

Abstract
Prefoldin is a molecular chaperone found in the domains eukarya and archaea that acts in conjunction with Group II chaperonin to correctly fold other nascent proteins. Previously, our group identified a putative single subunit of prefoldin, γ PFD, that was up-regulated in response to heat stress in the hyperthermophilic archaeon Methanocaldococcus jannaschii. In order to characterize this protein, we subcloned and expressed it and the other two prefoldin subunits from M. jannaschii, α and β PFD, into Eschericia coli and characterized the proteins. Whereas α and β PFD readily assembled into the expected hexamer, γ PFD would not assemble with either protein. Instead, γ PFD forms long filaments of defined dimensions measuring 8.5 nm × 1.7–3.5 nm and lengths exceeding 1 μm. Filamentous γ PFD acts as a molecular chaperone through in vitro assays, in a manner comparable to PFD. A possible molecular model for filament assembly is discussed.
Keywords: prefoldin, molecular chaperones, protein filaments, heat-shock response
Living cells rely on a chaperone network comprising distinct protein families to establish protein-folding fidelity within the crowded intracellular environment. Molecular chaperones are essential for de novo folding of certain proteins and for surviving exposures to potentially lethal environmental stresses such as elevated temperatures or toxic substances (Gasch et al. 2000). One particular family of chaperones, found mainly in eukarya and archaea, is the prefoldins (PFDs). Under optimal growth conditions, archaeal PFDs are thought to function as de novo protein-folding chaperones in conjunction with group II chaperonins (Hartl and Hayer-Hartl 2002), although in eukaryotes PFDs may have acquired a more specific chaperone function in establishing correct tubulin assembly (Vainberg et al. 1998). Furthermore, unlike many chaperones, PFDs are not generally upregulated in response to heat shock (Shockley et al. 2003; Laksanalamai et al. 2004).
Prefoldins can be grouped into two main evolutionarily related classes: one consisting of 140 residues (α PFD), and the other of 120 residues (β PFD). α PFD has a two-β-strand insertion in the middle of the protein (Leroux et al. 1999). The crystal structure of PFD from Methanobacterium thermoautrophicum has been resolved to 2.3 Å resolution (Siegert et al. 2000). Based on this structure, PFD quaternary structure is composed of a hexamer of two α and four β PFD subunits assembled by hydrophobic packing of two β-barrels. Coiled-coil α-helices protrude downward from each of these subunits, creating a “jellyfish-like” appearance.
Methanocaldococcus jannaschii is a hyperthermophilic barophilic archaeaon that was first isolated from a black smoker chimney at a depth of 2600 m (Jones et al. 1983). M. jannaschii has two distinct α PFD subunits and one β PFD subunit encoded in its genome (Leroux et al. 1999). Most other archaeal organisms encode one α PFD subunit and one β subunit (Leroux et al. 1999), raising questions about the role of the second α PFD in M. jannaschii. Recently, we examined the response of M. jannaschii to a lethal heat shock from 85°C to 95°C (Boonyaratanakornkit et al. 2005). It was discovered that the mRNA transcript of open reading frame (ORF) MJ0648, which encodes a protein homologous to α PFD, was upregulated over 20-fold in response to the heat shock. Prior to this finding PFDs had not been shown to be involved in an organism's heat-shock response. MJ0648 appears to have diverged from most other archaeal α PFDs (Leroux et al. 1999); phylogenetic analysis of MJ0648 shows that the most closely related PFD is from Aquifex aeolicus, a thermophilic bacterium. Because A. aeolicus does not have a gene corresponding to the β prefoldin in its genome, the implication is that this extra prefoldin has assumed a paralogous function within A. aeolicus and M. jannaschii.
The central aim of the present work is to ascertain how the paralogous α PFD of M. jannaschii assembles, and to determine whether this protein was a general molecular chaperone. The paralogous α PFD has been renamed γ prefoldin in this paper to highlight differences between it and its homologs α and β PFD. We show that γ PFD forms long filaments of definable dimensions in vitro and functions as a molecular chaperone. Further, γ PFD does not appear to associate with either α or β PFD to form heterooligomeric complexes. A possible molecular model for filamentous assembly is presented, incorporating structural constraints from TEM and AFM measurements of the filament. Our model is consistent with the ability of the filament to function as a molecular chaperone.
Results
α, β, γ Prefoldins have similar secondary structures
The α (MJ0952), β (MJ0987), and γ (MJ0648) PFD from M. jannaschii were heterologously expressed in Escherichia coli and purified. All proteins expressed as soluble proteins and could be purified by heat treatment at 80°C followed by two column chromatography steps. The far-UV circular dichroism scans of each protein conducted at room temperature were identical, indicating that each PFD has approximately the same secondary structure (Fig. 1A,B). Furthermore, each CD spectrum had pronounced relative minima at 222 nm and 208 nm, consistent with a predominantly α-helical secondary structure, as has been observed in previous studies of PFDs (Leroux et al. 1999). The secondary structure of γ PFD is stable from 25°C up to at least 97°C (Fig. 1C), encompassing the physiologically relevant temperature range for M. jannaschii, which has an optimal growth temperature of 85°C. In contrast, α PFD began to unfold at 85°C (Fig. 1C).
Figure 1.

Far-UV circular dichroism spectra at 25°C of M. jannaschii prefoldin subunits (A, B) and their melting profiles based on [θ] at 222 nm (C). For further details see Materials and Methods. (A) — α PFD, - - - β PFD; (B) - - - γ PFD (C) ▴ α PFD, ○ β PFD, □ γ PFD. Error bars refer to one standard deviation.
γ Prefoldin does not associate with α or β prefoldin
α and β PFD from M. jannaschii, when incubated at 80°C in a minimal salt buffer, form the expected hexameric quaternary assembly found in other archaeal prefoldins (Leroux et al. 1999; Okochi et al. 2002). This assembly was confirmed by comigration on a size-exclusion column (Fig. 2A). α PFD alone migrates in two peaks, which presumably correspond to a monomer and dimer. Densitometric analysis of the complex on a denaturing gel showed the expected α:β stoichiometry of 1:2 (Fig. 2A). Sedimentation equilibrium measurements of the α/β complex indicated a molecular mass of 87 kDa, consistent with the molecular weight expected for a prefoldin hexamer (data not shown). α/β PFD assembled at incubation temperatures of 20°C or 80°C, in the presence or absence of 0.5 M KCl, in the presence or absence of 20 mM MgCl2, or in the presence or absence of 1 mM ATP (data not shown).
Figure 2.
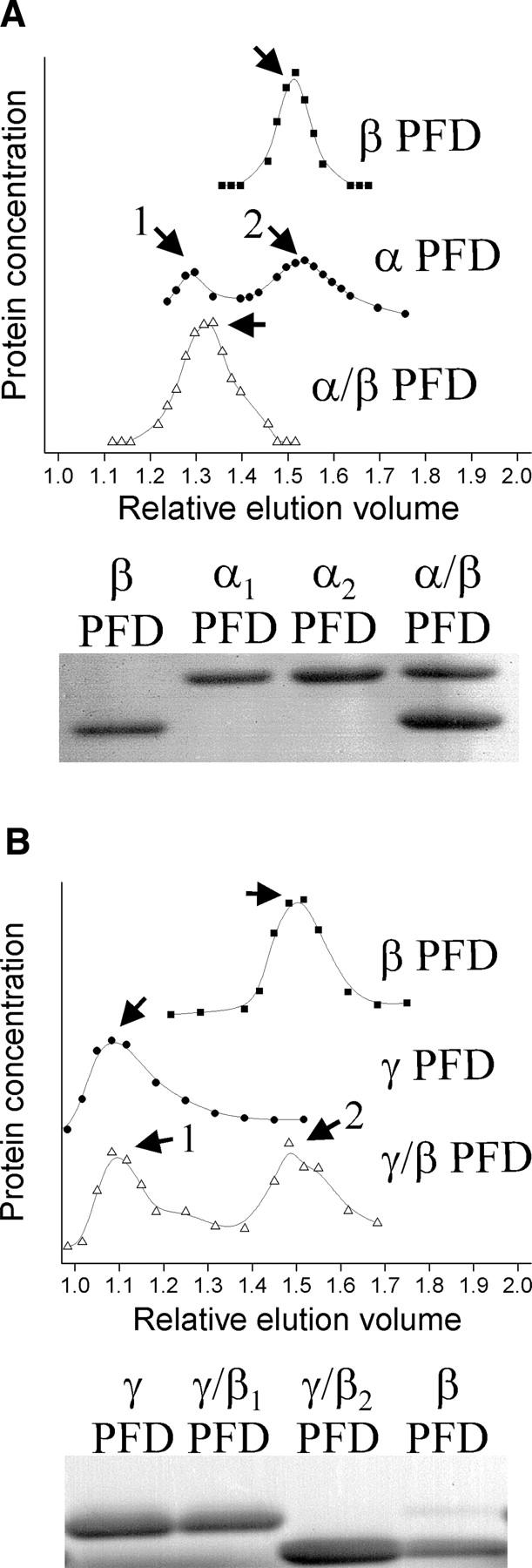
Size-exclusion chromatography and SDS-PAGE of individual and combined prefoldin subunits. (A) Chromatograms and gels indicating that α and β subunits assemble into α/β PFD. (B) Chromatograms and gels indicating that α and γ subunits do not assemble. Samples indicated by numbers and arrows were collected and run on the denaturing gels shown along the bottom. The relative elution volume is the elution volume divided by the void volume.
In contrast, γ PFD would not associate with either α or β PFD under the aforementioned assembly conditions (Fig. 2B, data not shown). Instead, γ PFD eluted in the void space of a size-exclusion column with a 600-kDa exclusion limit, showing that it forms a high molecular weight oligomer. Native gel electrophoresis gave qualitatively similar results as the size exclusion results (data not shown).
Dynamic light-scattering measurements on γ PFD gave a translational diffusion coefficient of 4.5 × 10−8 cm2 · s−1 (data not shown). A spherical protein with this translational diffusion coefficient would have a remarkably large Stokes radius of ∼48 nm (data not shown). Furthermore, it is unlikely that the γ PFD is in an aggregate state since the CD spectrum shows a predominantly helical protein with a secondary structure indistinguishable from either the α and β PFD.
γ Prefoldin assembles as a homo-oligomeric filament
Examination of γ PFD by transmission electron microscopy (TEM) revealed protein filaments from 200 nm to over 2 μm in length (Fig. 3A-C). The filament width, however, was uniform: 8.4 ± 0.4 nm (Fig. 3C). This width corresponds to the length of the α PFD subunit in the crystal structure of prefoldin from M. thermoautrophicum (8.0 nm). Because the sequence of γ PFD is globally alignable to this α PFD subunit, it is reasonable to assume that the observed filament width corresponds to the length of the coiled-coiled portion of γ PFD.
Figure 3.

Transmission electron microscopy of γ PFD filaments plated at 5 μM for 2 min at 21°C. (A, B) Scale bar is 200 nm. Note filament lengths are over 1 μm. (C) Scale bar is 50 nm. Magnification shows a uniform width of 8.4 ± 0.4 nm.
The height of the γ PFD filament was probed using atomic force microscopy (AFM) in the tapping mode. The lengths of the plated filaments remained comparable to those seen under TEM. The height, however, is calculated to be 1.8–2.6 nm, showing the aspect ratio between the width and the height to be ∼3–4 (Fig. 4A,B). However, compression by the probe tip during tapping mode AFM is expected to cause an underestimate of the filament height from our measurements (Shao et al. 1996). Based on the M. thermoautotrophicum α/β PFD crystal structure (Siegert et al. 2000), each coiled-coil of the α PFD dimer has a width of ∼1.8 nm; hence, a dehydrated α PFD dimer would have a height of ∼3.6 nm if the dimer was adsorbed to the surface along the axis of a single coiled coil. Our AFM measurements are therefore consistent with the 3.6 nm height expected for this dehydrated α PFD dimer. However, a possible filament structure composed of repeating γ PFD monomer units, rather than dimers, cannot be formally excluded by the present structural constraints.
Figure 4.

(A, B) AFM measurements show a height of the filament between 1.8–2.6 nm.
Ten of the last 15 residues on the C-terminal end of the γ PFD are glutamates. Thus, to determine whether divalent cation-mediated crosslinking of these acidic residues is responsible for the formation of the filament, we performed an experiment wherein 250 μM γ PFD unfolded in 8 M guanidine chloride (GndCl) was refolded in 40 mM Tris, pH 8.0 (final GndCl concentration of 1 M), in the presence or absence of 10 mM EDTA, for 3 h at room temperature. These samples were then separated on a 4%–12% Tris glycine gel by native gel electrophoresis. Migration of the samples was indistinguishable (data not shown), indicating that the glutamate residues are not responsible for the higher order assembly of γ PFD.
γ Prefoldin is a molecular chaperone
A hallmark of molecular chaperones is their ability to prevent aggregation of nonnative proteins. Accordingly, chaperone assays were performed with the γ and α/β PFD. To this end, the individual proteins were coincubated with bovine heart citrate synthase (CS), an aggregation-prone enzyme, at 42°C. Based on absorbance measurements at 505 nm, γ PFD prevented aggregation of thermally denatured CS at a 4:1 γ PFD:CS monomer-to-monomer stoichiometric ratio, and partially prevented aggregation at a 2:1 ratio (Fig. 5A). Neither α nor β PFD was able to prevent aggregation of CS at a 5:1 ratio (Fig. 5B). However, the assembled α/β PFD did prevent aggregation of CS at a 1:1 hexamer-to-monomer ratio (Fig. 5B).
Figure 5.

Chaperone assays of M. jannaschii prefoldins showing reduced aggregation quantity of 150 nM pig-heart citrate synthase (CS) at 42°C (A, B) and improved refolding of His-GFP at 22°C following denaturation in 6 M GndCl (C). (A) • CS, □ 5:1 γ PFD:CS, ▴ 2:1 γ PFD:CS; (B) • CS, ○ α PFD:CS 5:1, ▴ β PFD:CS 5:1, □ α/β PFD: CS 1:1; (C) α/β PFD (○) or γ PFD (▴). The GFP results illustrate that both α/β PFD and γ PFD increased the amount of folded His-GFP.
The ability of α/β and γ PFD to assist in refolding of chemically denatured GFP was also investigated. The chaperones were coincubated with a chemically denatured His-GFP variant in a refolding buffer at 22°C. GFP refolding can be measured by 490/520 nm (ex/em) fluorescence (Fukuda et al. 2000). Although neither α/β nor γ PFD increased the folding rate of the His-GFP variant (data not shown), endpoint assays at 16 h showed that the amount of GFP correctly folded increased with increasing concentrations of either γ PFD or α/β PFD (Fig. 5C). GFP refolding experiments at 37°C yielded similar results (data not shown). Although PFDs are not generally thought to be able to actively refold proteins in a manner similar to ATP-dependent chaperones such as HSP70 or HSP60, an apparent refolding effect for ATP-independent chaperones has been observed in cases where a proportion of the nonnative protein tends to aggregate rather than refold properly (Lee et al. 1995). We should note that such diagnostic chaperone assays were all done at temperatures much lower than the optimal growth temperature for M. jannaschii.
In vivo assembly of γ PFD
All of the experiments described so far were performed in vitro on recombinant proteins; it was thus unknown under what culture conditions and at what concentration γ PFD is produced in M. jannaschii, and whether the native quaternary assembly of γ PFD is a filament. Previous proteomic studies (Giometti et al. 2002; Zhu et al. 2004) failed to detect γ PFD by standard proteomic methodologies under optimal growth conditions. Thus, antibodies were raised against recombinant γ PFD in order to begin testing for production of γ PFD. Western blots were performed against supernatants of cell lysates prepared from nonheat-shocked M. jannaschii cells and M. jannaschii cells subjected to a 1-h 95°C heat shock. Despite upregulation of the γ PFD gene previously observed by (Boonyaratanakornkit et al. 2005), γ PFD was ∼0.05 wt % of the total protein for both heat-shocked and nonheat-shocked samples (Fig. 6A). Assuming a cytosolic protein concentration of 200–400 mg/mL (Ellis 2001), this corresponds to an intracellular γ PFD concentration of 6–12 μM. At 80°C, which is close to the optimal growth temperature of M. jannasachii, recombinant γ PFD forms filaments at ∼3 μM in 50 mM Tris-HCl (pH 8.0) (data not shown).
Figure 6.

Western blots showing production of γ PFD under both heat-shock and nonheat-shock conditions (A). Elution profile of recombinant γ PFD from a Superdex 200 size-exclusion column (plot) correlates with the elution profile of γ PFD from heat-shocked and nonheat-shocked M. jannaschii cells (Westerns) (B).
To investigate whether γ PFD is filamentous in vivo, Western blots were also performed against supernatant of cell lysate from heat-shocked M. jannaschii and of recombinant γ PFD that were passed through a size exclusion column. In both samples, γ PFD elutes at or near the void space of the size exclusion column, consistent with it forming a filament (Fig. 6B). However, because of the time required to process samples prior to separation on the size exclusion column, it is possible that filament formation occurred in vitro.
Discussion
Based on a width of 8.4 nm from TEM measurements, a height of >3 nm from AFM measurements, and a length on the order of several hundred nanometers, we constructed a model for the quaternary structure of the γ PFD filament (Fig. 7A–D). The α/β PFD hexamer assembles from an α PFD dimer core, in which the central region of each α PFD monomer is responsible for hexamer formation (Fandrich et al. 2000; Siegert et al. 2000). In our model, the homologous central region from γ PFD self-associates, forming a filament composed of repeating dimer units along the length of the filament. This assembly would then orient the open ends of the coiled coils from each monomer along the same direction. Because the distal ends of the coiled coils have been shown to be essential for chaperone activity in α/β PFD (Okochi et al. 2004), it is probable that correct orientation of these ends along one direction is necessary to maintain chaperone activity. While our model is consistent with all of the present data, other quaternary structures could also fit the dimensional constraints imposed by the TEM and AFM measurements. However, it is difficult to envisage alternative models that are consistent with the chaperone function of the γ PFD filament.
Figure 7.

Ribbon diagrams of (A) α/β PFD from M. thermoautotrophicum, based on the crystal structure (Siegert et al. 2000) showing the α PFD helices in green, the β PFD helices in red, and the double β-barrel interface in orange; (B, C) approximate dimensions of an α PFD dimer from M. thermoautotrophicum; (D) potential assembly of the γ PFD filament (top view), based on homology modeling, showing the stacking of β-strands in the same manner as for the α/β PFD.
Our model for γ PFD is a long filament of protein with binding sites at precisely spaced distances. This “molecular flypaper” presumably binds proteins along its multivalent linear binding sites. Because many heat-shock proteins from hyperthermophiles are extremely large oligomeric complexes (Kim et al. 1998; Rockel et al. 2002), the high-MW oligomeric nature of γ PFD is not unexpected. What is less obvious is why a linear, as opposed to a globular, filament would be a capable heat-shock protein. Leroux and coworkers convincingly showed that the mechanism of action for PFD entails polyvalent interactions at the distal ends of the coiled coils (Lundin et al. 2004). We postulate that the γ PFD functions in a similar way in utilizing polyvalent binding toward partially denatured proteins. Because γ PFD exhibits such a high degree of polyvalency, it is unlikely that it will be a useful chaperone under nonstress conditions because of the potential to trap partially folded intermediates for long time periods. Thus, it is unresolved how this protein, if filamentous in vivo, can function effectively as a heat-shock protein.
Trent and coworkers observed a filamentous structure for the heat-shock protein HSP60 in the hyperthermophile Sulfolobus shibatae (Trent et al. 1997; Yaoi et al. 1998). However, the filaments of γ PFD and HSP60 differ in two important ways. First, the TEM images of γ PFD suggest a persistence length (a measure of filament stiffness) on the order of 0.1–1 μm. Based on the same visual criterion of the approximate distance between bends in the filament, the HSP60 persistence length appears to be at least an order of magnitude longer. The relatively low room-temperature persistence length argues strongly against the γ PFD filament acting as a cytoskeletal structural element because it is not rigid over μm-long length scales. Second, based on TEM images of the HSP60 filament, the active site of potential chaperone action is buried within the protein–protein interface. In our model of γ PFD the active site is distinct and distant from the intersubunit interface region of the protein, which is consistent with chaperone functionality as a filament.
In addition to the unusual function of γ PFD filaments, self-assembling proteins are of interest for various applications in nanobiotechnology (McMillan et al. 2002; Mao et al. 2004). The γ PFD is notable in this regard because the modular nature of its coiled-coil regions should allow amino acid insertions without compromising the filamentous assembly. Thus, the γ PFD offers the potential for engineering specific biomolecular functions into a highly stable nano-scale filamentous structure.
Materials and Methods
Reagents
Enzymes for DNA manipulation were from New England Biolabs. Oligonucleotides were from Operon Biotechnologies. Plasmids were purchased from Novagen. All other reagents were purchased from Sigma.
Plasmid construction
The oligonucleotides 5′-CCATGGAAAATATGGCTGAAGATTTA-3′ (forward primer), 5′-GGATCCTTATTGTTTCTTATCTTGAGCTTGTTG-3′ (reverse primer), 5′-CATATGACTGTTATGGAATTACCACCAC-3′ (forward primer), 5′-GGATCCTTATTGTGCTGTAGGTATCATTTTTTGA-3′ (reverse primer), 5′-CCATGGTAAATGAAGTCATAGACATAAATG-3′ (forward primer), and 5′-GGATCCTTATTCAGCTTTTTCTTCATTTTCTTC-3′ (reverse primer) were used to rescue the genes encoding the α prefoldin (MJ0952), β prefoldin (MJ0987), and γ prefoldin (MJ0648), respectively, from genomic M. jannaschii DNA by PCR. These PCR fragments were cloned into TOPO sequencing vectors and sequences were verified by DNA sequencing. The DNA sequence containing GFP was rescued off the plasmid clPIT-GFP (Yu and Schaffer 2006) using the oligonucleotides 5′-GGGAATTCCATATGCTACCGGTCGCC-3′ (forward primer) and 5′-TCCGTTTAAACTCGAGATCTGAGTCC-3′ (reverse primer) with NdeI (5′) and XhoI (3′) restriction sites. α PFD, γ PFD, and GFP were subcloned into pET-19, while β PFD was subcloned into pET-30. These plasmids were subsequently transformed into E. coli strain BL21 (DE3).
Protein expression and purification
Cultures were grown in TB in 1-L shake flasks at 37°C and protein expression was induced by addition of 1 mM IPTG at OD600 = 0.6. Cultures were then shaken at 26oC for 12 h, after which time the cells were harvested by centrifugation at 4500g for 10 min in a Jouan centrifuge (Thermo Electronic, Waltham, MA). Cell pellets were resuspended in 50 mM Tris-HCl (pH 8.0) and recentrifuged at 4500g for 10 min. Cell pellets were stored at –80°C until further purification.
Cell pellets were resuspended in 50 mM Tris-HCl (pH 8.0), 10 mM DTT. Cells were lysed by sonication and then centrifuged at 20,000g for 20 min to remove cellular debris. Supernatant was heat treated at 80°C for 1 h and then centrifuged to remove most endogenous precipitated proteins. The resulting solution was applied to a Mono Q anion exchange column (Amersham Biosciences, Piscataway, NJ) and eluted with a linear gradient from 0–1 M NaCl. The subunits were concentrated with Amicon Ultra-15 centrifuge filters with a 5-kDa cutoff (Millipore) and applied to a Superdex 200 HR 16/60 size-exclusion column (Amersham Biosciences).
His-tagged GFP was recovered from insoluble fractions of the cell pellet by repeated washing with 1% Triton X-100 and 5 mM EDTA in 50 mM Tris-HCl (pH 8.0). The fractions were collected and diluted and then applied to a Sepharose-NTA affinity column with Ni2+ as the chelating metal. The protein was eluted with a linear gradient of 0–0.2 M imidazole. Fractions containing His-tagged GFP were collected, concentrated, and stored in 20 mM HEPES buffer, pH 7.2.
Protein concentration was determined by the Bradford method (Bradford 1976) using bovine serum albumin (BSA) as the standard. Protein identification and verification of molecular weight were performed using MALDI-TOF mass spectrometry. All protein preparations were pure based on visual inspection of a Coomassie Blue stained denaturing gel.
M. jannaschii cell extract preparation
M. jannaschii cells were grown in a 1-L bioreactor as previously described (Park and Clark 2002). Cells were either grown at 88°C with 7.8 atm H2:CO2 (4:1 v/v) gas substrate until mid-exponential growth phase or heat shocked from 88°C to 98°C in mid-exponential growth phase and held at 98°C for at least 30 min (Boonyaratanakornkit et al. 2005). Cells were lysed upon exposure to 50 mM Tris, pH 8.0, for 5 min. After centrifugation at 15,000g for 10 min to clarify the supernatant, protein extract was concentrated using Amicon Ultra 15 filters and run on a Superdex 200 HR 16/60 at a flow rate of 1 mL/min with 50 mM Tris-HCl (pH 8.0) and 2 mM DTT as the mobile phase.
Size-exclusion chromatography
α PFD, β PFD, and γ PFD were initially unfolded in 8 M guanidine chloride (GndCl) for 1 h at 60°C. Differing combinations of proteins (α PFD, β PFD, γ PFD, α/β PFD, α/γ PFD, β/γ PFD) were then reconstituted for 1 h at either 80°C or 21°C in refolding buffer. Refolding buffers all included 50 mM Tris pH 8.0 and 10 mM DTT, but differed in KCl concentration (0, 0.5 M), MgCl2 concentration (0, 20 mM), or ATP concentration (0, 1 mM). After centrifugation at 20,000g for 1 min, samples were individually applied to a Superdex200 HR 16/60 size-exclusion column with 50 mM Tris-HCl (pH 8.0), 10 mM DTT, as the mobile phase. The protein concentration of each fraction was determined by the Bradford method. Individual fractions were also assessed by SDS-PAGE and Coomassie Blue staining.
Circular dichroism
Circular dichroism measurements were performed on an Aviv Circular Dichroism spectrometer Model 62DS (Aviv) with ∼50 μg/mL protein in 10 mM sodium phosphate buffer pH 7.2. For far-UV wavelength CD scans, measurements were taken from 200–250 nm at 1-nm resolution at 25°C. Data were averaged over three scans. For the temperature melt, ellipticity was measured at 222 nm at 3°C intervals between 25°C and 97°C with a 45-sec equilibration time.
Aggregation assays
Thermal aggregation of bovine heart citrate synthase (CS) was monitored as previously described (Buchner et al. 1998). Briefly, CS was diluted into 40 mM HEPES-KOH buffer, pH 7.2, at 45°C and an appropriate amount of α, β, or γ PFD to reach a final concentration of 0.15 μM CS monomer under stirring. Turbidity at 505 nm was measured for 20 min on an Aviv 14NT-UV-Vis spectrophotometer equipped with a stirrable, thermostatted cell holder. Control experiments without chaperone were also performed. All assays were performed at least in triplicate.
GFP refolding assays
Recombinant His-tagged GFP (His-GFP) (44 μM) was denatured in 6 M guanidinium chloride (GndCl) for 2 h at 22°C, then diluted 66-fold into renaturation buffer (50 mM Tris-HCl [pH 8.0], 10 mM DTT). Refolding assays were performed at 22°C and at 37°C. Endpoints of refolded GFP fluorescence were based on emission at 520 nm from an excitation wavelength of 490 nm (bandwidth = 5 nm) normalized to internal standards of fully folded His-GFP and fully unfolded His-GFP. Endpoint fluorescent measurements were taken after 16 h. We monitored fluorescence for at least 30 min prior to this endpoint, during which time no further refolding was observed. All measurements were taken on a SpectraMax Fluorescent plate reader (Molecular Devices) at least in triplicate. Stoichiometric amounts of γ PFD or α/β PFD were added to the renaturation buffer before the addition of His-GFP.
Transmission electron microscopy
γ PFD at 10 μg/mL in 20 mM sodium phosphate buffer, pH 7.5, was plated for 2 min at 21°C on glow-discharged 400-mesh Ni grids (SPI supplies). The sample was wicked off of the grids, and the grids were washed thoroughly with nanopure water. A negative stain of 1% uranyl acetate was applied for 2 min. Grids were visualized using an FEI Tecnai 12 120KV transmission electron microscope (FEI).
Atomic force microscopy
γ PFD at 5 μg/mL was plated onto mica sheets for 2 min. The sample was wicked off, and the mica was washed thoroughly with nanopure water. The mica sheet was placed under a nitrogen stream to dry. The sample was probed by a Digital Instruments Nanoscope III instrument in tapping mode in ambient air and at room temperature.
Western blotting
Chicken antibodies were raised against purified recombinant γ PFD (Aves Labs). Western blotting was performed according to established procedures (Sambrook et al. 1989). Control experiments verified that there was no cross-reactivity against α, β PFD.
Molecular graphics
Molecular graphics images were generated using the UCSF Chimera package from the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (Pettersen et al. 2004).
Acknowledgments
We thank Dr. Susan Marqusee for use of the CD spectrophotometer, Dr. Howard Schachman for use of the analytical ultracentrifuge, Reena Zalpuri for assistance with TEM, Brad Olsen and Calvin Darosa for assistance with AFM, Enrigue Gomez for assistance with dynamic light scattering, Dr. David Schaffer for the gift of the clPIT-GFP plasmid, and Dr. Troy Cellmer for helpful discussions and suggestions. This research was funded by the National Science Foundation (BES-0224733), and T.A.W. was supported by an NIH Training Grant RR-01081.
Footnotes
Reprint requests to: Douglas S. Clark, 201 Gilman Hall, Department of Chemical Engineering, University of California, Berkeley, Berkeley, CA 94720, USA; e-mail: clark@berkeley.edu; fax: (510) 643-1228.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.062599907.
References
- Boonyaratanakornkit B.B., Simpson, A.J., Whitehead, T.A., Fraser, C.M., El-Sayed, N.M., and Clark, D.S. 2005. Transcriptional profiling of the hyperthermophilic methanarchaeon Methanococcus jannaschii in response to lethal heat and non-lethal cold shock. Environ. Microbiol. 7: 789–797. [DOI] [PubMed] [Google Scholar]
- Bradford M.M. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72: 248–254. [DOI] [PubMed] [Google Scholar]
- Buchner J., Grallert, H., and Jakob, U. 1998. Analysis of chaperone function using citrate synthase as nonnative substrate protein. Methods Enzymol. 290: 323–338. [DOI] [PubMed] [Google Scholar]
- Ellis R.J. 2001. Macromolecular crowding: An important but neglected aspect of the intracellular environment. Curr. Opin. Struct. Biol. 11: 114–119. [DOI] [PubMed] [Google Scholar]
- Fandrich M., Tito, M.A., Leroux, M.R., Rostom, A.A., Hartl, F.U., Dobson, C.M., and Robinson, C.V. 2000. Observation of the noncovalent assembly and disassembly pathways of the chaperone complex MtGimC by mass spectrometry. Proc. Natl. Acad. Sci. 97: 14151–14155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda H., Arai, M., and Kuwajima, K. 2000. Folding of green fluorescent protein and the cycle3 mutant. Biochemistry 39: 12025–12032. [DOI] [PubMed] [Google Scholar]
- Gasch A.P., Kao, C.M., Carmel-Harel, O., Eisen, M.B., Storz, G., Botstein, D., and Brown, P.O. 2000. Genomic expression programs in the response of yeast cells to environmental changes. Mol. Biol. Cell 11: 4241–4257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giometti C.S., Reich, C., Tollaksen, S., Babnigg, G., Lim, H., Zhu, W., Yates, J., and Olsen, G. 2002. Global analysis of a “simple” proteome: Methanococcus jannaschii . J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 782: 227–243. [DOI] [PubMed] [Google Scholar]
- Hartl F.U. and Hayer-Hartl, M. 2002. Molecular chaperones in the cytosol: From nascent chain to folded protein. Science 295: 1852–1858. [DOI] [PubMed] [Google Scholar]
- Jones W.J., Leigh, J.A., Mayer, F., Woese, C.R., and Wolfe, R.S. 1983. Methanococcus jannaschii sp. nov. an extremely thermophilic methanogen from a submarine hydrothermal vent. Arch. Microbiol. 136: 254–261. [Google Scholar]
- Kim R., Kim, K.K., Yokota, H., and Kim, S.H. 1998. Small heat shock protein of Methanococcus jannaschii, a hyperthermophile. Proc. Natl. Acad. Sci. 95: 9129–9133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laksanalamai P., Whitehead, T.A., and Robb, F.T. 2004. Minimal protein-folding systems in hyperthermophilic archaea. Nat. Rev. Microbiol. 2: 315–324. [DOI] [PubMed] [Google Scholar]
- Lee G.J., Pokala, N., and Vierling, E. 1995. Structure and in vitro molecular chaperone activity of cytosolic small heat shock proteins from pea. J. Biol. Chem. 270: 10432–10438. [DOI] [PubMed] [Google Scholar]
- Leroux M.R., Fandrich, M., Klunker, D., Siegers, K., Lupas, A.N., Brown, J.R., Schiebel, E., Dobson, C.M., and Hartl, F.U. 1999. MtGimC, a novel archaeal chaperone related to the eukaryotic chaperonin cofactor GimC/prefoldin. EMBO J. 18: 6730–6743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundin V.F., Stirling, P.C., Gomez-Reino, J., Mwenifumbo, J.C., Obst, J.M., Valpuesta, J.M., and Leroux, M.R. 2004. Molecular clamp mechanism of substrate binding by hydrophobic coiled-coil residues of the archaeal chaperone prefoldin. Proc. Natl. Acad. Sci. 101: 4367–4372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao C., Solis, D.J., Reiss, B.D., Kottmann, S.T., Sweeney, R.Y., Hayhurst, A., Georgiou, G., Iverson, B., and Belcher, A.M. 2004. Virus-based toolkit for the directed synthesis of magnetic and semiconducting nanowires. Science 303: 213–217. [DOI] [PubMed] [Google Scholar]
- McMillan R.A., Paavola, C.D., Howard, J., Chan, S.L., Zaluzec, N.J., and Trent, J.D. 2002. Ordered nanoparticle arrays formed on engineered chaperonin protein templates. Nat. Mater. 1: 247–252. [DOI] [PubMed] [Google Scholar]
- Okochi M., Yoshida, T., Maruyama, T., Kawarabayasi, Y., Kikuchi, H., and Yohda, M. 2002. Pyrococcus prefoldin stabilizes protein-folding intermediates and transfers them to chaperonins for correct folding. Biochem. Biophys. Res. Commun. 291: 769–774. [DOI] [PubMed] [Google Scholar]
- Okochi M., Nomura, T., Zako, T., Arakawa, T., Iizuka, R., Ueda, H., Funatsu, T., Leroux, M., and Yohda, M. 2004. Kinetics and binding sites for interaction of the prefoldin with a group II chaperonin: Contiguous non-native substrate and chaperonin binding sites in the archaeal prefoldin. J. Biol. Chem. 279: 31788–31795. [DOI] [PubMed] [Google Scholar]
- Park C.B. and Clark, D.S. 2002. Rupture of the cell envelope by decompression of the deep-sea methanogen Methanococcus jannaschii . Appl. Environ. Microbiol. 68: 1458–1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen E.F., Goddard, T.D., Huang, C.C., Couch, G.S., Greenblatt, D.M., Meng, E.C., and Ferrin, T.E. 2004. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 25: 1605–1612. [DOI] [PubMed] [Google Scholar]
- Rockel B., Jakana, J., Chiu, W., and Baumeister, W. 2002. Electron cryo-microscopy of VAT, the archaeal p97/CDC48 homologue from Thermoplasma acidophilum . J. Mol. Biol. 317: 673–681. [DOI] [PubMed] [Google Scholar]
- Sambrook J., Fritsch, E.F., and Maniatis, T. 1989. Molecular cloning: A laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Shao Z.F., Mou, J., Czajkowsky, D.M., Yang, J., and Yuan, J.Y. 1996. Biological atomic force microscopy: What is achieved and what is needed. Adv. Phys. 45: 1–86. [Google Scholar]
- Shockley K.R., Ward, D.E., Chhabra, S.R., Conners, S.B., Montero, C.I., and Kelly, R.M. 2003. Heat shock response by the hyperthermophilic archaeon Pyrococcus furiosus . Appl. Environ. Microbiol. 69: 2365–2371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegert R., Leroux, M.R., Scheufler, C., Hartl, F.U., and Moarefi, I. 2000. Structure of the molecular chaperone prefoldin: Unique interaction of multiple coiled coil tentacles with unfolded proteins. Cell 103: 621–632. [DOI] [PubMed] [Google Scholar]
- Trent J.D., Kagawa, H.K., Yaoi, T., Olle, E., and Zaluzec, N.J. 1997. Chaperonin filaments: The archaeal cytoskeleton? Proc. Natl. Acad. Sci. 94: 5383–5388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vainberg I.E., Lewis, S.A., Rommelaere, H., Ampe, C., Vandekerckhove, J., Klein, H.L., and Cowan, N.J. 1998. Prefoldin, a chaperone that delivers unfolded proteins to cytosolic chaperonin. Cell 93: 863–873. [DOI] [PubMed] [Google Scholar]
- Yaoi T., Kagawa, H.K., and Trent, J.D. 1998. Chaperonin filaments: Their formation and an evaluation of methods for studying them. Arch. Biochem. Biophys. 356: 55–62. [DOI] [PubMed] [Google Scholar]
- Yu J.H. and Schaffer, D.V. 2006. Selection of novel vesicular stomatitis virus glycoprotein variants from a peptide insertion library for enhanced purification of retroviral and lentiviral vectors. J. Virol. 80: 3285–3292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu W., Reich, C.I., Olsen, G.J., Giometti, C.S., and Yates 3rd, J.R. 2004. Shotgun proteomics of Methanococcus jannaschii and insights into methanogenesis. J. Proteome Res. 3: 538–548. [DOI] [PubMed] [Google Scholar]