

Abstract
The exchange of residues 67 and 205 of the S2 pocket of human cysteine cathepsins K and L induces a permutation of their substrate specificity toward fluorogenic peptide substrates. While the cathepsin L-like cathepsin K (Tyr67Leu/Leu205Ala) mutant has a marked preference for Phe, the Leu67Tyr/Ala205Leu cathepsin L variant shows an effective cathepsin K-like preference for Leu and Pro. A similar turnaround of inhibition was observed by using specific inhibitors of cathepsin K [1-(N-Benzyloxycarbonyl-leucyl)-5-(N-Boc-phenylalanyl-leucyl)carbohydrazide] and cathepsin L [N-(4-biphenylacetyl)-S-methylcysteine-(D)-Arg-Phe-β-phenethylamide]. Molecular modeling studies indicated that mutations alter the character of both S2 and S3 subsites, while docking calculations were consistent with kinetics data. The cathepsin K-like cathepsin L was unable to mimic the collagen-degrading activity of cathepsin K against collagens I and II, DQ-collagens I and IV, and elastin-Congo Red. In summary, double mutations of the S2 pocket of cathepsins K (Y67L/L205A) and L (L67Y/A205L) induce a switch of their enzymatic specificity toward small selective inhibitors and peptidyl substrates, confirming the key role of residues 67 and 205. However, mutations in the S2 subsite pocket of cathepsin L alone without engineering of binding sites to chondroitin sulfate are not sufficient to generate a cathepsin K-like collagenase, emphasizing the pivotal role of the complex formation between glycosaminoglycans and cathepsin K for its unique collagenolytic activity.
Keywords: cathepsin, collagen, cysteine protease, peptide inhibitor, substrate specificity
Papain-like enzymes, of which 11 cysteine cathepsins are encoded in the human genome, represent a major family of peptidases (family C1, clan CA) (Rawlings et al. 2006) (Merops: the peptidase database: http://merops.sanger.ac.uk). Besides maintenance of endosomal/lysosomal protein turnover, cysteine cathepsins (CPs) are involved in specific processes, including bone remodeling, immunity, apoptosis, or prohormone processing (for review, see Lecaille et al. 2002a). Moreover, they participate in a variety of pathological processes such as tumor progression, rheumatoid arthritis, osteoporosis, emphysema, atherosclerosis, or adiposity (Chapman et al. 1997; Turk et al. 2001; Joyce and Hanahan 2004; Taleb et al. 2005). Therefore, these enzymes represent attractive targets for the design of site-specific inhibitors of therapeutic interest (Brömme and Kaleta 2002).
Despite some functional redundancy between cathepsins K and L and their similarities of their catalytic domains and 3D structures (Nägler and Menard 2003), these two enzymes participate in different specialized functions in living organisms. For instance, cathepsin L is involved in the production of [Met] enkephalin, a peptide neurotransmitter (Yasothornsrikul et al. 2003), and may act as a kininogenase (Puzer et al. 2005). Lack of cathepsin L interferes with hair cycle control (Roth et al. 2000) and prevents proper antigen presentation in thymus (Riese and Chapman 2000). On the other hand, cathepsin K, which plays a pivotal role in bone remodeling, lung homeostasis, and in the proteolysis of thyroglobulin (Tepel et al. 2000; Bühling et al. 2004), has been proposed as a new kinin-degrading peptidase (Godat et al. 2004; Lecaille et al. 2007). Cathepsin K, which is predominantly expressed in osteoclasts, is a potent elastolytic enzyme with a unique collagenolytic activity among CPs (Chapman and Shi 2000). Indeed, cathepsin K degrades collagens I and II within covalently cross-linked triple helices, while cathepsin L cleaves only in their nonhelical telopeptide regions (Chapman et al. 1997; Garnero et al. 1998; Kafienah et al. 1998; Yasuda et al. 2004). Contrary to cathepsin L, which accommodates hydrophobic or aromatic residues in its S2 subsite (Phe ≫ Leu), analyses of cleavage sites of cathepsin K in type I and II collagens as well as profiling of its substrate specificity using positional scanning peptide libraries have confirmed that cathepsin K has a more restricted pocket and exhibits an exclusive preference for Pro at P2 among mammalian cathepsins (Garnero et al. 1998; Lecaille et al. 2002b; Choe et al. 2006). Comparison of structures of cathepsins K and L showed sequence differences at residues 67 and 205 (papain numbering) in the S2 binding pocket, forming, respectively, part of the wall and the bottom of the pocket. We demonstrated previously that mutation of residues Tyr67 and Leu205 in cathepsin K by those of cathepsin L (Tyr67Leu/Leu205Ala) impaired its collagenolytic, but not its gelatinolytic activity (Lecaille et al. 2002b).
In this study, a S2 cathepsin K-like (Leu67Tyr/Ala205Leu) variant of human cathepsin L was engineered and expressed in Pichia pastoris as a zymogen, processed into its mature form, and purified. The substrate specificity of wild-type cathepsins K and L and their respective S2 mutants were compared on the basis of a kinetic study by using peptidyl inhibitors and fluorogenic MCA-derived substrates. Experimental findings were supported by molecular analysis and binding calculations of the modeled complexes. Finally, proteolytic activities of cathepsin K, its S2 cathepsin L-like cathepsin K mutant, cathepsin L, and its S2 cathepsin K-like cathepsin L mutant were further evaluated by using model components of the extracellular matrix, that is, Congo Red elastin, collagens I and II, and DQ-collagens I and IV.
Results and Discussion
Generation and expression of the S2 cathepsin K-like (Leu67Tyr/Ala205Leu) mutant of cathepsin L
Molecular modeling studies and site-directed mutagenesis of cathepsin K have revealed some differences with cathepsin L in terms of size and shape of the side chains of their S2 pocket residues, especially in positions 67 and 205. Accordingly the S2 pocket of human cathepsin K was engineered into a cathepsin L-like variant (Y67L/L205A) (Lecaille et al. 2002b). A similar approach was used here to design the recombinant L67Y/A205L cathepsin L mutant. Briefly, the mutant was expressed in P. pastoris as a secreted zymogene (Mr 42,000) using the pPIC9K vector (see Fig. 1, lane 2), processed in the presence of pepsin at pH 4.0. After concentrating the supernatant (2 L) to a final volume of 15 mL, the L67Y/A205L cathepsin L variant was purified by chromatography (Butyl-Sepharose), which resulted in a single band of Mr ∼29,000 (Fig. 1, lane 3). Concentration of the purified mutant protein was 215 μM (final volume = 200 μL), as determined by E-64 titration.
Figure 1.
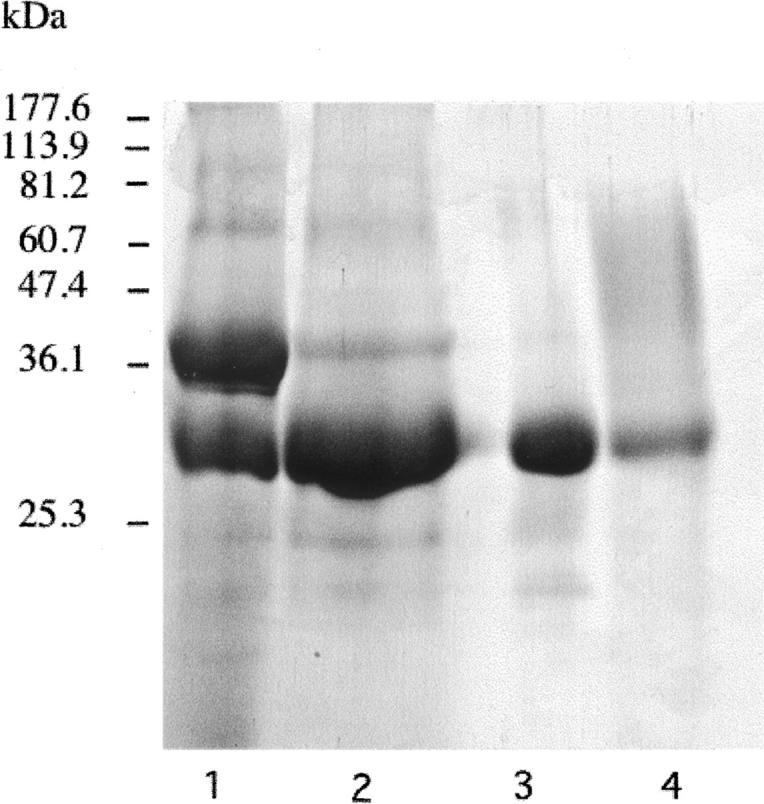
SDS-PAGE (12% Tris-glycine) of the purified Leu67Tyr/Ala205Leu cathepsin L mutant. (Lane 1) Crude supernatant fraction of the human Leu67Tyr/Ala205Leu mutant from P. pastoris; (lane 2) activation of the precursor mutant by treatment with pepsin; (lane 3) purified Leu67Tyr/Ala205Leu mutant after passage through the Butyl-Sepharose column; (lane 4) purified recombinant wild-type human cathepsin L. Molecular mass standards are reported in the left lane (Coomassie-blue staining).
Characterization of L67Y/A205L cathepsin L mutant using fluorogenic substrates
To examine if the exchange of residues 67 and 205 of the S2 pockets of cathepsins K and L could induce a switch of their respective substrate specificity, the hydrolysis of two representative dipeptide substrates (Z-Phe-Arg-MCA, Z-Leu-Arg-MCA) and a tripeptide substrate (Z-Gly-Pro-Arg-MCA) was evaluated. Steady-state kinetics was performed as described in Materials and Methods (Brömme et al. 1996), and the experimental data are summarized in Table 1. The values of k cat and K m were determined graphically from a Hanes linear plot, and their correctness confirmed by nonlinear regression analysis, while second-order rate constants (k cat/K m) were measured under pseudo-first-order conditions. In agreement with results of the profiling of the substrate specificity using positional scanning peptide libraries (Choe et al. 2006), wild-type cathepsin K exhibits a significant preference for Pro at P2 compared to wild-type cathepsin L, and mutations introduced in cathepsin K (Tyr67Leu/Leu205Ala) dramatically changed its S2 specificity (Lecaille et al. 2002b). Conversely, proline in the P2 position is well accepted by the L67Y/A205L cathepsin L mutant with a K m value identical to that of wild-type cathepsin K. Despite a fivefold lower second-order rate constant (due to a fivefold difference of k cat value), when compared to that of cathepsin K, this result underlines the importance of Tyr67 in the S2 subsite for the accommodation of a proline residue at P2 (Aibe et al. 1996; Xia et al. 1999; Smooker et al. 2000; Lecaille et al. 2002b). The mutation introduced in the S2 subsite of cathepsin L is also reflected by an inversion of the K m values between Z-LR-MCA and Z-FR-MCA (Table 1). Unlike wild-type cathepsin L and the S2 mutant of cathepsin K (Y67L/L205A), which readily hydrolyze Z-Phe-Arg-MCA, the L67Y/A205L cathepsin L mutant prefers Z-Leu-Arg-MCA with an approximately sevenfold increase in k cat/K m. An analogous preference for this substrate is observed for wild-type cathepsin K. This difference in specificity for Leu in the P2 position was even greater for wild-type cathepsin L (∼22-fold reduction) and the Y67L/L205A cathepsin K mutant (∼35-fold reduction).
Table 1.
Hydrolysis of peptidyl-MCA substrates by Leu67Tyr/Ala205Leu cathepsin L mutant, Tyr67Leu/Leu205Ala cathepsin K mutant, and wild-type cathepsins L and K

Inhibition by peptide-based inhibitors
N-(4-Biphenylacetyl)-S-methylcysteine-(D)-Arg-Phe-β-phenethylamide (also called cat L inh. 7), a noncovalent cathepsin L inhibitor designed from its propeptide (Chowdhury et al. 2002), selectively inhibited cathepsin L (nanomolar range). Compared to the initial report (Chowdhury et al. 2002), apparent discrepancies in Ki values depend, in fact, on differences between the buffers used for in vitro enzymatic assays (see Table 2). Its inhibitory profile was reversed for the two mutants: Indeed, an ∼100-fold increase in the Ki value was observed for the L67Y/A205L cathepsin L variant, while an ∼100-fold decrease occurred for the Y67L/L205A cathepsin K mutant when compared to its wild-type form (Table 2). Taking into account that the Phe residue from the cat L inh. 7 interacts with the S2 subsite of cathepsin L, while methylcysteine and (D)-Arg interact with the S1 and the S′1 subsites, respectively (Chowdhury et al. 2002), our data suggest that the selectivity of cat L inh. 7 for cathepsin L mostly depends on its preference for Phe at P2, in agreement with its substrate specificity. Moreover, the calculated binding affinities for cat L inh. 7 with cathepsins L and K and their mutants are consistent with the experimentally determined values (Table 3), providing confidence in interpreting the structural features of the predicted binding modes. It has to be noticed that the L67Y and A205L mutations in cathepsin L not only altered the S2 pocket, but also the shape of the S3 subsite, resulting in significant changes in the bound inhibitor conformation (Fig. 2A). The center of the benzene ring in the phenylalanine side chain is displaced by 1.0 Å in the S2 pocket of the mutant compared to the wild-type cathepsin L. The phenyl ethyl moiety in the S3 subsite is completely displaced from the subsite in the mutant. The centroid of the benzene ring moves a distance of 6.5 Å. The arginine side chain also moves significantly, resulting in a loss of favorable electrostatic interactions between the side-chain guanidinium group and Asp 162 of the enzyme. All these changes result in a calculated relative change in binding free energy of 2.24 kcal/mol between the L67Y/A205L mutant of cathepsin L and its wild-type form (Table 3), corresponding to an experimental ∼100-fold difference in Ki values. The opposite feature is observed in cathepsin K and its Y67L/L205A mutant (Fig. 2B). In the cathepsin K mutant, the binding mode of the inhibitor resembles that in wild-type cathepsin L, while the binding mode in wild-type cathepsin K resembles that in mutant cathepsin L. The calculated relative change in binding free energy is −1.45 kcal/mol between the mutant cathepsin K and its wild-type form (Table 3), corresponding to an experimental 88-fold improvement in binding affinity. The modeled binding modes of the cathepsin L inh. 7 in these four complexes support that the L67Y/A205L cathepsin L mutant acts like wild-type cathepsin K, and, conversely, the Y67L/L205A cathepsin K mutant acts like wild-type cathepsin L.
Table 2.
Inhibition of cathepsins L, K, and their respective S2 pocket mutants by selective peptide-based inhibitors

Table 3.
Modeled binding free energy of the cat L inh. 7 [N-(4-biphenylacetyl)-S-methylcysteine-(D)-Arg-Phe-β-phenethylamide] with cathepsins L and K and their respective S2 pocket mutants

Figure 2.

Binding mode of N-(4-biphenylacetyl)-S-methylcysteine-(D)-Arg-Phe-β-phenethylamide (Cat L inh. 7) with (A) cathepsin L and its Leu67Tyr/Ala205Leu variant, and with (B) cathepsin K and its Tyr67Leu/Leu205Ala variant. Wild-type (green) and mutant (yellow) binding modes are superimposed. (Dashed lines) The shortest distances between the P3 phenylethyl moiety (phenyl ring) and the S3 subsite, and the phenyl ring of Phe at P2 and the S2 subsite, respectively. The inhibitor is represented in stick mode and in ball-and-stick mode for the mutated protein side chains. Other selected nearby protein residues are show as thin lines.
1-(N-Benzyloxycarbonyl-leucyl)-5-(N-Boc-phenylalanyl-leucyl) carbohydrazide (i.e., cat K inh. Boc-I) is a potent selective cathepsin K inhibitor, derived from 1,5-bis(Cbz-Leu) carbohydrazide (Thompson et al. 1997). Previous crystallographic studies of the enzyme–inhibitor complex at 2.2 Å resolution [cathepsin K/1,5-bis(Cbz-Leu) carbohydrazide complex] revealed a mode of binding consistent with addition of the catalytic cysteine thiol to the central urea carbonyl (Thompson et al. 1997). Analysis of the initial rates of substrate hydrolysis as a function of inhibitor concentration yielded an apparent inhibition constant (K i,app) for a reversible association of the inhibitor with cathepsin prior to the time-dependent step, according to the nonlinear character of the progress curves of hydrolysis as previously detailed (Wang et al. 2002). The inactivation rate k obs and K i,app were determined as described in Materials and Methods. In agreement with a previous report (Wang et al. 2002), cat K inh. Boc-I is a potent and selective inhibitor of cathepsin K, as reflected by K i,app (= 3.5 nM) and second-order rate constant k obs/[I] (= 5.6 × 105 M−1 sec−1) values, while we were unable to measure K i,app for cathepsin L (Table 2). Additionally, cat K inh. Boc-I was also a potent inhibitor of the L67Y/A205L cathepsin L mutant (K i,app = 4.5 nM), while we failed to determine the apparent inhibition constant toward the Y67L/L205A cathepsin K mutant. Similarly to that observed with cat L inh. 7, the inhibition profile of cat K inh. Boc-I was altered by mutations of residues 67 and 205, suggesting that its selectivity is mainly due to P2/S2 interactions. Taken together, the kinetic analysis using MCA-derived substrates (see previous paragraph) and our inhibitor analysis confirm unambiguously that the peptide selectivity of cathepsins K and L relies mostly on their respective residues 67 and 205 (papain numbering) of the S2 pocket.
Proteolytic activity of L67Y/A205L cathepsin L mutant on collagens, gelatin, and elastin
Collagens, which are predominantly located in the extracellular matrix (ECM), are built up of three polypeptide chains containing the repeating triplet sequence Gly-X-Y, where Pro is commonly found in the X position and Hyp (4-hydroxyproline) in the Y position. Collagen degradation is a key step in connective tissue/bone remodeling, angiogenesis, and organ development (Ortega et al. 2003). Fibrillar collagens I and II with their triple helical structural arrangement are highly resistant to general proteolysis and require specific proteinases for their degradation (Prockop and Kivirikko 1995). In contrast, type IV collagen, which is a major component of the basal lamina, is characterized by the presence of nonhelical domains. Among mammalian papain-like enzymes, cathepsin K, but not cathepsin L, possesses unique collagen I- and collagen II-degrading properties by forming a highly collagenolytic complex with bone and cartilage resident chondroitin-4-sulfate (C-4S) (Li et al. 2000, 2002, 2004). Cathepsin K with a double mutation in its S2 binding pocket (Y67L/L205A) lost its ability to accept Pro from the repeating triplet sequence Gly-X-Y at P2, leading to the full abolition of its collagenolytic activity (Lecaille et al. 2002b). Conversely, we assayed the L67Y/A205L cathepsin L mutant to address the effect of the exchange of residues 67 and 205 on its collagenolytic activity. Bovine type I collagen was incubated at 28°C in the presence or absence of C-4S with cathepsins L and K, and with L67Y/A205L cathepsin L (Fig. 3A). Coomassie-stained gels were evaluated by densitometry. As previously shown (Li et al. 2000, 2004), the stability of cathepsin K is dramatically improved in the presence of C-4S, resulting in a significantly increased collagenolytic activity. In contrast, cathepsin L displayed only a weak triple-helical collagenolytic activity that was suppressed after addition of 0.15% (w/v) C-4S, although cathepsin L does not form an active complex with C-4S as cathepsin K does (Li et al. 2004). Remaining amounts of the β11 band, and to a lesser extent β12 and γ bands, were faintly decreased in the presence of L67Y/A205L cathepsin L with respect to its wild-type form (Fig. 3A), according to densitometric analysis (data not shown). However, this minor difference did not reflect a significantly enhanced collagenase activity within the triple helix, but may indicate a small improvement of the overall activity of the cathepsin L mutant. While wild-type cathepsin K, in the presence of C-4S, fully degraded collagen II, as previously reported (Li et al. 2004), both wild-type and mutant cathepsins L did not reveal any proteolytic activities against bovine collagen II from cartilage (Fig. 3B), since no change in the relative amount of the α, β, and γ bands of collagen II was observed. On the other hand, cleavage of gelatin (denatured type I collagen) was similar for both wild-type and the S2 mutant of cathepsin L (Fig. 3C), which is in agreement with previous reports demonstrating that the gelatinolytic activities of cathepsins L and K are comparable (Lecaille et al. 2002b; Li et al. 2004). Elastinolytic and collagenolytic activities of the enzymes were further evaluated using Congo Red-labeled elastin and fluorescein-conjugated collagens. The Y67L/L205A cathepsin K mutant poorly degraded chromogenic elastin-Congo Red and fluorogenic DQ-collagens I and IV, thus comparing to wild-type cathepsin L (Fig. 4). In contrast, the L67Y/A205L cathepsin L mutant hydrolyzed DQ-collagen I and elastin-Congo Red somewhat more efficiently, although this activity remained weak. No significant improvement of its DQ-collagen IV degrading activity was observed. Taken together, present results indicate that the switch of the S2 subsite of cathepsin L into a S2 cathepsin K-like substrate specificity is insufficient to mimic the collagen-degrading properties of cathepsin K. Furthermore, they corroborate the hypothesis that only cathepsin K in conjunction with its complex formation with glycosaminoglycans is a potent collagenase; indeed, cathepsin L does not form a complex with glycosaminoglycans (Li et al. 2004).
Figure 3.
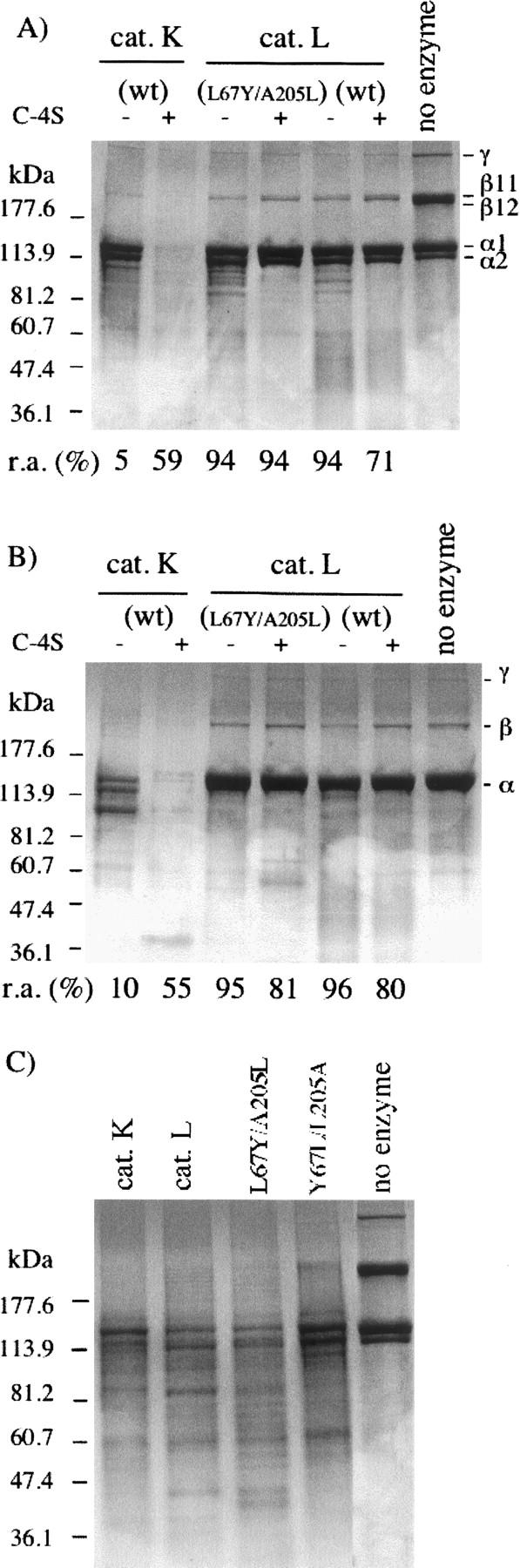
Collagenolytic and gelatinolytic activities of cathepsin K, cathepsin L, and Leu67Tyr/Ala205Leu cathepsin L mutant. (A) Cathepsins K, L, and L67Y/A205L cathepsin L mutant (600 nM) were incubated with skin collagen I (0.4 mg/mL) in 100 mM sodium acetate buffer (pH 5.5) containing 2 mM dithiothreitol and 2 mM EDTA, for 8 h at 28°C, in the presence or absence of 0.15% (w/v) C-4S. Samples were further analyzed by SDS-polyacrylamide electrophoresis using 4%–20% Tris/glycine gels (Coomassie-blue staining). Undigested bovine type I collagen was used as a standard. Molecular mass standards are indicated in the left lane. (B) Collagenolytic assays with soluble collagen II from articular joints (0.6 mg/mL).(C) Heat-denatured collagen I (gelatin; 0.4 mg/mL) was incubated with cathepsins K, L, and the two S2 mutants (10 nM), respectively, for 30 min at 28°C.
Figure 4.

Hydrolysis of Congo Red-labeled elastin and fluorescein-conjugated collagens. Bovine skin DQ-collagen I, human placenta DQ-collagen IV, and elastin-Congo Red were incubated with cathepsins K, L, and their respective S2 mutants as described in Materials and Methods. Collagenolytic activities were measured by monitoring the fluorescence release (excitation wavelength: 395 nm; emission wavelength: 415 nm). After removal of the uncleaved substrate, the elastinolytic activity was deduced from the absorbance of soluble released dye (λ = 490 nm). Proteolytic activities were expressed as normalized values (%), using wild-type cathepsin K as reference. The results (triplicate assays) are means ± SEM.
In summary, substitutions of residues 67 and 205 induce an exchange of the respective S2 substrate specificity of cathepsins K and L toward small peptidyl substrates and inhibitors. Nevertheless, the S2 cathepsin K-like (L67Y/A205L) cathepsin L mutant is unable to generate a collagen-degrading activity similar to that of wild-type cathepsin K. The present data support our previous findings that the triple-helical collagenolytic activity of cathepsin K largely depends on its ability to form complexes with glycosaminoglycans (Li et al. 2004).
Materials and Methods
Enzymes and inhibitors
Wild-type human cathepsin K and the Tyr67Leu/Leu205Ala cathepsin K mutant were expressed in P. pastoris as described elsewhere (Lecaille et al. 2002b). Human cathepsin L was supplied by Calbiochem (VWR International). The buffer for enzyme assays was 100 mM sodium acetate buffer (pH 5.5), containing 2 mM dithiothreitol and 2 mM EDTA. The concentration of active cathepsins was determined by titration with E-64 (Sigma-Aldrich) (Barrett et al. 1982). 1-(N-Benzyloxycarbonyl-leucyl)-5-(N-Boc-phenylalanyl-leucyl) carbohydrazide (Wang et al. 2002) and N-(4-biphenylacetyl)-S-methylcysteine-(D)-Arg-Phe-β-phenethylamide (Chowdhury et al. 2002) were supplied by Calbiochem.
Site-directed mutagenesis of the S2 cathepsin K-like variant of cathepsin L
Human procathepsin L cDNA cloned into the AvrII and NotI sites of the pBluescript SK (+) phagemid (Stratagene) was used as the template for in vitro site-directed mutagenesis (Dieter Brömme, unpubl.). Each point mutation was introduced individually into the cDNA using the PCR ligation method with Pfu polymerase (New England Biolabs Inc.). The oligonucleotides used for the mutagenesis were as follows (the underlined bases code for the mutated amino acids at positions 67 and 205): Leu 67 → Tyr, 5′-TGCAATGGTGGCTACATGGATTAT-3′; Ala 205 → Leu, 5′-GGAATTGCCTCACTAGCCAGCTACCCC-3′. The nucleotide sequence was determined to ensure integrity of the construct. Unique AvrII and NotI sites, introduced at the 5′- and 3′-ends of each amplified mutant proenzyme PCR product, were used to ligate each construct into the pPIC9K expression vector (Invitrogen Corp.). The double mutant construct was subsequently sequenced using a series of primers derived from either the vector or internal cDNA sequences. Sequence analyses were performed using an Applied Biosystems model 3777 automated sequencer. The expression construct for the Leu67Tyr/Ala205Leu mutant proenzyme was linearized with SalI and then electroporated into P. pastoris GS115 cells (Invitrogen Corp.). After phenotype screening according to the manufacturer's instructions, several clones were obtained. Clones were grown up in shaker flasks, and the liquid culture media was concentrated using an YM10 ultrafiltration membrane with cutoff size >10,000 Da (Amicon Inc.), and washed with 100 mM sodium acetate, 2.5 mM EDTA (pH 5.5). The presence of the mutant was detected by Western Blot, using a rabbit polyclonal antibody raised against human cathepsin L. Activation of the precursor protein and the purification of the active cathepsin L mutant were performed according to Lecaille et al. (2007).
Kinetic parameters
Assays with peptidyl MCA-derived substrates
Z-Phe-Arg-MCA, Z-Leu-Arg-MCA, and Z-Gly-Pro-Arg-MCA were purchased from Bachem. Steady-state kinetics were performed with fluorogenic substrates as previously described (Brömme et al. 1996). The enzymatic activity was followed by monitoring the fluorescence release (Kontron SFM 25 spectrofluorimeter; excitation wavelength: 350 nm; emission wavelength: 460 nm). Cathepsins were assayed at 28°C at fixed final concentration (1–5 nM) and variable substrate concentrations (1–200 μM) in 100 mM sodium acetate buffer (pH 5.5) containing 2 mM dithiothreitol and 2 mM EDTA. The values of k cat and K m were determined graphically from Hanes linear plot using various concentrations of substrate (0.1–10 μM), and the accuracy of K m and k cat values was confirmed by nonlinear regression analysis (substrate concentration: 0.2–6 μM). Experiments were performed in triplicate. Second-order rate constants (k cat/K m) were first measured under pseudo-first-order conditions, that is, using a substrate concentration far below the K m. Kinetic data were determined using the Enzfitter software (Biosoft) and were reported as means ± SD.
Pseudopeptidyl inhibitors
Cathepsin L inhibitor [N-(4-biphenylacetyl)-S-methylcysteine-(D)-Arg-Phe-β-phenethylamide]
Wild-type cathepsin L (2.5 nM) was incubated in the presence of varying concentrations (1–1000 nM) of N-(4-biphenylacetyl)-S-methylcysteine-(D)-Arg-Phe-β-phenethylamide (also called cat L inh. 7), and the inhibition constant (Ki) was determined, as previously described (Chowdhury et al. 2002), using Z-Phe-Arg-MCA (2.5 μM) as substrate. Inhibition assays were repeated with wild-type cathepsin K, L67Y/A205L cathepsin L mutant, and Y67L/L205A cathepsin K mutant using the same conditions.
Cathepsin K inhibitor [Z-L-NHNHCONHNH-LF-Boc; 1-(N-Benzyloxycarbonyl-leucyl)-5-(N-Boc-phenylalanyl-leucyl) carbohydrazide]
Standard enzyme assays and inhibition studies were performed with 1-(N-Benzyloxycarbonyl-leucyl)-5-(N-Boc-phenylalanyl-leucyl) carbohydrazide (cat K inh. Boc-I), assuming that the inhibitor is purely competitive (Wang et al. 2002). Experiments were performed in triplicate. The apparent inhibition constant Ki (app.) was obtained by following the progress curve of the hydrolysis of Z-Phe-Arg-MCA (1–10 μM) by cathepsin K (0.5 nM), in the absence of inhibitor (v 0) or by premixing with different concentrations of inhibitor (1–100 nM) (vi). The substrate concentration used was equivalent to K m, and the total enzyme concentration was at least 10-fold l< Ki (app.). Finally Ki (app.) was obtained by plotting (v 0/vi − 1) against [I] (Nicklin and Barrett 1984), according to Equation 1: v 0/vi = 1 + [I]/Ki (app.). The rate constant for inactivation (k obs) was calculated according to (Williams and Morrison 1979), and the apparent second-order rate constant, k obs/[I], was deduced using the k obs values and the appropriate inhibitor concentrations. Similar experiments were repeated with cathepsin L and the two S2 mutants.
Molecular modeling
The crystal structure of the complex of mature cathepsin L with a dimer of inhibitor 13 (Chowdhury et al. 2002) (pdb: 1mhw) was used as a template for docking the inhibitors in this study. The inhibitor in the crystal structure was converted to cat L inh. 7 [N-(4-biphenylacetyl)-S-methylcysteine-(D)-Arg-Phe-β-phenethylamide] by truncating the inhibitor dimer into a monomer and mutating the P2 tyrosine to phenylalanine. All histidine residues were protonated, and the catalytic cysteine was modeled as a thiolate. The crystallographic waters were removed, and hydrogen atoms were added to the crystal structure using REDUCE (Word et al. 1999). Both N and C termini of the protein were modeled in the ionized state. A version of the complex with the L67Y and A205L mutations was also constructed by structure manipulation in SYBYL 6.6 (Tripos, Inc.). Cat L inh. 7 was manually docked into the crystal structure of cathepsin K (pdb: 1ayw) using the modeled complex with cathepsin L as a guide to orient the inhibitor. Protonation of the protein was carried out in the same manner as with cathepsin L. A version of the complex with the Y67L and L205A mutations was constructed as well. The resulting four enzyme-inhibitor complexes were subjected to conjugate gradient energy minimization using a distance-dependent dielectric function (e = 4r) and an 8 Å nonbonded cutoff to an RMS gradient of 0.01 kcal/mol·Å. Minimizations were carried out in three stages, using the AMBER force field supplemented with parameters for the nonnatural amino acids (Cornell et al. 1995). First, only the hydrogen positions were minimized, followed by the side-chain atoms and finally the whole protein–ligand complex including the backbone. Each of the energy-minimized enzyme-inhibitor complexes was then subjected to a conformational search using a Monte Carlo with energy minimization (MCM) procedure (Li and Scheraga 1987; Chowdhury et al. 2002). The starting structures of each cycle of minimization were obtained by randomly perturbing one or more side-chain dihedral angles as well as crankshaft rotations of the peptide units of the inhibitor. Protein residues including the backbone and side-chain atoms within 8 Å of the inhibitor were allowed to move during minimization. All other protein residues were kept fixed. A total of 1000 MCM cycles were carried out for each inhibitor. Using the lowest-energy structures from the MCM search, we calculated theoretical binding affinities of cat L inh. 7 with the wild-type and mutant cathepsins L and K. The absolute binding free energies were calculated using a solvated interaction energy (SIE) approach (Naim et al. 2007). The SIE is calculated as:
where
E coul and E vdw are the intermolecular Coulomb and van der Waals interaction energies, respectively, calculated without nonbonded cutoffs. ΔG rf, the reaction field energy, and ΔG cav, the cavity term, are the electrostatic and nonelectrostatic components of the solvation energy, respectively. ΔSA is the change in molecular surface area upon binding. The parameter C is a regression constant for fitting the absolute and not just the relative free energies. Empirical parameters for the SIE expression were obtained by calibrating against a database of about 100 protein–ligand complexes (Naim et al. 2007) that did not include the cathepsins. The optimized parameter values used were β = 0.040, γ = 0.007 kcal/mol·Å2, and C = −2.61 kcal/mol. The electrostatic terms also depend on internal and external dielectric constants, D in and D out, respectively. We used D in = 26.5 and D out = ∞, that is, a conductor-limit approximation for the solvent. The reaction field energy, ΔG rf, was calculated with a continuum model using a boundary element solution to the Poisson equation using the BRI BEM program (Purisima and Nilar 1995, Purisima 1998).
Collagenolytic, gelatinolytic, and elastinolytic activities of cathepsins
Collagens I and II, gelatin
Cathepsins K, L, and L67Y/A205L cathepsin L mutant (600 nM) were incubated with 0.4 mg/mL of soluble calf skin type I collagen (U.S. Biochemical Corp.) in 100 mM sodium acetate buffer (pH 5.5) containing 2 mM dithiothreitol and 2 mM EDTA, for 8 h at 28°C, in the presence or absence of 0.15% (w/v) C-4S. Collagenolytic assays were repeated with 0.6 mg/mL of collagen type II from calf articular joints (Amersham Pharmacia Biotech). Heat-denatured type I collagen (gelatin; 0.4 mg/mL) was incubated with cathepsins K, L, and the two S2 mutants (10 nM), respectively, for 30 min at 28°C. Residual cathepsin activity (0, 4, and 8 h) was assayed, after withdrawal of an aliquot of each sample, by using Z-Phe-Arg-MCA as substrate (2.5 μM). After reactions were stopped by the addition of E-64 (10 μM), samples were subjected to SDS-polyacrylamide electrophoresis using 4%–20% Tris/glycine gels (Novex), and degradation products were visualized by Coomassie-blue staining. The NIH Image 1.62 software (http://rsb.info.nih.gov/nih-image/) was used for densitometric determination of protein bands.
Assays with DQ collagens
The fluorogenic bovine skin DQ collagen I (10 μg/mL) (Molecular Probes) was incubated with cathepsins K, L, and their respective mutants (10 nM) for 45 min at 25°C in 100 mM sodium acetate buffer (pH 5.5), 2 mM dithiothreitol, and 2 mM EDTA. Collagenolytic activity was followed by monitoring the fluorescence release (excitation wavelength: 395 nm; emission wavelength: 415 nm) (spectromicrofluorimeter SpectraMax Gemini; Molecular Devices), and slopes were calculated with the software Softmaxpro (Molecular Devices). Assays were performed in triplicate. Similar experiments were performed with human placenta DQ collagen IV (Molecular Probes).
Assays with Elastin Congo Red
The elastinolytic activity of cathepsins K, L, and their corresponding mutants was monitored in the presence of Elastin Congo Red (Sigma) according to the protocol of the manufacturer. Briefly, Elastin Congo Red suspension (10 mg/mL in 0.1 M phosphate buffer at pH 5.5, dithiothreitol/EDTA 2 mM) was incubated for 12 h at 28°C with each cathepsin (250 nM) under harsh agitation. After addition of E-64 (1 mM) to block the reaction, uncleaved substrate was eliminated by centrifugation (10,000 rpm for 5 min at 4°C), and absorbance of soluble released dye was measured at 490 nm.
Acknowledgment
We thank Nadia Tabalit for her technical assistance.
Footnotes
Reprint requests to: Gilles Lalmanach, INSERM U618 Protéases et Vectorisation Pulmonaires, Equipe Protéases et Pathologies Pulmonaires, Université François Rabelais–Faculté de Médecine, 10 Boulevard Tonnellé, F-37032 Tours cedex, France; e-mail: gilles.lalmanach@univ-tours.fr; fax: (33) 2 47 36 60 46.
Abbreviations: CPs, cysteine proteinases; C-4S, chondroitin-4-sulfate; DTT, dithiothreitol; GAG, glycosaminoglycan; E-64, L-3-carboxy-trans-2,3-epoxypropionyl-leucylamido-(4-guanidino) butane; MCA, 7-amino-4 methylcoumarin; MCM, Monte Carlo energy minimization; SIE, solvated interaction energy; Z, Benzyloxycarbonyl.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.062666607.
References
- Aibe K., Yazawa, H., Abe, K., Teramura, K., Kumegawa, M., Kawashima, H., and Honda, K. 1996. Substrate specificity of recombinant osteoclast-specific cathepsin K from rabbits. Biol. Pharm. Bull. 19: 1026–1031. [DOI] [PubMed] [Google Scholar]
- Barrett A.J., Kembhavi, A.A., Brown, M.A., Kirschke, H., Knight, C.G., Tamai, M., and Hanada, K. 1982. L-Trans-epoxysuccinyl-leucylamido(4-guanidino)butane (E-64) and its analogues as inhibitors of cysteine proteinases including cathepsins B, H and L. Biochem. J. 201: 189–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brömme D. and Kaleta, J. 2002. Thiol-dependent cathepsins: Pathophysiological implications and recent advances in inhibitor design. Curr. Pharm. Des. 8: 1639–1658. [DOI] [PubMed] [Google Scholar]
- Brömme D., Klaus, J.L., Okamoto, K., Rasnick, D., and Palmer, J.T. 1996. Peptidyl vinyl sulphones: A new class of potent and selective cysteine protease inhibitors: S2P2 specificity of human cathepsin O2 in comparison with cathepsins S and L. Biochem. J. 315: 85–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bühling F., Rocken, C., Brasch, F., Hartig, R., Yasuda, Y., Saftig, P., Brömme, D., and Welte, T. 2004. Pivotal role of cathepsin K in lung fibrosis. Am. J. Pathol. 164: 2203–2216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman H.A. and Shi, G.P. 2000. Protease injury in the development of COPD: Thomas A. Neff Lecture. Chest 117: 295S–299S. [DOI] [PubMed] [Google Scholar]
- Chapman H.A., Riese, R.J., and Shi, G.P. 1997. Emerging roles for cysteine proteases in human biology. Annu. Rev. Physiol. 59: 63–88. [DOI] [PubMed] [Google Scholar]
- Choe Y., Leonetti, F., Greenbaum, D.C., Lecaille, F., Bogyo, M., Brömme, D., Ellman, J.A., and Craik, C.S. 2006. Substrate profiling of cysteine proteases using a combinatorial peptide library identifies functionally unique specificities. J. Biol. Chem. 281: 12824–12832. [DOI] [PubMed] [Google Scholar]
- Chowdhury S.F., Sivaraman, J., Wang, J., Devanathan, G., Lachance, P., Qi, H., Menard, R., Lefebvre, J., Konishi, Y., Cygler, M., et al. 2002. Design of noncovalent inhibitors of human cathepsin L. From the 96-residue proregion to optimized tripeptides. J. Med. Chem. 45: 5321–5329. [DOI] [PubMed] [Google Scholar]
- Cornell W.D., Cieplak, P., Bayly, C.I., Gould, I.R., Merz Jr, K.M., Ferguson, D.M., Spellmeyer, D.C., Fox, T., Caldwell, J.W., and Kollman, P.A. 1995. A second generation force field for the simulation of proteins, nucleic acids and organic molecules. J. Am. Chem. Soc. 117: 5179–5197. [Google Scholar]
- Garnero P., Borel, O., Byrjalsen, I., Ferreras, M., Drake, F.H., McQueney, M.S., Foged, N.T., Delmas, P.D., and Delaisse, J.M. 1998. The collagenolytic activity of cathepsin K is unique among mammalian proteinases. J. Biol. Chem. 273: 32347–32352. [DOI] [PubMed] [Google Scholar]
- Godat E., Lecaille, F., Desmazes, C., Duchene, S., Weidauer, E., Saftig, P., Brömme, D., Vandier, C., and Lalmanach, G. 2004. Cathepsin K: A cysteine protease with unique kinin-degrading properties. Biochem. J. 383: 501–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joyce J.A. and Hanahan, D. 2004. Multiple roles for cysteine cathepsins in cancer. Cell Cycle 3: 1516–1519. [DOI] [PubMed] [Google Scholar]
- Kafienah W., Brömme, D., Buttle, D.J., Croucher, L.J., and Hollander, A.P. 1998. Human cathepsin K cleaves native type I and II collagens at the N-terminal end of the triple helix. Biochem. J. 331: 727–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecaille F., Kaleta, J., and Brömme, D. 2002a. Human and parasitic papain-like cysteine proteases: Their role in physiology and pathology and recent developments in inhibitor design. Chem. Rev. 102: 4459–4488. [DOI] [PubMed] [Google Scholar]
- Lecaille F., Choe, Y., Brandt, W., Li, Z., Craik, C.S., and Brömme, D. 2002b. Selective inhibition of the collagenolytic activity of human cathepsin K by altering its S2 subsite specificity. Biochemistry 41: 8447–8454. [DOI] [PubMed] [Google Scholar]
- Lecaille F., Vandier, C., Godat, E., Hervé-Grépinet, V., Brömme, D., and Lalmanach, G. 2007. Modulation of hypotensive effects of kinins by cathepsin K. Arch. Biochem. Biophys 459: 129–136. [DOI] [PubMed] [Google Scholar]
- Li Z. and Scheraga, H.A. 1987. Monte Carlo-minimization approach to the multiple-minima problem in protein folding. Proc. Natl. Acad. Sci. 84: 6611–6615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z., Hou, W.S., and Brömme, D. 2000. Collagenolytic activity of cathepsin K is specifically modulated by cartilage-resident chondroitin sulfates. Biochemistry 39: 529–536. [DOI] [PubMed] [Google Scholar]
- Li Z., Hou, W.S., Escalante-Torres, C.R., Gelb, B.D., and Brömme, D. 2002. Collagenase activity of cathepsin K depends on complex formation with chondroitin sulfate. J. Biol. Chem. 277: 28669–28676. [DOI] [PubMed] [Google Scholar]
- Li Z., Yasuda, Y., Li, W., Bogyo, M., Katz, N., Gordon, R.E., Fields, G.B., and Brömme, D. 2004. Regulation of collagenase activities of human cathepsins by glycosaminoglycans. J. Biol. Chem. 279: 5470–5479. [DOI] [PubMed] [Google Scholar]
- Nägler D.K. and Menard, R. 2003. Family C1 cysteine proteases: Biological diversity or redundancy? Biol. Chem. 384: 837–843. [DOI] [PubMed] [Google Scholar]
- Naim M., Bhat, S., Rankin, K.N., Dennis, S., Chowdhury, S.F., Siddiqi, I., Drabik, P., Sulea, T., Bayly, C.I., Jakalian, A., et al. 2007. Solvated interation Energy (SIE) for scoring protein-ligand binding affinities. I. Exploring the parameter space. J. Chem. Inf. Model. 47: 122–133. [DOI] [PubMed] [Google Scholar]
- Nicklin M.J. and Barrett, A.J. 1984. Inhibition of cysteine proteinases and dipeptidyl peptidase I by egg-white cystatin. Biochem. J. 223: 245–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortega M.M., Nascimento, H., Costa, F.F., Teori, M.T., and Lima, C.S. 2003. A polymorphism in the angiogenesis inhibitor, endostatin, in multiple myeloma. Leuk. Res. 27: 93–94. [DOI] [PubMed] [Google Scholar]
- Prockop D.J. and Kivirikko, K.I. 1995. Collagens: Molecular biology, diseases, and potentials for therapy. Annu. Rev. Biochem. 64: 403–434. [DOI] [PubMed] [Google Scholar]
- Purisima E.O. 1998. Fast summation boundary element methods for calculating solvation free energies of macromolecules. J. Comput. Chem. 19: 1494–1505. [Google Scholar]
- Purisima E.O. and Nilar, S.H. 1995. A simple yet accurate boundary element method for continuum dielectric calculations. J. Comput. Chem. 16: 681–689. [Google Scholar]
- Puzer L., Vercesi, J., Alves, M.F., Barros, N.M., Araujo, M.S., Aparecida Juliano, M., Reis, M.L., Juliano, L., and Carmona, A.K. 2005. A possible alternative mechanism of kinin generation in vivo by cathepsin L. Biol. Chem. 386: 699–704. [DOI] [PubMed] [Google Scholar]
- Rawlings N.D., Morton, F.R., and Barrett, A.J. 2006. MEROPS: The peptidase database. Nucleic Acids Res. 34: D270–D272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riese R.J. and Chapman, H.A. 2000. Cathepsins and compartmentalization in antigen presentation. Curr. Opin. Immunol. 12: 107–113. [DOI] [PubMed] [Google Scholar]
- Roth W., Deussing, J., Botchkarev, V.A., Pauly-Evers, M., Saftig, P., Hafner, A., Schmidt, P., Schmahl, W., Scherer, J., Anton-Lamprecht, I., et al. 2000. Cathepsin L deficiency as molecular defect of furless: Hyperproliferation of keratinocytes and perturbation of hair follicle cycling. FASEB J. 14: 2075–2086. [DOI] [PubMed] [Google Scholar]
- Smooker P.M., Whisstock, J.C., Irving, J.A., Siyaguna, S., Spithill, T.W., and Pike, R.N. 2000. A single amino acid substitution affects substrate specificity in cysteine proteinases from Fasciola hepatica . Protein Sci. 9: 2567–2572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taleb S., Lacasa, D., Bastard, J.P., Poitou, C., Cancello, R., Pelloux, V., Viguerie, N., Benis, A., Zucker, J.D., Bouillot, J.L., et al. 2005. Cathepsin S, a novel biomarker of adiposity: Relevance to atherogenesis. FASEB J. 19: 1540–1542. [DOI] [PubMed] [Google Scholar]
- Tepel C., Brömme, D., Herzog, V., and Brix, K. 2000. Cathepsin K in thyroid epithelial cells: Sequence, localization and possible function in extracellular proteolysis of thyroglobulin. J. Cell Sci. 113: 4487–4498. [DOI] [PubMed] [Google Scholar]
- Thompson S.K., Halbert, S.M., Bossard, M.J., Tomaszek, T.A., Levy, M.A., Zhao, B., Smith, W.W., Abdel-Meguid, S.S., Janson, C.A., D'Alessio, K.J., et al. 1997. Design of potent and selective human cathepsin K inhibitors that span the active site. Proc. Natl. Acad. Sci. 94: 14249–14254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turk V., Turk, B., and Turk, D. 2001. Lysosomal cysteine proteases: Facts and opportunities. EMBO J. 20: 4629–4633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D., Pechar, M., Li, W., Kopeckova, P., Brömme, D., and Kopecek, J. 2002. Inhibition of cathepsin K with lysosomotropic macromolecular inhibitors. Biochemistry 41: 8849–8859. [DOI] [PubMed] [Google Scholar]
- Williams J.W. and Morrison, J.F. 1979. The kinetics of reversible tight-binding inhibition. Methods Enzymol. 63: 437–467. [DOI] [PubMed] [Google Scholar]
- Word J.M., Lovell, S.C., Richardson, J.S., and Richardson, D.C. 1999. Asparagine and glutamine: Using hydrogen atom contacts in the choice of side-chain amide orientation. J. Mol. Biol. 285: 1735–1747. [DOI] [PubMed] [Google Scholar]
- Xia L., Kilb, J., Wex, H., Lipyansky, A., Breuil, V., Stein, L., Palmer, J.T., Dempster, D.W., and Brömme, D. 1999. Localization of rat cathepsin K in osteoclasts and resorption pits: Inhibition of bone resorption cathepsin K-activity by peptidyl vinyl sulfones. Biol. Chem. 380: 679–687. [DOI] [PubMed] [Google Scholar]
- Yasothornsrikul S., Greenbaum, D., Medzihradszky, K.F., Toneff, T., Bundey, R., Miller, R., Schilling, B., Petermann, I., Dehnert, J., Logvinova, A., et al. 2003. Cathepsin L in secretory vesicles functions as a prohormone-processing enzyme for production of the enkephalin peptide neurotransmitter. Proc. Natl. Acad. Sci. 100: 9590–9595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda Y., Li, Z., Greenbaum, D., Bogyo, M., Weber, E., and Brömme, D. 2004. Cathepsin V, a novel and potent elastolytic activity expressed in activated macrophages. J. Biol. Chem. 279: 36761–36770. [DOI] [PubMed] [Google Scholar]