

Abstract
The superfamily of eye lens βγ-crystallins is highly modularized, with Greek key motifs being used to form symmetric domains. Sequences of monomeric γ-crystallins and oligomeric β-crystallins fold into two domains that pair about a further conserved symmetric interface. Conservation of this assembly interface by domain swapping is the device adopted by family member βB2-crystallin to form a solution dimer. However, the βB1-crystallin solution dimer is formed from an interface used by the domain-swapped dimer to form a tetramer in the crystal lattice. Comparison of these two structures indicated an intriguing relationship between linker conformation, interface ion pair networks, and higher assembly. Here the X-ray structure of recombinant human βB2-crystallin showed that domain swapping was determined by the sequence and not assembly conditions. The solution characteristics of mutants that were designed to alter an ion pair network at a higher assembly interface and a mutant that changed a proline showed they remained dimeric. X-ray crystallography showed that the dimeric mutants did not reverse domain swapping. Thus, the sequence of βB2-crystallin appears well optimized for domain swapping. However, a charge-reversal mutation to the conserved domain-pairing interface showed drastic changes to solution behavior. It appears that the higher assembly of the βγ-crystallin domains has exploited symmetry to create diversity while avoiding aggregation. These are desirable attributes for proteins that have to exist at very high concentration for a very long time.
Keywords: βB1-crystallin, cataract, crystal structure, domain swapping, eye lens, Greek key, oligomer assembly
It is the high concentrations of α, β, and γ-crystallin proteins that provide both refraction and transparency of the vertebrate eye lens (Bloemendal et al. 2004). As well as solubility, the crystallins need stability, as they last a lifetime inside cells with little capacity for protein renewal or disposal (Bron et al. 2000). However, as human lifespan increases and with the elderly forming an increasing proportion of the population, age-related cataract becomes an ever-increasing burden (Klein et al. 2002). Crystallins are built from two types of β-sheet sandwich folds: the α-crystallin domain and the βγ-crystallin domain. Whereas α-crystallins (and the larger family of metazoan small heat shock chaperone proteins) are large, polydisperse multimers with ill-defined dynamic assembly characteristics (Horwitz 2003), the β- and γ-crystallins are examples of modular assembly. All lens β- and γ-crystallin polypeptide chains fold into two βγ-crystallin domains, and each βγ-crystallin domain is built from two similar Greek key motifs (Wistow et al. 2005). The modular assembly is reflected in their genes: Whereas γ-crystallins have each domain encoded by an exon, in β-crystallins an exon encodes each Greek key motif (Lubsen et al. 1988; Wistow and Piatigorsky 1988). Recently, it has been shown that vertebrate βγ-crystallins evolved from a single-domain protein present in urochordates, before the evolution of an image-forming lens, and with an exon pattern indicating that β-crystallins are ancestral (Shimeld et al. 2005). The main difference at the protein level between β- and γ-crystallins is that β-crystallins have sequence extensions and associate into dimers and higher oligomers, whereas γ-crystallins have no long extensions and are monomeric (Bloemendal et al. 2004; Hejtmancik et al. 2004). All lens βγ-crystallin 3D structures have a “conserved domain-paring interface” between N- and C-terminal domains. The two-domain arrangement of γ-crystallins appears to recapitulate an ancient dimer as the domains pair about an interface showing vestiges of dyad symmetry, which is now supported by a covalent polypeptide linker in the monomer (Fig. 1A).
Figure 1.
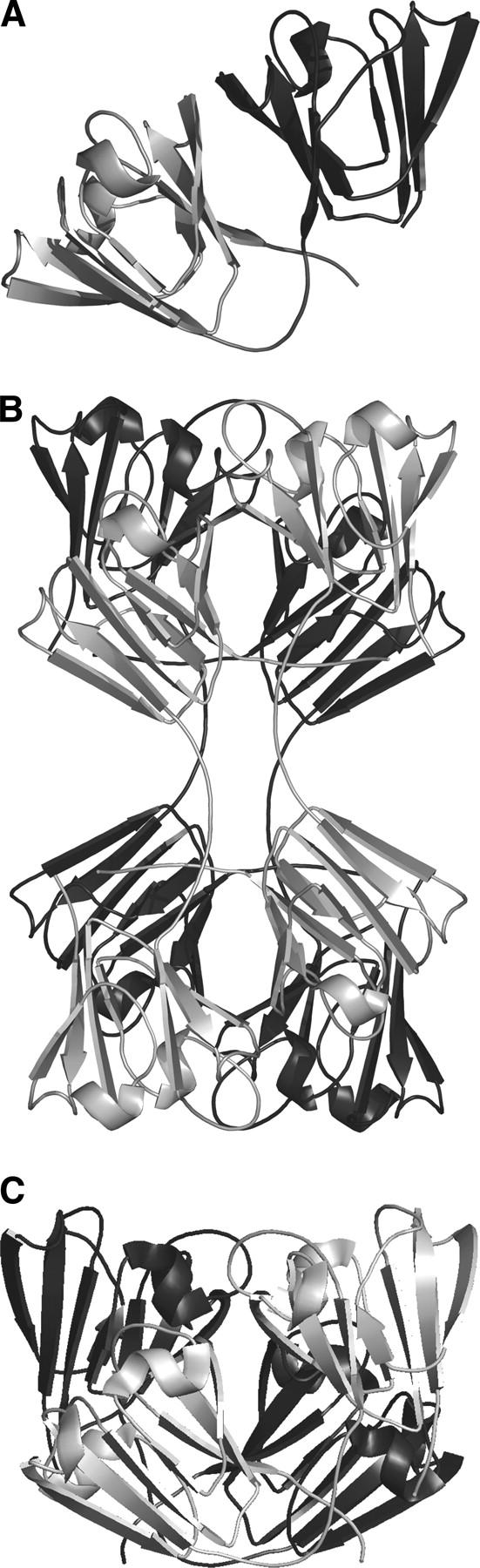
An overview of the symmetry of domain assemblies in β- and γ-crystallins. (A) A monomeric γ-crystallin with the N-terminal domain (dark) and C-terminal domain (light gray) shown. The interface between the two domains is described in the text as “the conserved domain-pairing interface.” In earlier papers this was called the PQ interface (Bax et al. 1990). (B) In the crystal lattice βB2-crystallin assembles as a dimer of dimers, using a “higher-assembly” interface that was previously called the QR interface. Here, the dimers are colored in uniform color. In the (front) light dimer, the N-terminal domains are upper right and lower left. The conserved domain-pairing interface between the upper right N-terminal domain of one chain and its partner C-terminal domain from another chain is in a similar orientation as in A above. (C) A dimer of truncated human βB1-crystallin. (Light) Monomer in front, (dark gray) monomer to the rear. The orientation of the domains in the light monomer is similar to the γ-crystallin monomer in A. The orientation of the dimer is similar to the top half of the βB2-tetramer in B. The arrangement of four domains in the two chains of the βB1-dimer is similar to four domains from four chains in a βB2-tetramer.
The first X-ray structure of a β-crystallin showed that dimerization is governed by domain swapping (Bax et al. 1990). The β-crystallins can be subdivided into basic (βB1, βB2, βB3) and acidic branches, and it was the structure of bovine βB2-crystallin that showed that conservation of the domain-pairing interface was the key to dimerization (Bax et al. 1990). It exemplified how the mechanism of domain swapping (Bennett et al. 1995; Newcomer 2001) can promote rapid evolution of a dimer by a simple change in linker conformation rather than the slow evolution of a new interface (dark or gray dimer, Fig. 1B). Although bovine and human lens βB2-crystallins are dimers in solution (Bateman et al. 2001), the X-ray structure of βB2-crystallin isolated from bovine lens showed that at the high protein concentration found in the crystal lattice they are organized as a dimer of dimers (dark dimer and gray dimer, Fig. 1B). In this higher quaternary state, therefore, a new interface is buried between the domain-swapped dimers. Surprisingly, the crystal structure of a truncated form of human βB1-crystallin (sequence identity with βB2-crystallin 58% across domains), which is a dimer in solution, was found not to domain swap, yet it still resembled one-half of the lattice tetramer of bovine βB2-crystallin (van Montfort et al. 2003; Fig. 1C). In effect, the two types of basic β-crystallin homodimers had different linker conformations, with one using a γ-crystallin conformation burying an additional interface in the dimer assembly. The evolutionary drive to build these genetically economic, modular devices may stem from selection pressure to provide the lens with structural polydispersity to maintain solubility over concentration gradients.
This study aims to decipher the sequence determinants that drive the selection of the interface for dimer assembly. It is not likely to be found in the very similar linker sequences (Fig. 2), in line with previous linker transplantation experiments between β- and γ-crystallins (Mayr et al. 1994; Trinkl et al. 1994). βB2-crystallin was used as the model sequence, as the full-length sequence is readily crystallized, unlike the human βB1 paralog (Bateman et al. 2001). In the original crystal structure determination, purification of the bovine βB2 ortholog necessitated the use of denaturing conditions followed by reassociation to a dimer (Bax and Slingsby 1989). Here, we repeat the crystal structure determination on recombinant human βB2-crystallin in order to test the effect of folding conditions as well as specific residues on the quaternary arrangement of domains. A comparison of the ion pair networks of bovine βB2-crystallin and human truncated βB1-crystallin has previously shown that topologically equivalent residues make ion pairs across different domain interfaces (van Montfort et al. 2003). These residues were targeted for mutagenesis, as well as a proline residue distal from an interface region but which correlated with intramolecular domain pairing in γ-crystallins and truncated human βB1-crystallin. These mutants did not reveal any change to the assembly size; however, a charge reversal to an ion pair at the “conserved domain-pairing interface” caused demonstrable changes to biophysical properties of βB2-crystallin.
Figure 2.
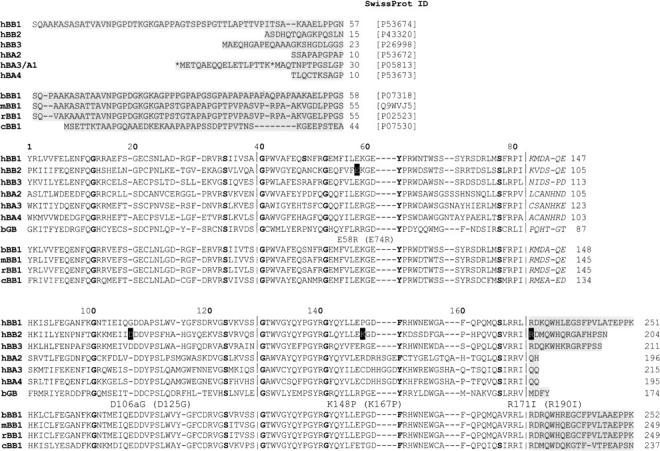
Sequence alignment of the βγ-crystallin domains. The sequences of the human family of β-crystallins are aligned with bovine γB-crystallin, with other mammalian βB1 ortholog sequences indicated below. The sequences are broken into their modules (domains, linkers, and extensions), with the boundaries of the Greek key motifs indicated on the domains. The domains are aligned topologically with the motif conserved glycines and serines in bold, as are the corner tyrosines of motifs 2 and 4. The numbering above the columns is that of γB-crystallin (P02526), and the numbers on the right-hand side of the other sequences correspond to SwissProt numbering. The mutation sites (black outline) are labeled underneath using both numbering systems.
Two numbering systems are used in this paper. The first citation refers to the topological numbering based on the sequence alignment with bovine γB-crystallin. This numbering is used preferentially because the aim of this paper is to compare topologically equivalent interface residues between three different two-domain crystallin polypeptide chains (βB2-crystallin, βB1-crystallin, and γB-crystallin), even though their connections and hence quaternary assemblies vary. In addition, residues are also given their numbering as given in the SwissProt database. Both of these numbering schemes are used in Figure 2.
Results
Characterization of the wild-type human βB2-crystallin
The typical yield from 1 L of culture was 6–9 mg of protein. The identity of the protein was confirmed by electron spray mass spectrometry and shown to be chemically unmodified. Based on far-UV circular dichroism spectroscopy, the purified protein showed elements of β-strand secondary structure and some unordered structure, as shown previously (Reddy et al. 2004) and consistent with the crystal structure of bovine βB2-crystallin. On gel filtration, human βB2-crystallin has an elution volume of 13.32 mL, which was very similar to bovine βB2-crystallin (13.35 mL). Based on standard proteins the molecular weight was estimated to be 40.7 kDa, compared with a calculated mass of 46,498 Da for a dimer based on the sequence. The molecular weight of bovine βB2-crystallin has been measured by ultracentrifugation and shown to be a dimer (Zarina et al. 1994). The molecular mass of human βB2-crystallin at 1 mg/mL measured by light scattering was 45.63 kDa and changed little with protein concentration up to 10 mg/mL. It can therefore be concluded that wild-type βB2-crystallin is a dimer in solution.
Crystal structure of wild-type human βB2-crystallin
Recombinant human βB2-crystallin crystallized in the same space group (I222) and with very similar cell dimensions as the urea-unfolded/refolded lens bovine βB2-crystallin (Table 1). The human protein crystallized only in space group I222; bovine βB2-crystallin crystallized in both C222 and I222 lattices. The structure was solved by molecular replacement using the bovine domains as a search model, taking care not to make assumptions concerning the connectivity of the linker between N- and C- terminal domains. The majority of residues fit into the electron density, but, as with the bovine βB2 structure, the first 13 and last nine residues were not visible and can be assumed to be flexible. The asymmetric unit of the crystal contained one polypeptide chain, with the N- and C-terminal domains not making any intramolecular interactions. Using the symmetry of the space group, the domain-swapped dimer was generated, as well as a lattice tetramer (Fig. 1B) as in the bovine crystal structure (Bax et al. 1990). After refinement the R-factor and R-free converged to 18% and 20%, respectively (Table 1). The RMSD between human and the I222 bovine βB2-crystallin coordinates was 0.58 Å for the main-chain atoms and 1.13 Å for all the atoms (main and side chains). The average B-factor for the protein main chain is 18.3 Å2 and for the solvent is 34.7 Å2.
Table 1.
Statistics of data collection of wild-type human βB2-crystallin collected at the ESRF and the three mutants collected in-house

The X-ray structure shows that the topology of human βB2-crystallin is identical to the bovine ortholog, and, furthermore, a comparison of the inter- and intramolecular contacts of the two orthologs, both solved in the I222 space group, showed they were very similar. Crystals were also grown with the same cell dimensions and space group from a sample of unfolded/refolded human βB2-crystallin. Thus, domain swapping in human βB2-crystallin does not depend on unfolding/refolding of the protein but is encoded in the sequence. A view of the human βB2-crystallin crystal lattice tetramer is shown that highlights selected ion pairs present at the various domain interfaces (Fig. 3A). Of particular note in the domain-swapped βB2-crystallin is residue R171 (190) which ion pairs with D108 (127) across an interface indicated by the dotted vertical line, whereas in the intramolecular domain-paired βB1-crystallin (Fig. 3B), R171 (232) ion pairs with D107 (126) across the interface indicated by the horizontal line.
Figure 3.

A comparison of domain assembly in human βB2-crystallin and βB1-crystallin. (A) The structure of the human βB2-crystallin tetramer viewed from the top when compared with Figure 1B showing that chains A and B form one domain-swapped dimer, and chains C and D form the other dimer. The conserved domain-pairing interface is between the N-terminal domain of A (green) and the C-terminal domain of B (orange), and between the N-terminal domain of C (oyster) and the C-terminal domain of D (magenta). Residues participating in the ion pairs network between the C-terminal domain and the C-terminal hinge of chains B and D are shown interacting about an interface (vertical yellow dotted line) and are labeled with the topological γ-numbering. (B) In human βB1-crystallin the similar arrangement of four domains is from two polypeptide chains. The topologically equivalent charged residues interact across the dimer interface (horizontal dashed line) that is equivalent to the dimer–dimer interface in βB2-crystallin.
Selection of mutants for crystallographic analysis
D106aG
There is an insertion of an aspartic acid that is characteristic of the basic β-crystallins when aligned to γ-crystallin sequences: In βB2-crystallin it is D106a (125) (Fig. 2). Comparison of the tetramer of human βB2-crystallin formed in the lattice and human βB1-crystallin dimer structures show that the side chains topologically equivalent in γB-crystallin to H88, D107, D108, R169, and R171 are (H106/148, D126/168, D127/169, R188/230, and R190/232) using the topologically equivalent/continuous numbering systems of βB2 and βB1, respectively (Fig. 2). These side chains are involved in networks of ion pair interactions in the vicinity of D106a that are different depending on the domain pairing assembly (van Montfort et al. 2003; see also Fig. 3). In the domain-swapped bovine and human βB2-crystallin crystal structures, D106a (125) ion pairs with H88 (106) in the hinge region between the C-terminal domain and the linker, and may have an effect on the linker conformation. Sequence alignments indicate that among βB1-crystallin sequences (bovine, chick, mouse, rat), the human ortholog is unusual in having a glycine instead of an aspartate (glutamate) at position 106a (167). It is possible that this change to the ion pair network causes intramolecular domain pairing specifically in human βB1-crystallin. Therefore, we mutated D106a (125) with glycine (D106aG) in order to see if this would promote intramolecular domain pairing (Fig. 2).
R171I
In βB2-crystallin, R171 (190) is located in the hinge between the C-terminal extension and the C-terminal domain and makes an intramolecular salt bridge with D108 (127) across the interface of the N- and C-terminal domains (Fig. 3A). In the crystal structure of human βB1-crystallin, the equivalent residue R171 (232) makes an intermolecular ion pair with D107(168) (Fig. 3B); however, in γ-crystallins residue 171 is always hydrophobic. R171 (190) was therefore mutated in human βB2-crystallin to isoleucine, R171I. A double mutant was also created, D106aG/R171I.
K167P
A tyrosine corner is a conserved topological feature of many β-sheet proteins and occurs in the second Greek key motif in each domain of the lens βγ-crystallin family (Hemmingsen et al. 1994). In βB2-crystallin, K148 (167) is located in a loop before the conserved tyrosine corner motif. Alignment with other basic β-crystallins and γ−crystallins shows this residue is predominantly a proline, and, as the other solved members of the βγ-crystallin family display intramolecular domain pairing, mutation K167P in βB2-crystallin was created.
Characterization of mutant human βB2-crystallins
The mutants outlined above were expressed and harvested under identical conditions to wild-type βB2-crystallin. They were purified from the soluble fraction after cell lysis, and 1 L of culture yielded ∼8 mg of purified protein. The identity of the mutant proteins was confirmed by electrospray mass spectroscopy and shown to be chemically unmodified. CD analysis of the mutants resulted in spectra very similar to wild-type βB2-crystallin. Gel filtration of all the mutants yielded similar elution volumes in the range of 13.32–13.36 mL, which were comparable to wild-type human βB2-crystallin and consistent with the mutants being dimeric. Dynamic light scattering also indicated that all the mutants had similar hydrodynamic radii between 2.85 and 3.1, corresponding to molecular masses of 39.5–47 kDa. As all of the mutants were indistinguishable from the wild type based on these solution measurements, crystal structures were necessary in order to investigate the quaternary arrangement of domains. Although the double mutant (D106aG and R171I) indicated no major difference in secondary structure, oligomer size, or pI from the native protein, it failed to crystallize when screened using a wide range of conditions, including extensive trial conditions around those that yielded crystals for the wild-type and the single mutants.
Crystal structures of mutant βB2-crystallins
D106aG
The mutant crystallized under similar conditions to wild type, in the same space group and with the same cell dimensions (Table 1). Diffraction data were collected to 2.5 Å, and the structure was solved by molecular replacement using the human wild-type N- and C-terminal domains separately as search models, without the linker peptide. The maps showed a continuous chain of electron density linking domains in the domain-swapped position. The refined electron density confirmed the absence of a side chain at the mutated residue. The average B-factor for the protein main chain is 25.8 Å2 and for the solvent is 32.2 Å2.
Visual inspection of the refined structure superimposed on the native structure and analysis with the program CONTACT (Collaborative Computational Project No. 4 1994) revealed no significant difference to the local arrangement of the side chain orientations across the domains. The mutant did not ion pair as in truncated human βB1-crystallin; it resembled the domain-swapped arrangement. In comparison with wild-type βB2-crystallin (Figs. 3A, 4A), the D106a mutation (Fig. 4B) removes a negative charge that interacts with H88, in the hinge region of βB2-crystallin, and R169 in the C-terminal extension (Fig. 3A). There is now no negative charge in the charged cluster, yet the only noticeable change is an increase in distance between H88 to R169 in the mutant when compared with wild-type βB2-crystallin. The histidine has moved slightly away from the interior of the tetramer, though not as far as the equivalent residue in the dimer of human βB1 (His148). The difference in orientation of the histidine residues in βB1 when compared with βB2 is due primarily to the conformation of the linker. The extended linker has closer interaction with R169 (188) than the βB1 linker, which is bent.
Figure 4.
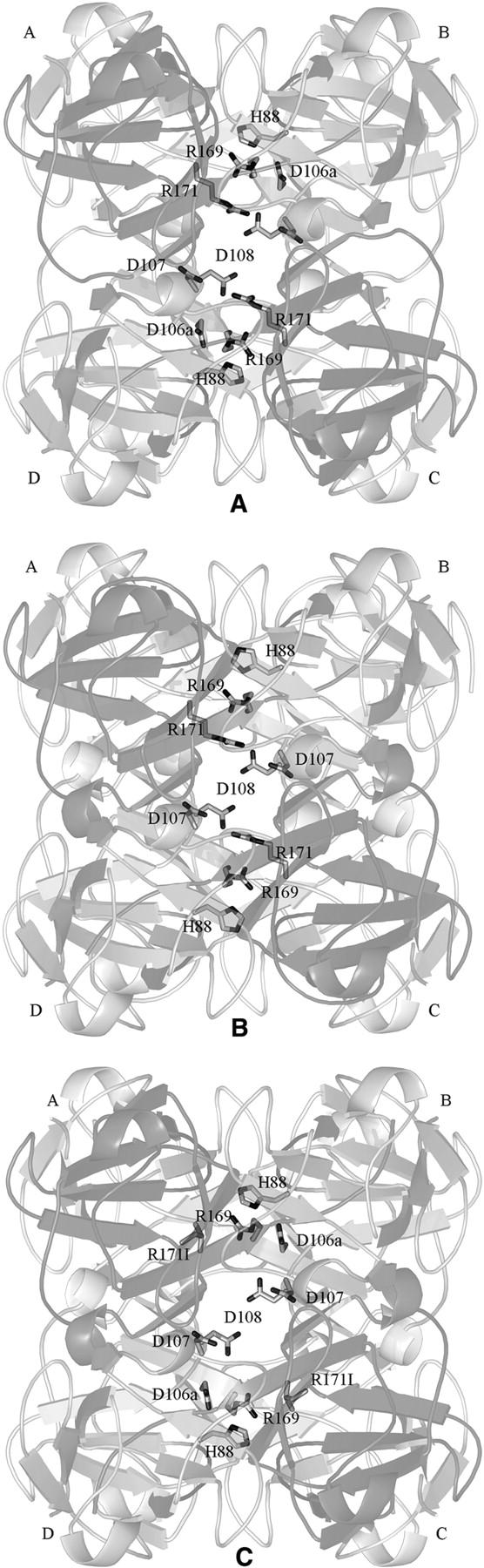
The crystal structures of the interface network mutants. (A) A view of the wild-type βB2-crystallin tetramer shown to emphasize the ion pairs network. (B) The crystal structure of the D106aG mutant showing the positively charged R169 and R171 are still in the same orientation as wild type; however, there is a slight shift of H88 away from the center of the assembly. (C) The crystal structure of the R171I mutant showing that although the ion pair with D108 is disrupted, the other charged residues are largely unaffected.
R171I
The mutant also crystallized under similar conditions to wild type with similar crystal characteristics (Table 1). Diffraction data were collected to 2.5 Å, and the structure was solved by molecular replacement and refined using similar protocols as described for the above mutant. The average B-factor for the protein main chain is 26.9 Å2 and for the solvent is 32.8 Å2. The refined electron density confirmed the presence of the new side chain. However, even though its ion pair is absent (Fig. 4C), D108 is in exactly the same position, pointing toward the interior of the tetramer, and there is no measurable difference in the distances between the remaining ionic interactions. The structure shows that even with the arginine residue missing from the network of ion pairs, the other side chain interactions are maintained.
K148P
K148 (167) is part of the tyrosine corner (Fig. 2), an important topological determinant of the βγ-crystallin domain fold. Sequences with proline at this position, such as γ-crystallins and human βB1-crystallin, engage in intramolecular domain pairing, whereas in domain-swapped human and bovine βB2-crystallin this residue is lysine. It is possible that a residue with restricted torsion angles might affect the rate of folding of the C-terminal domain and have an effect on domain pairing. The crystals of the K148P (167) mutant had the morphology of the wild type and diffracted beyond 2.5 Å (Table 1). The average B-factor for the protein main chain is 22.9 Å2 and for the solvent is 34.2 Å2. The electron density map around the mutation site showed an excellent fit with proline. However, there was no difference in the local environment around the proline, and the structure remained domain-swapped. One factor that causes some proteins to have a propensity to domain swap is strain in a loop or turn (O'Neill et al. 2001). Analysis of torsion angles of P148 in structures that are not domain-swapped such as human γD (1hk0), bovine γB (1amm), and human βB1 (1oki) showed they were within the core β-sheet region, whereas in human domain-swapped wild-type βB2-crystallin and bovine βB2-crystallin, lysine 148 is outside the core β-sheet region (Table 2). In the K148P mutant the φ ψ torsion angles are now within the core β-sheet region. Thus, in βγ-crystallins the absence of proline in this position introduces some strain in this region, but this is not correlated with domain swapping.
Table 2.
The torsion angles of a residue in a tyrosine corner as a function of presence/absence of a proline and of domain swapping

Mutation at the conserved interface
As little perturbation of the quaternary assembly of domain-swapped βB2-crystallin had been achieved, a mutation was also made at the “conserved domain-pairing interface,” between N- and C-terminal domains (Fig. 5). Residue 58 (74) in the N-terminal domain is close in space to residue 168 (187) in the C-terminal domain, and because this interface is organized about an approximate twofold axis, the topologically equivalent residue 147 (166) in the C-terminal domain is close to residue 79 (97) in the N-terminal domain (topologically equivalent to residue 168). In monomeric γ-crystallins (except intermediary types like γS and γN), all four residues are arginine (Bloemendal et al. 2004; Wistow et al. 2005). However, in oligomeric β-crystallins, regardless of whether domains are paired intra- or intermolecularly, residues topologically equivalent to positions 58 and 147 are glutamates, resulting in two ion pairs at the borders of the hydrophobic interface patch (Fig. 5). An E58R (E74R) mutation would thus change a stabilizing ion pair into a repulsive interdomain contact.
Figure 5.

The conserved domain-pairing interface of human βB2-crystallin. The N-terminal domain (light gray) of one chain interacts with the C-terminal domain (dark gray) of a separate chain about a hydrophobic interface. Surrounding this core are ion pairs with this view showing that the pseudo twofold symmetry allows the topologically equivalent acidic residues (E58 and E147, and see alignment in Fig. 2) in the N- and C-terminal domains, respectively, to interact with topologically equivalent basic residues R168 and R79.
The E58R mutation had a drastic effect on solubility. The wild-type protein and all the other mutants were expressed in the soluble fraction of Escherichia coli, and most are soluble upwards of 100 mg/mL. The E58R mutant, however, was always found in inclusion bodies, despite screening of different expression conditions, and had to be refolded. It also precipitated at concentrations of ∼2–3 mg/mL. The molecular weight was estimated at this protein concentration both by gel filtration and light scattering. The elution volume of the E58R mutant at 14.55 mL was closer to bovine γB-crystallin (14.80 mL) than to the wild-type βB2-dimer. The results show that the mutant no longer behaves like a dimer, but appears more monomeric. This mutant demonstrates that human βB2-crystallin is sensitive to mutations at the conserved domain-pairing interface: It becomes much less soluble, prone to aggregation, and does not behave as a solution dimer.
Discussion
The evolution of the βγ-crystallin domains into molecules of different sizes, shapes, and surfaces is presumably driven by the selective requirement of high solubility in the crowded, high refractive index lens cytoplasm. Crystal structures of monomeric γ-crystallins and two homodimeric β-crystallins have shown that in all cases the similar N- and C-terminal domains engage in an approximately symmetric interaction that we call the “conserved domain-pairing interface.” However, at the dimer level there are two options: domain swapping involving mainly a conformational switch to a linker region (βB2-crystallin), whereas the other requires the burial of a new interface (βB1-crystallin). It is intriguing that the new dimeric interface observed in the solution dimer of βB1-crystallin is the equivalent interface used for dimer–dimer assembly by the domain-swapped βB2-crystallin in the crystal lattice (van Montfort et al. 2003). In this discussion we call this “the higher-assembly interface,” although in the case of βB1-crystallin it is a monomer–monomer interface and in the case of βB2-crystallin it is the dimer–dimer interface. The several members of the lens β-crystallins readily exchange subunits and thus create solution polydispersity (Slingsby and Bateman 1990; Bloemendal et al. 2004; Hejtmancik et al. 2004). Having two types of geometric homodimers has implications for their heterodimeric assembly: The option of the “higher-assembly interface” would appear to provide the potential to interchange solvent-facing surfaces with buried interfaces (see Fig. 1). Here, we attempted to determine a sequence code for this assembly switch.
Analysis of the side chain interactions showed that certain charged residues occupying topologically equivalent positions in the domains were found to use different ion pair partners depending on which interface was buried in the assembly (van Montfort et al. 2003). Mutations were made to residues in these ion pair networks in an attempt to switch the assembly. The model sequence used was human βB2-crystallin, and outcomes may result in monomer, domain-swapped dimer, or the use of the “the higher-assembly interface.” Although it is straightforward to distinguish monomer from dimer, it is more taxing to distinguish the two kinds of four-domain dimer. It was therefore necessary to use X-ray crystallography to visualize the mutant dimers. However, first of all it was important to validate that the sequence alone was the cause of domain swapping in βB2-crystallin.
Experimental conditions that result in a protein being denatured or partially unfolded for some period of time can create an optimal environment in vitro for domain swapping (Bennett et al. 1995). Wild-type human βB2-crystallin, produced by recombinant expression as opposed to harvesting from lens cells under denaturing conditions followed by reassociation, was shown to be dimeric in solution. The crystal structure showed that the dimer is domain-swapped like refolded bovine βB2-crystallin. Therefore, the mode of association is determined by the amino acid sequence.
In all βγ-crystallins the “conserved domain-pairing interface” is mediated through a hydrophobic core that is surrounded by a set of polar and charged residues characteristic of either a β- or a γ-crystallin. These interface residues in γ-crystallins have been shown by mutagenesis to influence folding and stability (Palme et al. 1998a,b; Flaugh et al. 2005). However, the “loose ends,” such as the linker and adjacent C-terminal extension, participate in subtly different ways (Norledge et al. 1996). In this work we are probing the role of charged residues across two assembly interfaces. In one example, in dimeric βB1-crystallin, R171 (232) on the C-terminal extension ion pairs with D107 (168) across the “higher-assembly” interface, whereas in domain-swapped βB2-crystallin this residue makes an intramolecular ion pair with D108 (Fig. 3), and in monomeric γ-crystallins residue 171 is hydrophobic. When R171 was changed in βB2-crystallin to isoleucine, the resulting protein remained dimeric, the interaction between H88 (at the hinge between the linker and the C-terminal domain) and R169 (at the hinge between the C-terminal domain and the C-terminal extension) was maintained, and the dimer remained domain-swapped.
Basic β-crystallin sequences have an insertion compared with γ-crystallins, D106a, which is part of an acidic cluster that includes D107 and D108. In human βB1-crystallin, D107 forms an ion pair network that crosses the higher-assembly interface. However, human βB1-crystallin is unusual in having a glycine at this position, and this may have contributed to the formation of this mode of assembly. When the equivalent residue D106a in human βB2-crystallin was replaced with glycine, the crystal structure showed there was no change in this mode of association, and although there was some movement in H88, all of the other charged residues occupied similar space and orientations as in domain-swapped βB2-crystallin. The double mutant (R171I and D106aG) remained dimeric, but unfortunately did not crystallize.
As mutations at these insertion and hinge-extension regions failed to switch the assembly to the “higher-assembly interface,” it would appear that the sequence of βB2-crystallin is well optimized for domain swapping rather than forming the more entropically favorable monomer. This means that elusive crucial sequence differences are possibly widely distributed across the sequence and/or are exerting their effect early in the folding and assembly process. By contrast, a mutation that caused a charge reversal at the conserved domain-pairing interface changed the biophysical properties of βB2-crystallin. There has been considerable interest in a pair of glutamine residues that also participate in flanking the hydrophobic core residues of this conserved interface. In βB2-crystallin these glutamines are deamidated in human cataractous lens (Searle et al. 2005), and replacement with glutamates leads to destabilization in human βB2-crystallin (Lampi et al. 2006), human βB1-crystallin (Kim et al. 2002), and human γD-crystallin (Flaugh et al. 2006). It is likely that ion pairs and polar contacts across this conserved interface in oligomeric β-crystallins stabilize the “local” symmetry of the paired-domain interface. In the case of the domain-swapped βB2-crystallin, this kind of stabilization is likely to ensure the geometry for the dimer “dyad” symmetry. Any “slippage” at the hydrophobic interface could lead to endless aggregation.
Materials and Methods
Expression, mutagenesis, and purification of human βB2-crystallin
The human βB2-crystallin clone in a pET3a plasmid (Novagen), a gift from Dr. N.H. Lubsen, Radboud University, Nijmegen, was expressed in BL21 DE3 competent cells. Ten milliliters of an overnight culture were used to inoculate 1 L of 2yt with 50 μg/mL ampicillin. The culture was grown at 37°C with shaking at 200 rpm until an O.D. of 0.4–0.6 was reached followed by induction with 25 μM IPTG, then the culture was grown for an additional 4 h. Cells were harvested by centrifugation at 3000 rpm for 20 min, the supernatant was discarded, and the cell pellet was resuspended in Bugbuster (Novagen). After sonication and overnight dialysis with 25 mM Tris–HCl (pH 8.0), 1 mM DTT (buffer A), the purification protocol involved three chromatographic steps. In the first step, the whole-cell lysate was subjected to anion exchange on a Q-sepharose Hiprep 16/10 column (Amersham Biosciences) equilibrated with buffer A and eluted on a 0–1 M NaCl gradient. Samples containing βB2-crystallin as indicated by SDS-PAGE were desalted by pressure-filtration through a 30-kDa cutoff Amicon membrane, and buffer-exchanged into 25 mM BTP (pH 7.0), 1 mM DTT (buffer B). This sample was chromatographed on a second ion-exchange column, 8 mL of monoQ (Pharmacia) equilibrated with buffer B and a 0–1 M NaCl gradient, the protein fractions were collected and concentrated, then subjected to gel filtration on a Superose 12 column (Pharmacia). The concentration of purified protein, calculated using an extinction coefficient of 1.7 for a 1 mg/mL solution, showed that a typical yield from 1 L of culture was 6–9 mg of protein. All mutagenesis experiments were done using the Quickchange mutagenesis kit (Stratagene) and were sequenced at the University of Cambridge Department of Biochemistry DNA Sequencing Facility. Expression and purification of βB2-crystallin mutants was performed using the native protocol as a model that was optimized for each mutant. Protein identity was confirmed by electrospray mass spectrometry as described previously (Bateman et al. 2001).
Unfolding and refolding of wild-type human βB2-crystallin
A solution of the purified wild-type protein was unfolded by addition of solid urea to make a final concentration of 6 M urea. The unfolded protein was refolded by adding dropwise to an excess of a cold, stirred solution of 25 mM Tris-HCl buffer, 1 mM DTT, at pH 8.0, and equilibrated into the native buffer using an Amicon stirred ultrafiltration cell fitted with a YM 30 membrane.
Unfolding and refolding of E58R (E74R)
Expression of the E58R mutant was lower than wild type, and was found in the insoluble fraction. The pellet was washed several times with 50 mM Tris-HCl pH 8.0, 100 mM NaCl, 5 mM DTT, and 1 mM EDTA and was spun at 5000g for 10 min. The inclusion bodies were solubilized in the wash buffer, which had 8 M urea added, gently shaken for 1 h at 4°C, then spun at 5000g for 10 min. The unfolded protein was refolded by adding dropwise to a 10× excess volume of a cold, stirred solution of 25 mM Tris-HCl buffer, 4 mM DTT, 1 mM EDTA, at pH 8.0, and equilibrated into the native buffer using an Amicon stirred ultrafiltration cell fitted with a YM 30 membrane.
Size determination
Gel filtration
Two-hundred-microliter samples of wild-type human βB2-crystallin at 4 mg/mL were loaded onto a Superose 12 10/30 HR column at 22°C and eluted in 25 mM BTP, 200 mM NaCl, 1 mM DTT pH 7.5, at 0.5 mL/min. The column was calibrated with standard protein markers (catalase, 232 kDa; aldolase, 158 kDa; albumin, 67 kDa; ovalbumin, 43 kDa; and Rnase A, 13.7 kDa; Amersham Biosciences). The mutants were analyzed under the same conditions as wild type.
Light scattering
One-hundred-microliter sample volumes of wild-type βB2-crystallin in 25 mM Tris-HCl, pH 8.0 at various concentrations between 1 and 10 mg/mL were injected into a Dynapro 810 dynamic Right Angle Laser Light Scattering (RALLS) instrument (DynaPro International) after it was calibrated with distilled water and a steady count rate was observed. Twenty to 50 measurements were recorded depending on the baseline. The average over at least 15 readings was taken. The oligomer size was estimated from the calculated hydrodynamic radius based on a plot (provided by the manufacturer) of standard globular proteins. Identical sample volumes and buffers were used for the wild type and mutants, but the protein concentration of E58R was lower at 1.5 mg/mL in comparison to 2.5 and 5 mg/mL for the other crystallin samples.
Crystallization of wild-type and mutant human βB2-crystallin
Numerous crystallization conditions were screened, although good diffracting crystals were obtained only under conditions similar, but not identical, to the bovine ortholog. Conditions were optimized so that crystals diffracting beyond 2.0 Å were grown at 4°C by hanging drop vapor diffusion from wells containing 42% methyl pentane diol (MPD), 50 mM Tricine buffered at pH 8.0, 0.1 M MgCl2, 2 mM DTT, and 0.1 M NaCacodylate and from a 4-μL drop volume with a 1:1 ratio of 14 mg/mL protein with precipitant well solution. The crystals were harvested and cryoprotected in 45% MPD, flash frozen in liquid nitrogen, and stored for transport to the synchrotron. Crystals of the mutants were obtained by screening around the conditions under which wild-type human βB2-crystallin protein crystallized. Diffraction quality crystals were obtained and data were collected in-house on a Rigaku RU-H3R rotating anode and Mar Research image plate.
Circular dichroism spectroscopy
The wild-type human βB2-crystallin was passed through a desalting column equilibrated with 20 mM sodium phosphate, pH 7.5 for secondary structure estimation using far-UV circular dichroism spectroscopy. Spectra were recorded on an AVIV 62DS spectrometer and were collected over the wavelength range 190–300 nm at 0.2-nm intervals. Spectra were derived from three repeat scans of 20 min.
Structure determination
Wild type
Two data sets, one to 3.0-Å and one to 1.7-Å resolution, were collected on beam-line ID14–3 at the ESRF, Grenoble. Because a lot of the low-resolution reflections in the high-resolution images were overloaded, both data sets were processed, scaled, and merged using MOSFLM and SCALA (CCP4 1994), and both datasets were indexed in space group I222, indicating one molecule in the asymmetric unit. Initial phases were obtained by molecular replacement with MOLREP (CCP4 1994) using data 30–3.0 Å from the 3-Å data set. The disconnected bovine βB2-crystallin N- and C-terminal domains were used as search models. A continuous chain of density was visible from the N- to the C-terminal domain, revealing that the linker was in an extended conformation. The molecular replacement procedure was repeated using the complete polypeptide chain of the bovine coordinates (PDB ID: 2bb2). After replacing the human sequence changes, the structure was refined using REFMAC5 (Murshudov et al. 1997) combined with manual model building using XFIT (McRee 1999), and ARP/wARP (Perrakis et al. 1999) was used to automatically generate water molecules. A Ramachandran plot of the refined structure showed that 86.2% of residues were in the most favored region, and 13.2% were in the additionally allowed region. Residue D106a (125) was in a disallowed region and is an insertion when the sequence is aligned with a γ-crystallin (Fig. 2). The coordinates have been deposited in the Protein Data Bank with ID 1ytq.
Mutants
Diffraction data for the mutants were collected in-house, and each of the mutants diffracted to 2.5 Å. All data sets were processed, scaled, and merged using MOSFLM and SCALA (CCP4 1994), and were indexed in space group I222 like the native crystal. The N- and C-domains of the wild-type structure (minus the wild-type side chain) were initially used for molecular replacement; however, as the linker maintained its conformation, the whole wild-type molecule was used for molecular replacement and refinement.
Programs for analysis of contacts
The structures were validated with PROCHECK (Laskowski et al. 1993), and the interatomic distances were calculated by CONTACT (CCP4 1994).
Acknowledgments
We thank Dr. Nicolette H. Lubsen for discussions and the gift of the human βB2-crystallin clone. We are grateful to Daresbury SRS and the ESRF Grenoble for beam time. The work has been supported by an EU BioMed Grant (BMH4-CT98-3895). C.S. and O.A.B. are grateful for the financial support of the Medical Research Council (London).
Footnotes
Reprint requests to: Christine Slingsby, Birkbeck College, Department of Crystallography, Malet Street, London WC1E 7HX, UK; e-mail: C.Slingsby@mail.cryst.bbk.ac.uk; fax: 44-(0)20-7631 6803.
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.062659107.
Abbreviations: MPD, 2-Methyl-2, 4-pentanediol; DTT, dithiothreitol; BTP, 1,3-bis[tris(hydroxymethyl)methylamino] propane; IPTG, isopropyl-beta-D-thiogalactopyranoside.
References
- Bateman O.A., Lubsen, N.H., and Slingsby, C. 2001. Association behaviour of human βB1-crystallin and its truncated forms. Exp. Eye Res. 73: 321–331. [DOI] [PubMed] [Google Scholar]
- Bax B. and Slingsby, C. 1989. Crystallization of a new form of the eye lens protein βB2-crystallin. J. Mol. Biol. 208: 715–717. [DOI] [PubMed] [Google Scholar]
- Bax B., Lapatto, R., Nalini, V., Driessen, H., Lindley, P.F., Mahadevan, D., Blundel, T.L., and Slingsby, C. 1990. X-ray analysis of βB2-crystallin and evolution of oligomeric lens proteins. Nature 347: 776–780. [DOI] [PubMed] [Google Scholar]
- Bennett M.J., Schlunegger, M.P., and Eisenberg, D. 1995. 3D Domain swapping—A mechanism for oligomer assembly. Protein Sci. 4: 2455–2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloemendal H., de Jong, W., Jaenicke, R., Lubsen, N.H., Slingsby, C., and Tardieu, A. 2004. Ageing and vision: Structure, stability and function of lens crystallins. Prog. Biophys. Mol. Biol. 86: 407–485. [DOI] [PubMed] [Google Scholar]
- Bron A.J., Vrensen, G.F.J.M., Koretz, J., Maraini, G., and Harding, J.J. 2000. The ageing lens. Ophthalmologica 214: 86–104. [DOI] [PubMed] [Google Scholar]
- Collaborative Computational Project No. 4 1994. The CCP4 suite: Programs for protein crystallography. Acta Cryst. D50: 760–763. [DOI] [PubMed] [Google Scholar]
- Flaugh S.L., Kosinski-Collins, M.S., and King, J. 2005. Interdomain side-chain interactions in human γ D crystallin influencing folding and stability. Protein Sci. 14: 2030–2043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flaugh S.L., Mills, I.A., and King, J. 2006. Glutamine deamidation destabilizes human γ D-crystallin and lowers the kinetic barrier to unfolding. J. Biol. Chem. 281: 30782–30793. [DOI] [PubMed] [Google Scholar]
- Hejtmancik J.F., Wingfield, P.T., and Sergeev, Y.V. 2004. β-crystallin association. Exp. Eye Res. 79: 377–383. [DOI] [PubMed] [Google Scholar]
- Hemmingsen J.M., Gernert, K.M., Richardson, J.S., and Richardson, D.C. 1994. The tyrosine corner: A feature of most Greek key β-barrel proteins. Protein Sci. 3: 1927–1937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horwitz J. 2003. α-crystallin. Exp. Eye Res. 76: 145–153. [DOI] [PubMed] [Google Scholar]
- Kim Y.H., Kapfer, D.M., Boekhorst, J., Lubsen, N.H., Bächinger, H.P., Shearer, T.R., David, L.L., Feix, J.B., and Lampi, K.J. 2002. Deamidation, but not truncation, decreases the urea stability of a lens structural protein, βB1-crystallin. Biochemistry 41: 14076–14084. [DOI] [PubMed] [Google Scholar]
- Klein B.E.K., Klein, R., and Lee, K.E. 2002. Incidence of age-related cataract over a 10-year interval: The Beaver Dam Eye Study. Ophthalmology 109: 2052–2057. [DOI] [PubMed] [Google Scholar]
- Lampi K.J., Amyx, K.K., Ahmann, P., and Steel, E.A. 2006. Deamidation in human lens β B2-crystallin destabilizes the dimer. Biochemistry 45: 3146–3153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski R.A., MacArthur, M.W., Moss, D.S., and Thornton, J.M. 1993. PROCHECK—A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26: 283–291. [Google Scholar]
- Lubsen N.H., Aarts, H.J.M., and Schoenmakers, J.G.G. 1988. The evolution of lenticular proteins: The β- and γ-crystallin super gene family. Prog. Biophys. Mol. Biol. 51: 47–76. [DOI] [PubMed] [Google Scholar]
- Mayr E.-M., Jaenicke, R., and Glockshuber, R. 1994. Domain interactions and connecting peptides in lens crystallins. J. Mol. Biol. 235: 84–88. [DOI] [PubMed] [Google Scholar]
- McRee D.E. 1999. XtalView/Xfit—A versatile program for manipulating atomic coordinates and electron density. J. Struct. Biol. 125: 156–165. [DOI] [PubMed] [Google Scholar]
- Murshudov G.N., Vagin, A.A., and Dodson, E.J. 1997. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53: 240–255. [DOI] [PubMed] [Google Scholar]
- Newcomer M.E. 2001. Trading places. Nat. Struct. Biol. 8: 282–284. [DOI] [PubMed] [Google Scholar]
- Norledge B.V., Mayr, E.-M., Glockshuber, R., Bateman, O.A., Slingsby, C., Jaenicke, R., and Driessen, H.P.C. 1996. The X-ray structures of two mutant crystallin domains shed light on the evolution of multi-domain proteins. Nat. Struct. Biol. 3: 267–274. [DOI] [PubMed] [Google Scholar]
- O'Neill J.W., Kim, D.E., Johnsen, K., Baker, D., and Zhang, K.Y. 2001. Single-site mutations induce 3D domain swapping in the B1 domain of protein L from Peptostreptococcus magnus . Structure 9: 1017–1027. [DOI] [PubMed] [Google Scholar]
- Palme S., Jaenicke, R., and Slingsby, C. 1998a. Unusual domain pairing in a mutant of bovine lens γB-crystallin. J. Mol. Biol. 279: 1053–1059. [DOI] [PubMed] [Google Scholar]
- Palme S., Jaenicke, R., and Slingsby, C. 1998b. X-ray structures of three interface mutants of γB-crystallin from bovine eye lens. Protein Sci. 7: 611–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrakis A., Morris, R., and Lamzin, V.S. 1999. Automated protein model building combined with iterative structure refinement. Nat. Struct. Biol. 6: 458–463. [DOI] [PubMed] [Google Scholar]
- Reddy A.M., Bateman, O.A., Chakarova, C., Ferris, J., Berry, V., Lomas, E., Sarra, R., Smith, M.A., Moore, A.T., Bhattacharya, S.S., et al. 2004. Characterisation of the G91del CRYBA1/3-crystallin protein: A cause of human inherited cataract. Hum. Mol. Genet. 13: 945–953. [DOI] [PubMed] [Google Scholar]
- Searle B.C., Dasari, S., Wilmarth, P.A., Turner, M., Reddy, A.P., David, L.L., and Nagalla, S.R. 2005. Identification of protein modifications using MS/MS de novo sequencing and the OpenSea alignment algorithm. J. Proteome Res. 4: 546–554. [DOI] [PubMed] [Google Scholar]
- Shimeld S.M., Purkiss, A.G., Dirks, R.P.H., Bateman, O.A., Slingsby, C., and Lubsen, N.H. 2005. Urochordate βγ-crystallin and the evolutionary origin of the vertebrate eye lens. Curr. Biol. 15: 1684–1689. [DOI] [PubMed] [Google Scholar]
- Slingsby C. and Bateman, O.A. 1990. Quaternary interactions in eye lens β-crystallins: Basic and acidic subunits of β-crystallins favor heterologous association. Biochemistry 29: 6592–6599. [DOI] [PubMed] [Google Scholar]
- Trinkl S., Glockshuber, R., and Jaenicke, R. 1994. Dimerization of βB2-crystallin: The role of the linker peptide and the N- and C-terminal extensions. Protein Sci. 3: 1392–1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Montfort R.L.M., Bateman, O.A., Lubsen, N., and Slingsby, C. 2003. Crystal structure of truncated human βB1-crystallin. Protein Sci. 12: 2606–2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wistow G.J. and Piatigorsky, J. 1988. Lens crystallins: The evolution and expression of proteins for a highly specialized tissue. Annu. Rev. Biochem. 57: 479–504. [DOI] [PubMed] [Google Scholar]
- Wistow G., Wyatt, K., David, L., Gao, C., Bateman, O., Bernstein, S., Tomarev, S., Segovia, L., Slingsby, C., and Vihtelic, T. 2005. γ N-crystallin and the evolution of the βγ-crystallin superfamily in vertebrates. FEBS J. 272: 2276–2291. [DOI] [PubMed] [Google Scholar]
- Zarina S., Slingsby, C., Jaenicke, R., Zaidi, Z.H., Driessen, H., and Srinivasan, N. 1994. Three-dimensional model and quaternary structure of the human eye lens protein γS-crystallin based on β- and γ-crystallin X-ray coordinates and ultracentrifugation. Protein Sci. 3: 1840–1846. [DOI] [PMC free article] [PubMed] [Google Scholar]