement could be used as a phasing method. A molecular replacement model of Np275 consisting of residues 20–98 (approximately four coils) was built utilizing the N-terminal coils of the pentapeptide repeat protein MfpA since this is the region with the highest sequence identity (34% over 78 residues), particularly at the i and i−2 positions (see Fig. 1A for nomenclature). In addition, the structure of MfpA was useful for modeling side chains, especially those that conform to the consensus sequence and were observed to be in definable conformations. Molecular replacement solutions assuming a monomer per asymmetric unit produced incomplete crystal packing, while the top solution assuming two molecules per asymmetric unit yielded reasonable packing; however, attempts to refine this solution did not produce a reasonable drop in the Rfree. Rigid body refinement after conversion to polyalanine resulted in electron density maps with obvious electron density for side chains consistent with the general “correctness” of the molecular replacement solution. A closer inspection of the maps indicated that one of the monomers from the molecular replacement solutions was rotated 90° incorrectly. Correcting the misalignment and further modeling of side chain conformations based on the polyalanine-phased electron density resulted in vastly improved maps and permitted completion of the structure. Examination of “incorrect” solutions, many of which had very similar molecular replacement scores, indicates that both rotational ambiguity resulting in incorrect placement of the RHQBH faces and translational ambiguity resulting in misalignment of coils will be serious complications in the determination of RHQBHs by molecular replacement, and that in construction of a molecular replacement model every attempt should be made to make individual faces and coils unique by including the side chains.
Figure 1.
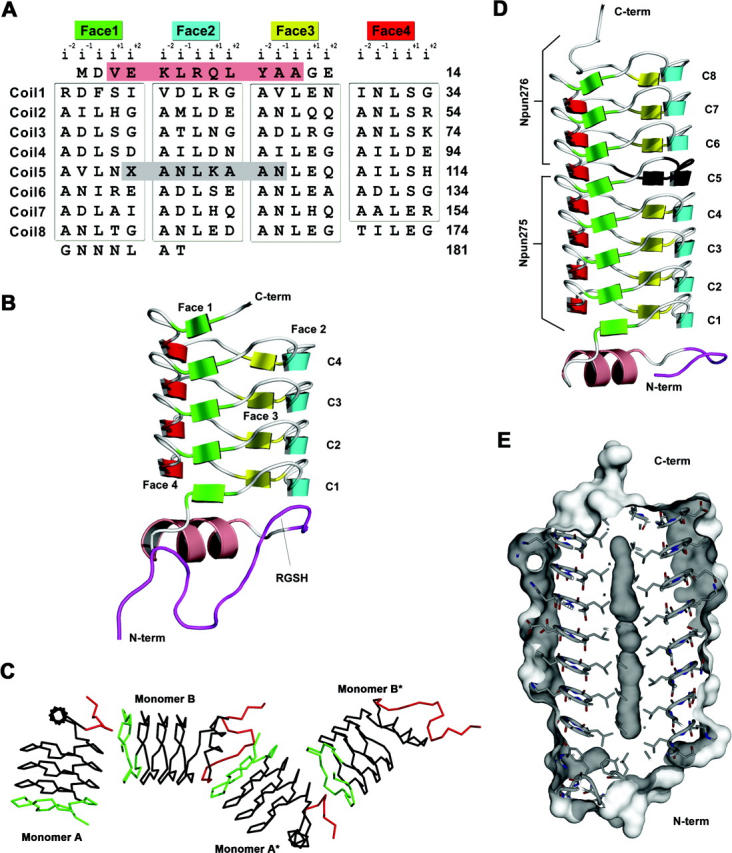
Primary and three-dimensional structures of Np275 and Np275/276. (A) Structure-based sequence alignment of Np275/276. The 31 pentapeptide repeats that are in the RHQBH conformation (boxed) are organized by their location on the faces of the quadrilateral. (Salmon) The single α-helix, (dark gray) the eight residues encoded by the DNA located between Np275 and Np276. For Np275/276 the stop codon (X) was mutated to Gln. (B) Ribbon representation of Np275. (Maroon) Residues originating from the N-terminal cleavable 6×-His-tag. (C) Np275 crystal contacts between monomers involving the N-terminal 6×-His-tag (red) and the exposed C-terminal coil (green). (D) Ribbon representation of Np275/276. (Black) The “stitching” residues between Np275 and Np276, (maroon) residues originating from the N-terminal cleavable 6×-His-tag. (E) Connolly surface of Np275/276.
Structure of Np275
The structure of Np275 as a single domain, and in fusion with Np276, are very similar (RMSD of 0.29 Å over 83 Cα, Table 1), so the nonfusion protein will be discussed only briefly. The majority of Np275 forms a right-handed quadrilateral β-helix, similar to that observed for the mycobacterial fluoroquinolone-resistance peptide, MfpA (Fig. 1B). In the RHQBH fold, a pentapeptide repeat occupies each of the four faces of the quadrilateral-shaped coils with the i and i−2 residues’ side chains facing inward and the i−1, i+1, and i+2 residues’ side chains facing outward. Np275 has four complete coils, starting with residue 15 and ending at residue 98, the C terminus. The PRP domain is partially capped on the N-terminal end by an α-helix while the C-terminal coil is not capped, exposing the central hydrophobic residues and β-strands to external interactions. Interestingly, the hexahistidine tag used in the purification of Np275 is partially visible (16 and 10 amino acids) in both monomers in the asymmetric unit and each makes important crystal contacts, through both hydrophobic interactions and hydrogen bonds, with the open coil of the C terminus of the opposing monomer (Fig. 1C). In addition, in one of the monomers the thrombin cleavage sequence RGSH forms part of an additional coil at the N terminus by mimicking the conformation of the i+1, i+2, i−2, and i−1 residues, respectively (Fig. 1B,C). Glycine and serine residues are among the residue types that are seen most frequently at the i+2 and i−2 positions, respectively, of the typical pentapeptide. The interactions with the hexahistidine tag highlight the aggregation potential of β-helical proteins that are not fully capped (Richardson and Richardson 2002).
Table 1.
Diffraction data collection and refinement statistics

Structure of Np275/276
An analysis of the genomic environment of Np275 led to the observation that the genes for Np275 and Np276 are in the same frame and that the intervening 21 bases would code for a continuation of the pentapeptide repeat. The DNA encoding Np275 and Np276 was PCR amplified from Nostoc punctiforme as a single fragment and cloned into an overexpression vector. Sequencing of PCR-amplified DNA confirmed the validity of the stop codon, and, when overexpressed, only Np275 was apparent in crude extracts. In order to study the possibility that Np275 and Np276 may have once been a single larger PRP, the stop codon of Np275 was mutated to a Gln to create a fusion protein. The fusion protein could be overexpressed, purified by Ni-NTA chromatography, crystallized, and its structure determined by molecular replacement using Np275 as the starting model.
The fusion of Np275 to Np276 extends the four-coil β-helix of Np275 by an additional 3.75 coils, yielding a protein that has a cylindrical shape with dimensions of ∼22 Å in diameter by 55 Å in length (Fig. 1D). The sequence composition and structure of Np275/276 are consistent with observations previously made for the pentapeptide repeats of MfpA (Hegde et al. 2005). The central i residue is hydrophobic and points inward toward the center of the β-helix, while the internal facing i−2 residue is located in the corners of the β-helix and typically is a small hydrophobic or polar residue. The three external residues (i−1, i+1, and i+2) are much more variable; although, as observed for MfpA, there is a subset of amino acids that occurs more frequently at these positions. Firstly, the i+2 residue is in the left-handed α-helical conformation that favors glycine, in this case in 10 of 31 pentapeptides. Secondly, in Np275/276, 21 out of 31 pentapeptides have an Asn or Asp as the i−1 residue, and in all but one case the side chain is hydrogen bonded to the backbone amide of the i+1 residue. In addition, in 12 of these 21 coils there is a corresponding hydrogen bond to the side chain of the i+1 residue where the i+1 residue is a Ser, Thr, or Arg. Therefore, the propensity of the i−1 to be Asn or Asp is linked to the propensity of the i+2 to be a Ser, Thr, or Arg.
There are two “conformations” observed for the individual pentapeptides of MfpA; those utilizing a type II turn and those utilizing a type IV turn. In the type II turns, the backbone carbonyl of the i residue hydrogen bonds to the i−2 backbone amide of the following pentapeptide, and the i−1 residue is the only position that participates in full intercoil hydrogen bonding. In contrast, in the type IV turns there is a rotation of the peptide bond between the i and i+1 residue, and the i−1, i, and i+1 residues participate fully in intercoil hydrogen bonding. The propensity of pentapeptides to utilize type II versus type IV turns appears to be associated with the identity of the central i residues of the coil, with those coils that have a high proportion of phenylalanines as the i residue typically having type IV turns, while those coils with a high proportion of leucines as the central residue predominately utilizing type II turns. The type IV turns result in a larger coil area that can be occupied by the bulkier phenylalanine residue. Indeed, in Np275/276 the i residue is a leucine in 29 out of 31 pentapeptides, and all of the eight coils utilize type II turns.
The C-terminal 12 residues of Np275/276 project along the β-helical axis as a random coil, although only six of these residues were visible in electron density maps. The fusion protein therefore has capping features at both ends, with a random coil at the C terminus and an α-helix at the N terminus. This is in contrast to the open C-terminal face of Np275 and what would be predicted to be an open N-terminal face of Np276. These open faces are eliminated by the fusion of Np275 and Np276, with the single replacement of the stop codon by Gln99 and the remaining intervening genomically encoded residues seamlessly incorporated into the β-helix as a portion of coil5, where they adopt the same conformation as observed in all of the other coils. This is consistent with Np275 and Np276 once being a single pentapeptide protein, and suggests that pentapeptide repeat proteins may be exceptionally modular, with additional coils being appended or removed through recombinatorial and mutational events.
The major differences between MfpA and Np275/276 are their oligomeric state and the lack of sequence diversity in Np275/276. MfpA forms a dimer through the interaction of C-terminal α-helices, forming an approximately coincident β-helical axis ∼100 Å in length (Hegde et al. 2005). In contrast, Np275 and Np275/276 are monomeric in all crystal forms, and as determined by gel filtration (data not shown). The dimeric state of MfpA was proposed to be an important contributing factor to its interaction with DNA gyrase (Hegde et al. 2005). While MfpA utilizes phenylalanines and leucines at the i position and serines, threonines, cysteines, and alanines at the i−2 position, Np275/276 utilizes almost exclusively leucines and alanines, respectively, at these positions. This lack of sequence diversity leads to a highly uniform β-helix, much more so than observed for MfpA, whose dimensions are more variable. Comparison amongst the coils of Np275/276 yields RMSDs of <0.3 Å, except for coil1 and coil8, which are slightly distorted due to being initiating and terminating coils. The largest β-helix deviation in MfpA is a ∼12° bend in the helical axis between the N-terminal coils and the C-terminal coils due to disruption of interhelical hydrogen bonding between coils 4, 5, and 6 (Hegde et al. 2005). In contrast, the intercoil hydrogen bonding of Np275/276 is consistent throughout the structure, and there is no bend in the helical axis. Indeed, an examination of the sequences of pentapeptide repeat proteins in general indicates that most contain repeats with alanine and leucine at the i−2 and i positions, respectively; therefore, the structure of Np275/276 is more representative of PRP structures than MfpA.
MfpA was found to bind to DNA gyrase with a Kd of 450 nM, and a molecular model suggested that it may interact with the electropositive “saddle” of DNA gyrase (Hegde et al. 2005). MfpA has an electronegative surface potential carrying an excess of five negative to positive residues at its surface, for a total charge of −10 for the dimer (362 residues). Np275 and Np275/276 are even more electronegative with a total of −9 (98 residues) and −19 (181 residues) excess negative to positive residues at their surfaces, respectively. However, neither Np275 nor Np275/276 exhibited any significant inhibition of DNA gyrase supercoiling activity (data not shown). This suggests that MfpA is exhibiting DNA “mimicry” in binding to and inhibiting DNA gyrase by specific interactions rather than just by putting an electronegative surface potential on a right-handed helical scaffolding.
Like MfpA, Np275/276 has unoccupied internal molecular surface volumes along the helical axis; however, in Np275/276 these volumes are continuous (volume 281 Å3, Fig. 1E), mainly as a result of the smaller internal central residue, leucine. Interestingly, the pentapeptide protein HglK is proposed to be involved in glycolipid localization and/or assembly into heterocysts of Anabaena sp. Strain PCC 7120 (Black et al. 1995). The internal cavity of Np275/276 is not large enough to accommodate the linear hydrocarbon tail of a glycolipid; however, it may be accommodated by an expansion of the internal volume upon conversion of type II turns to type IV. Although this type of flexibility has not been observed in the conformation of individual pentapeptides in the structures of RHQBHs solved to date, the main factor determining the type of turn appears to be strictly related to the bulkiness of the internal residues, and perhaps a coil with predominately leucine residues and type II turns could transition to type IV turns upon forming hydrophobic interaction with an unbranched hydrocarbon.
Whether or not Np0275 and Np0276 are expressed in Nostoc punctiforme is not known, and it is therefore possible that one or both of these proteins are genomic “relics.” Neither protein is preceded by a ribosome binding site, although this is not uncommon as cyanobacterial proteins are typically preceded by a Shine-Dalgarno sequence <50% of the time (Sazuka and Ohara 1996; Starmer et al. 2006). Even if they are not expressed, they may be genetically stable and provide functional diversity when incorporated into future pentapeptide repeat proteins.
Further genetic and molecular characterizations of pentapeptide repeat proteins are required to determine the scope of the functionality of the right-handed quadrilateral β-helix. However, these structures confirm that primary sequence information can be used predictively in structural fold assignments for the PRP family. The robustness of the fold suggests that it may be possible to design, either de novo or through selection, RHQBH proteins with defined biochemical functions, including binding to DNA-binding proteins other than DNA gyrase.
Materials and Methods
Protein cloning, expression, and purification
The Np275 open reading frame was amplified using Nostoc punctiforme ATCC 29,133 genomic DNA by standard PCR techniques using the oligonucleotides NpPF (5′-ATCCCGCTCATATGGACGTAGAAAAACTCAGG-3′) and NpPR1 (5′-ATCCCGCTAAGCTTCTAATTTAAAACGGCTTCATC-3′) containing the underlined NdeI and HindIII restriction sites shown, respectively. The Np275 gene was cloned into the Nde1 and HindIII sites of pET-28a (Novagen). The DNA fragment containing the adjacent reading frames of Np275 and Np276 was PCR amplified and cloned in pET-28a as above except the reverse oligonucleotide was NpPR2 (5′-ATCCCGCTAAGCTTTTAGGTTGCAAGATTGTT-3′) to obtain both genes. The stop codon at the end of Np275 was mutated to Gln (TAG→CAG) using the QuikChange Site-Directed Mutagenesis Kit (Stratagene). All of the constructs were checked by DNA sequencing and found to be free of mutations.
The recombinant plasmids were transformed into Escherichia coli strain BL21 (DE3). For protein expression, 1 L of LB medium supplemented with kanamycin (30 μg/mL) was inoculated with 10 mL of overnight culture and incubated at 37°C. The culture was grown to mid-log phase (A600 ∼0.8), induced with 0.5 mM isopropyl thio-β-D-galactoside, and further incubated for 4–6 h. Recombinant proteins were purified by nickel affinity chromatography. Fractions containing pure protein (as determined by SDS-PAGE) were pooled, precipitated by ammonium sulfate at 85% saturation, and collected by centrifugation. For Np275, the pellet was resuspended in a minimal volume of 10 mM Tris pH 8.0, dialyzed overnight against the same buffer, concentrated to 25 mg/mL by centrifugal filtration in Amicon Ultra-15 concentrators (5-kDa cutoff), and stored at −80°C. The ammonium sulfate pellet of Np275/Np276 was treated similarly except the buffer was 15 mM Tris pH 8.0, 100 mM NaCl and an additional gel filtration chromatography step (Superdex S75) was used to obtain a homogeneous preparation.
Crystallization
Solution conditions that yielded crystals of Np275 and Np275/276 were identified using commercially available crystallization screens and vapor diffusion under oil. Typically, 2 μL of purified protein was combined with 2 μL of crystallization reagent under 150 μL of silicon oil (Fisher). The crystallization plates were stored at 18°C with the oil exposed to room humidity. Initial crystallization hits were refined using vapor diffusion under oil, and the resultant crystals were checked for suitable diffraction. All crystallographic data were collected on a MSC R-Axis IV++ image plate detector using CuKα radiation from a Rigaku RU-H3R X-ray generator and processed using MOSFLM (Leslie 2006). All protein preparations used in structure determination retained the 20-amino-acid hexahistidine thrombin-cleavable tag.
Np275 (25 mg/mL, 10mM Tris pH 8.0) crystallized in 20%–30% PEG 3350 (w/v), 100 mM NaCacodylate pH 6.8, 200 mM LiCl. Crystals grew as rods over 2–7 d with maximum dimensions of 0.3 × 0.1 × 0.1 mm. Crystals were soaked in 40% PEG3350 (w/v), 100 mM NaCacodylate pH 6.8, 200 mM LiCl prior to vitrification in liquid nitrogen. Crystals of Np275 belong to the orthorhombic space group P212121 with unit cell dimensions of a = 29.3, b = 63.2, c = 100.7 Å. Solvent content analysis suggested one (67.2% solvent) or two (34.3% solvent) molecules per asymmetric unit.
Np275/Np276 (10 mg/mL, 5 mM Tris pH 8.0, 33 mM NaCl) crystallized in 2.0–3.0 M (NH4)2SO4, 100 mM MES pH 6.5. Distorted bipyramidal shaped crystals grew over 1–2 wk in drops that had undergone a large depletion in volume by evaporation, and obtained maximum dimensions of 0.5 × 0.4 × 0.4 mm. Crystals were soaked in 3.5 M (NH4)2SO4, 100 mM MES pH 6.5 prior to vitrification in liquid nitrogen. Crystals of Np275/Np276 belong to the orthorhombic space group P212121 with unit cell dimensions of a = 49.6, b = 55.5, c = 59.0 Å. There is one molecule per asymmetric unit with a solvent content of 33.8%.
Structure determination
The structure of Np275 was determined by molecular replacement utilizing the molecular replacement program MOLREP and a molecular model of Np275 based on structurally homologous regions of MfpA (PDB ID 2BM5). Due to the symmetrical nature of the molecular replacement model, visual inspection of solutions and their resultant electron density maps was required to obtain correctly orientated initial models (see Results and Discussion for details). The resulting modified solution was improved using the molecular graphics program COOT (Emsley and Cowtan 2004) and refined with REFMAC (Murshudov et al. 1997). The final model consists of the complete Np275 polypeptide chain, 104 waters, and one chloride ion. In addition, a total of 27 residues from the N-terminal cleavable his-tag, nine from monomer A, and 18 from monomer B were modeled.
Initial phases for the Np275/Np276 data set were obtained utilizing residues 1–98 of Np275 as a molecular replacement model in the program AMORE (Navaza 1994). The majority of the model was built by the automated fitting and phasing program ARP/WARP (Perrakis 1997). The remainder of the structure was manually built with the molecular graphics program COOT (Emsley and Cowtan 2004), and refined in REFMAC (Murshudov et al. 1997). The final model contains 150 waters, one MES molecule, seven residues from the N-terminal cleavable his-tag, and all of the Np275/Np276 polypeptide chain except six C-terminal residues.
Atomic coordinates
The atomic coordinates and structure factors have been deposited at the Research Collaboratory for Structural Bioinformatics under PDB ID 2J8I and 2J8K.
Acknowledgments
This work was supported by National Institutes of Health Grants AI33696 (to J.S.B.) and T32 AI07501 (to M.W.V.).
Footnotes
Reprint requests to: John S. Blanchard, Department of Biochemistry, Albert Einstein College of Medicine, 1300 Morris Park Avenue, Bronx, NY 10461, USA; e-mail: blanchar@aecom.yu.edu; fax: (718) 430-8565.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.062637707.
References
- Bateman A., Murzin, A.G., and Teichmann, S.A. 1998. Structure and distribution of pentapeptide repeats in bacteria. Protein Sci. 7: 1477–1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black K., Buikema, W.J., and Haselkorn, R. 1995. The hglK gene is required for localization of heterocyst-specific glycolipids in the cyanobacterium Anabaena sp. strain PCC 7120. J. Bacteriol. 177: 6440–6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandler L.E., Bartsevich, V.V., and Pakrasi, H.B. 2003. Regulation of manganese uptake in Synechocystis 6803 by RfrA, a member of a novel family of proteins containing a repeated five-residues domain. Biochemistry 42: 5508–5514. [DOI] [PubMed] [Google Scholar]
- Emsley P. and Cowtan, K. 2004. Coot: Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60: 2126–2132. [DOI] [PubMed] [Google Scholar]
- Hegde S.S., Vetting, M.W., Roderick, S.L., Mitchenall, L.A., Maxwell, A., Takiff, H.E., and Blanchard, J.S. 2005. A fluoroquinolone resistance protein from Mycobacterium tuberculosis that mimics DNA. Science 308: 1480–1483. [DOI] [PubMed] [Google Scholar]
- Kolter R. and Moreno, F. 1992. Genetics of ribosomally synthesized peptide antibiotics. Annu. Rev. Microbiol. 46: 141–163. [DOI] [PubMed] [Google Scholar]
- Leslie A.G. 2006. The integration of macromolecular diffraction data. Acta Crystallogr. D Biol. Crystallogr. 62: 48–57. [DOI] [PubMed] [Google Scholar]
- Meeks J.C., Elhai, J., Thiel, T., Potts, M., Larimer, F., Lamerdin, J., Predki, P., and Atlas, R. 2001. An overview of the genome of Nostoc punctiforme, a multicellular, symbiotic cyanobacterium. Photosynth. Res. 70: 85–106. [DOI] [PubMed] [Google Scholar]
- Murshudov G.N., Vagin, A.A., and Dodson, E.J. 1997. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53: 240–255. [DOI] [PubMed] [Google Scholar]
- Navaza J. 1994. An automated package for molecular replacement. Acta Crystallogr. A50: 157–163. [Google Scholar]
- Perrakis A. 1997. wARP: Improvement and extension of crystallographic phases by weighted averaging of multiple-refined dummy atomic models. Acta Crystallogr. D53: 448–455. [DOI] [PubMed] [Google Scholar]
- Richardson J.S. and Richardson, D.C. 2002. Natural β-sheet proteins use negative design to avoid edge-to-edge aggregation. Proc. Natl. Acad. Sci. 99: 2754–2759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sazuka T. and Ohara, O. 1996. Sequence features surrounding the translation initiation sites assigned on the genome sequence of Synechocystis sp. strain PCC6803 by amino-terminal protein sequencing. DNA Res. 3: 225–232. [DOI] [PubMed] [Google Scholar]
- Starmer J., Stomp, A., Vouk, M., and Bitzer, D. 2006. Predicting Shine-Dalgarno sequence locations exposes genome annotation errors. PLoS Comput. Biol. 2: e57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran J.H., Jacoby, G.A., and Hooper, D.C. 2005. Interaction of the plasmid-encoded quinolone resistance protein Qnr with Escherichia coli DNA gyrase. Antimicrob. Agents Chemother. 49: 118–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetting M.W., Hegde, S.S., Fajardo, J.E., Fiser, A., Roderick, S.L., Takiff, H.E., and Blanchard, J.S. 2006. Pentapeptide repeat proteins. Biochemistry 45: 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]