

Abstract
A method to rapidly screen libraries of cyclic peptides in vivo for molecules with biological activity has been developed and used to isolate cyclic peptide inhibitors of the ClpXP protease. Fluorescence activated cell sorting was used in conjunction with a fluorescent reporter to isolate cyclic peptides that inhibit the proteolysis of tmRNA-tagged proteins in Escherichia coli. Inhibitors shared little sequence similarity and interfered with unexpected steps in the ClpXP mechanism in vitro. One cyclic peptide, IXP1, inhibited the degradation of unrelated ClpXP substrates and has bactericidal activity when added to growing cultures of Caulobacter crescentus, a model organism that requires ClpXP activity for viability. The screen used here could be adapted to identify cyclic peptide inhibitors of any enzyme that can be expressed in E. coli in conjunction with a fluorescent reporter.
Keywords: cyclic peptide, ClpXP protease, antibacterial, protease inhibitor, SICLOPPS
The discovery of antibiotics and pharmacological reagents requires identification of small molecules that can act in vivo. The use of in vivo screening methods to identify inhibitors of specific enzymes ensures that inhibitors will have biological as well as biochemical activity. To establish methods for rapidly screening a pool of possible inhibitors that will be effective in bacteria, inhibitors were sought from a library of cyclic peptides produced in bacteria using split intein circular ligation of proteins and peptides (SICLOPPS) technology (Scott et al. 1999). SICLOPPS uses the chemistry of inteins, naturally occurring protein-splicing sequences, to ligate the N and C termini of a peptide (Fig. 1A). The intein-catalyzed reaction is spontaneous in vivo and does not require extensive sequence conservation in the cyclic peptide, thus a wide variety of cyclic peptides and proteins can be efficiently produced. Because the cyclic peptides produced by SICLOPPS are genetically encoded, complex libraries can be produced by randomizing the DNA sequence encoding the peptide. Such libraries have been successfully screened for inhibitors of protein–protein interactions (Horswill et al. 2004).
Figure 1.
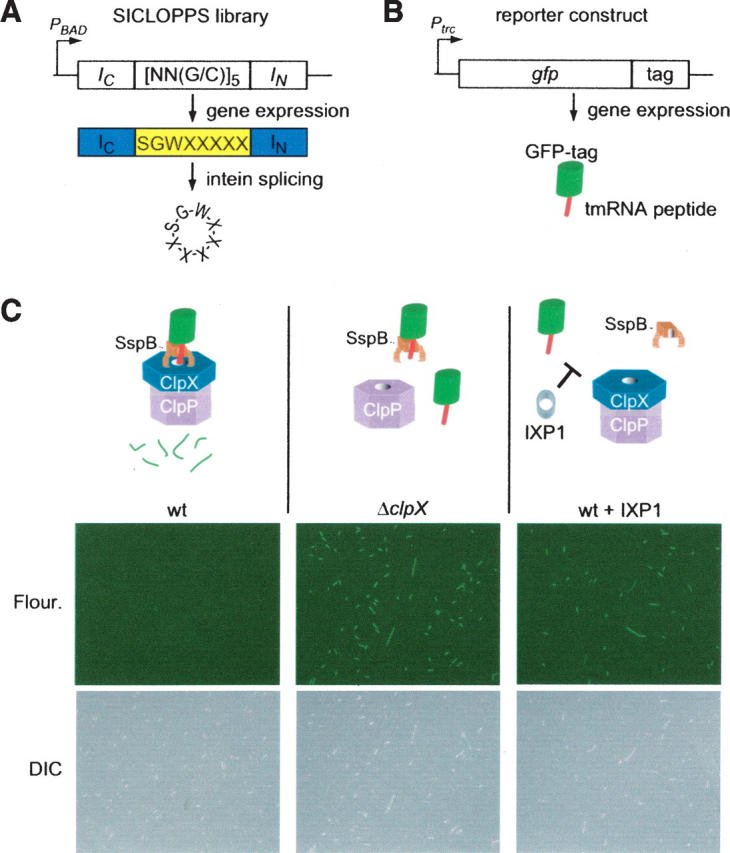
Selection for cyclic peptide inhibitors of ClpXP. (A) Schematic diagram of SICLOPPS library construction. Three fixed and five randomized codons were cloned in the coding region between the intein IC and IN domains. When this gene is expressed, the IC and IN domains promote circular ligation of the intervening peptide, resulting in 8-mer cyclic peptides with five random amino acids. (B) Schematic diagram of the GFP-tag reporter. The tmRNA peptide tag sequence was encoded at the 3′-end of the gfp gene under control of an IPTG-inducible promoter. All GFP produced from this gene will have the tmRNA peptide tag at the C terminus. (C) Result of expression of the GFP-tag reporter and inhibitory cyclic peptides in E. coli. Production of GFP-tag was induced in wild-type E. coli (wt), a strain lacking the clpX gene (ΔclpX), and a strain that was also producing the IXP1 cyclic peptide; the cells were imaged by immunofluorescence (fluor.) to see fluorescent cells and differential interference contrast microscopy (DIC) to see all cells. In wild type, SspB binds to the tmRNA peptide at the C terminus of GFP-tag and tethers the protein to the ClpXP protease, resulting in rapid degradation and no fluorescent cells. In the ΔclpX strain, the absence of active ClpXP protease results in stabilization of GFP-tag and highly fluorescent cells. In wild-type cells producing IXP1, the cyclic peptide inhibits degradation of GFP-tag, resulting in fluorescent cells. DIC images show that the ΔclpX cells and wild-type cells producing IXP1 are also slightly filamentous.
The tmRNA protein tagging and degradation pathway was chosen as a target for inhibition because this pathway is found in all bacteria and is essential for virulence in several pathogenic species, including species of Neisseria, Salmonella, and Yersinia (Huang et al. 2000; Julio et al. 2000; Okan et al. 2006). tmRNA is a specialized RNA that can enter a ribosome and add a peptide tag to the C terminus of the nascent protein (Keiler et al. 1996). The tmRNA-encoded peptide tag contains epitopes for several intracellular proteases, and most tagged proteins are rapidly degraded. In Escherichia coli, most cytoplasmic proteins tagged by tmRNA are recognized by a proteolytic specificity factor, SspB, that facilitates degradation by the ClpXP protease (Levchenko et al. 2000; Moore and Sauer 2005). ClpXP is a multi-subunit protease that degrades a variety of substrates in addition to tmRNA-tagged proteins (Flynn et al. 2003). Although there are no known inhibitors that are specific for ClpXP, activators of ClpP activity have antibacterial activity against Gram-positive bacteria (Brotz-Oesterhelt et al. 2005). To determine if inhibitors of ClpXP would have antibacterial activity against species that require this protease, a bacterial strain in which ClpXP is not essential was used to screen for cyclic peptides that block degradation of tmRNA-tagged proteins. Synthetic versions of these inhibitors were then tested for bactericidal activity against Caulobacter crescentus, a Gram-negative bacterium in which clpX and clpP are essential.
Results
Screen for cyclic peptide inhibitors of ClpXP
To identify inhibitors of proteolysis of tmRNA-tagged proteins, a reporter was engineered by encoding the tmRNA peptide tag at the 3′-end of the egfp gene, such that expression of this gene produces a variant of GFP containing the tmRNA peptide tag (GFP-tag) (Fig. 1B). When GFP-tag was produced in wild-type E. coli, the cells showed little fluorescence (Fig. 1C), presumably because the protein was recognized as a tmRNA-tagged protein and rapidly degraded by ClpXP. Degradation required both the tmRNA peptide tag and ClpX. When a variant of GFP with no tag or with a tag lacking the ClpXP recognition sequence at the C terminus (GFP-tagDD) was produced, the E. coli were highly fluorescent (data not shown). Likewise, in an E. coli strain deleted for clpX, production of GFP-tag resulted in cells that were highly fluorescent (Fig. 1C). These results suggested that cells producing GFP-tag would be fluorescent if an inhibitor of ClpXP was present.
A library of SICLOPPS plasmids was constructed that encodes the sequence SGW followed by five NN(G/C) codons. This SGWX5 library theoretically produces 3.2 × 106 different cyclic peptides. The SGW sequence allows efficient circular ligation, and the redundant codons can encode any of the 20 amino acids (Abel-Santos et al. 2003). The use of NN(G/C) instead of fully redundant codons reduces the probability of a stop codon and results in a more even distribution of encoded amino acids.
To isolate cyclic peptides that inhibit proteolysis of tmRNA-tagged proteins, the SGWX5 SICLOPPS library was expressed in E. coli containing GFP-tag, and fluorescent cells were selected from a population of ∼106 using FACS. Most cells producing a cyclic peptide had little fluorescence, indicating that most cyclic peptides do not inhibit ClpXP. Approximately 0.014% of the population had fluorescence over the background level, and 96 of these cells were isolated for clonal growth and characterization. To eliminate any clones that resulted from sorting errors or spurious accumulation of GFP, cells from each colony were cultured and examined by epifluorescence microscopy. All selected clones produced some fluorescent cells (cells with fluorescence intensity at least 0.5-fold the level observed in ΔclpX cells producing GFP-tag), and two clones, containing the peptides IXP1 and IXP2, produced cells with fluorescence indistinguishable from the ΔclpX strain (Fig. 1C; Table 1).
Table 1.
Cyclic peptides identified from in vivo screen

To determine if other libraries of cyclic peptides also contained inhibitors of GFP-tag degradation, a SICLOPPS library of 9-mer peptides with the sequence SGX5PL was engineered and screened in the same manner as the SGWX5 library. Three clones (IXP3, IXP4, and IXP5) producing GFP fluorescence of similar intensity to the ΔclpX strain were isolated (Table 1).
Cultures producing IXP1, IXP3, or IXP4 contained >70% fluorescent cells, indicating efficient inhibition of GFP-tag degradation (Table 1). In addition, the ΔclpX strain has a partially penetrant filamentous phenotype, and cells producing IXP1, IXP3, or IXP4 had a similar morphology (Fig. 1C), suggesting that the presence of these peptides mimics a genetic deletion of clpX. Fewer than 40% of the cells producing IXP2 or IXP5 were fluorescent, suggesting low intracellular concentrations of the cyclic peptide or inefficient inhibition of GFP-tag degradation in these clones. Although there is some sequence similarity between pairs of inhibitory cyclic peptides, there is no sequence conservation in the randomized region found in all of the peptides, indicating that they may inhibit the degradation of GFP-tag through different interactions.
Inhibition of ClpXP in vitro
To ensure that the selected cyclic peptides are inhibitors of ClpXP and do not cause accumulation of GFP-tag in vivo by some other mechanism, cyclic peptides were synthesized and purified to examine their effects on ClpXP activity in vitro. Proteolysis of GFP-tag in the presence of ClpXP and SspB was monitored by loss of GFP fluorescence in a continuous fluorometric assay. In the absence of cyclic peptide, GFP-tag was degraded with kinetic parameters k cat = 1.79 ± 0.08 min−1, K M = 0.74 ± 0.04 μM, similar to previously published values (Levchenko et al. 2000). No degradation was observed for GFP without a tmRNA tag or for GFP-tagDD when incubated with ClpXP and SspB (not shown). Likewise, no degradation was observed when ClpX or ClpP was omitted from the reaction (data not shown). These results confirm that proteolysis of GFP-tag in vitro requires ClpXP recognition of the tmRNA peptide tag.
Inclusion of purified IXP1 reduced the rate of GFP-tag proteolysis, demonstrating that this cyclic peptide is a bona fide inhibitor of ClpXP (Fig. 2). Increasing the concentration of IXP1 decreased both the apparent K M and the apparent k cat of the reaction, suggesting uncompetitive inhibition. Fitting the data to an uncompetitive model gave a K I value of 136 ± 35 μM (Fig. 2).
Figure 2.

Cyclic IXP1 inhibits ClpXP in vitro. GFP-tag was incubated with ClpXP, and proteolysis was monitored using a continuous fluorometric assay. Representative assays without inhibitor and with IXP1 are shown. The assays were repeated using different concentrations of substrate to determine the apparent kinetic parameters. Eadie-Hofstee plots (inset) for proteolysis with no inhibitor (solid line), 50 μM IXP1 (long dashes), and 100 μM IXP1 (short dashes), which are consistent with an uncompetitive inhibition model.
To exclude the possibility that IXP1 is a substrate for ClpXP, IXP1 was incubated with ClpXP in the absence of GFP-tag, and the amount of cyclic peptide was quantified by reverse-phase HPLC. The amount of cyclic peptide did not change over at least 1 h, and no linear peptide or smaller peptide products could be detected (Fig. 3). These results indicate that IXP1 is not degraded by ClpXP.
Figure 3.

Interaction of IXP1 with ClpX and ClpP in vitro. (A) IXP1 was incubated with ClpXP for 60 min, and samples before (0 min) and after (60 min) incubation were analyzed by reverse-phase HPLC. Plots of the absorbance at 280 nm versus time after injection are shown with arrows indicating the retention time for cyclic IXP1 and linear IXP1 as determined from control assays without ClpXP. The area under the cyclic peptide peaks was unchanged after 60 min. (B) The effects of IXP1 on the ATPase activity of ClpX with and without GFP-tag, and on the peptidase activity of ClpP, were measured. Each assay was normalized to the activity in the absence of IXP1. Error bars indicate the standard deviation at each IXP1 concentration.
The proteolytic mechanism of ClpXP involves both ATP-dependent unfolding of the substrate by ClpX and hydrolysis of peptide bonds by ClpP (Gottesman et al. 1997), thus the effects of IXP1 on these individual reactions were investigated. The rate of ClpX ATP hydrolysis was not inhibited by IXP1 at concentrations up to 200 μM (Fig. 3). When GFP-tag was included in the ATPase assay, the rate of hydrolysis increased by 1.6-fold, similar to previous reports (Burton et al. 2003), but IXP1 still had no effect on ClpX activity (Fig. 3). The peptidase activity of ClpP was assayed using the fluorogenic substrate Suc-Leu-Tyr-AMC (Maurizi et al. 1994), and addition of IXP1 decreased ClpP activity by <2% (Fig. 3). These results indicate that the individual activities of ClpXP are not affected by IXP1 and are consistent with an uncompetitive mechanism of inhibition.
Uncompetitive inhibition is characteristic of molecules that bind the enzyme–substrate complex, but not the free enzyme. In the in vitro and in vivo proteolysis reactions above, the proteolytic adaptor SspB binds GFP-tag and tethers it to ClpXP (Levchenko et al. 2000). In principle, IXP1 could act on the GFP-tag•ClpXP interaction or the SspB•ClpXP interaction. Because ClpXP can degrade GFP-tag in the absence of SspB, albeit at a slower rate (Levchenko et al. 2000), the proteolysis assays were repeated without the addition of SspB. The degradation of GFP-tag by ClpXP was still inhibited by IXP1 in the absence of SspB (data not shown), suggesting that IXP1 binds the substrate–ClpXP complex.
Because ClpXP recognizes substrates by at least five different motifs, it has been proposed that there are several substrate-binding sites on the protease (Flynn et al. 2003). To determine if IXP1 inhibits proteolysis of ClpXP substrates recognized by an epitope distinct from the tmRNA tag, λ O protein was used as an assay substrate. Sequences at the N terminus of λ O are recognized by ClpXP, and there is no interaction between λ O and SspB (Gonciarz-Swiatek et al. 1999; Levchenko et al. 2000). Degradation of λ O was assayed in the presence and absence of IXP1 by following the loss of intact λ O protein on SDS-polyacrylamide gels (Fig. 4). With no inhibitor, λ O was degraded with a half-life of 35 ± 2 min. Addition of 100 μM IXP1 increased the half-life to 73 ± 8 min, close to the value expected if the K I with λ O was the same as for GFP-tag. Therefore, IXP1 is a general inhibitor of ClpXP and affects degradation of substrates in addition to those tagged by tmRNA.
Figure 4.

Cyclic IXP1 inhibits degradation of λ O by ClpXP. λ O protein was incubated with ClpXP in the presence or absence of IXP1, and the loss of intact substrate was monitored by SDS-PAGE. Representative SDS-polyacrylamide gels stained with Coomassie blue showing the amount of λ O protein at various times after addition of ClpXP are shown. The amount of λ O protein remaining was plotted versus time and fit with a single exponential function to determine the substrate half-life. The average half-life for degradation of λ O was 35 ± 2 min in the absence of IXP1, and 73 ± 8 min in the presence of 100 μM IXP1.
Purified IXP3 and IXP4 also inhibited ClpXP in vitro but appeared to be competitive inhibitors of GFP-tag degradation (data not shown). IXP2 and IXP5 did not inhibit the reaction at the concentrations tested, consistent with the observation that fewer cells producing these peptides have high GFP-tag levels in vivo. The linear versions of IXP1, IXP2, IXP3, and IXP4 showed little inhibition of ClpXP in vitro at concentrations up to 1 mM, thus the cyclic architecture of the peptides is important for inhibition (Table 2). An arbitrary cyclic peptide from the SGWX5 library, Con62, also showed no detectable inhibition of ClpXP in vivo or in vitro at concentrations up to 1 mM (Tables 1 and 2). Therefore, both the sequence of the cyclic peptide and the cyclic architecture are important for inhibitory activity.
Table 2.
In vivo and in vitro properties of cyclic peptides

A linear peptide, XB, containing the 11 C-terminal residues of E. coli SspB, has been shown to bind to ClpX and inhibit the degradation of GFP-tag in the presence of SspB (Wah et al. 2003). IXP5 has some sequence similarity to the C terminus of SspB, suggesting that cyclic versions of the XB peptide might also be potent inhibitors. To test this hypothesis, linear and cyclic versions of XB were synthesized and assayed in vitro (Table 2). As previously reported (Wah et al. 2003), the linear XB peptide was a competitive inhibitor of ClpXP degradation of GFP-tag in the presence of SspB, and as expected, the linear XB peptide had no effect on GFP-tag degradation in the absence of SspB and did not inhibit the degradation of λ O by ClpXP (data not shown). Cyclic XB inhibited ClpXP proteolysis of GFP-tag in the presence of SspB with a K I = 8 ± 1 μM, sevenfold lower than the linear XB peptide. Like linear XB, cyclic XB did not inhibit the degradation of λ O (data not shown). Therefore, the circular ligation of the XB peptide increases the efficiency of inhibition, perhaps by decreasing the entropy of the free peptide, thereby increasing the energy of binding to ClpX.
Bactericidal activity of ClpXP inhibitors
ClpXP is essential in C. crescentus (Jenal and Fuchs 1998), thus the effect of adding purified inhibitory peptides to growing cultures was examined (Table 2). IXP1 killed C. crescentus with a minimum bactericidal concentration (MBC) of 279 ± 23 μM and a minimum inhibitory concentration (MIC) of 219 ± 42 μM, suggesting that IXP1 can both enter C. crescentus cells and inhibit C. crescentus ClpXP. The linear XB peptide had an MBC of 146 ± 11 μM and an MIC of 139 ± 44 μM, and the cyclic XB peptide was more effective, with an MBC of 40 ± 6 μM and an MIC of 29 ± 2 μM. It is important to note that although C. crescentus has an SspB protein that performs the same functions as E. coli SspB, the sequence that interacts with ClpX is highly diverged (Lessner et al. 2007). Nonetheless, all residues of E. coli ClpX that make hydrophobic or hydrogen-bonding contacts with the XB peptide (Park et al. 2007) are conserved in C. crescentus ClpX, thus the XB peptide might bind C. crescentus ClpX in the same manner as for E. coli ClpX. Cyclic peptides are not generally toxic to C. crescentus, because Con62 had no effect on bacterial growth. Thus, despite anticipated problems with transporting a peptide across the membrane of Gram-negative bacteria, peptides isolated from the screen had bactericidal activity.
Discussion
Using a high-throughput screen and SICLOPPS technology, efficient inhibitors of the degradation of tmRNA-tagged proteins were isolated. Kinetic assays and several lines of evidence suggest that IXP1 is an uncompetitive inhibitor of ClpXP. IXP1 is not a substrate for ClpXP but inhibits proteolysis of at least two ClpXP substrates that are recognized by different epitopes, and inhibition is independent of SspB, suggesting that IXP1 does not compete for binding to ClpX. IXP1 does not inhibit the ATPase activity of ClpX or the peptidase activity of ClpP, consistent with a mechanism in which IXP1 binds to the ClpXP–substrate complex. One step in the proteolytic mechanism, where IXP1 could act uncompetitively, is the translocation of the substrate through the central pore of ClpXP. Further structural and biochemical experiments will be required to understand exactly how IXP1 inhibits ClpXP, but it clearly does not use the same mechanism as the rationally designed peptide XB. Therefore, the screen described here can identify multiple inhibitors with diverse sequences and unexpected mechanisms of action. In principle, this screening technique could be used to identify cyclic peptide inhibitors for any pathway with a fluorescent reporter that can be expressed in E. coli. The method provides a set of lead compounds for reagent design or antibiotic development that includes diverse activities, and does not require any knowledge of molecular structures or cofactor requirements of the targeted pathway.
The cyclic architecture of the selected peptides was important for the inhibitory and bactericidal activities. Because libraries of linear peptides have not been screened, it is possible that there are linear peptides that would inhibit ClpXP, but each of the selected cyclic peptide sequences was less effective in a linear form. Even the XB peptide, which inhibits ClpXP by binding to the same site as the C-terminal tail of SspB, is more active as a cyclic peptide. The higher activity of cyclic peptides compared to linear versions could be the result of specific structural features, or of tighter binding of cyclic peptides due to decreased loss of entropy. In either case, cyclic peptides are likely to be more stable in vivo than linear peptides and are therefore more attractive for pharmacological and antibacterial agents.
Although the selected cyclic peptides are bactericidal, optimization of the sequence and length of the cyclic peptides might improve their bioactivity. In principle, further improvements could be made through modification or derivatization of the peptide, or the use of nonstandard amino acids. Finally, because the selected peptides appear to inhibit ClpXP through different interactions, using them in combination could have synergistic effects on the efficiency of inhibition. Even without improvements in efficiency, biologically active inhibitors such as IXP1 provide the ability to study the role of specific pathways in vivo without the drawbacks associated with the genetic deletion or depletion of essential activities.
Materials and Methods
Plasmids and bacterial strains
A GFP-based reporter for the proteolysis of tmRNA-tagged proteins was constructed by amplifying the egfp gene from pEGFP-N2 (BD Biosciences Clontech) using PCR with primers that add the codons for the tmRNA tag (AANDENYALAA) at the 3′-end of the gene before the stop codon, and cloning the product into pTrc99a. A similar strategy was used for the control reporters containing egfp with no tag and egfp with the DD tag (AANDENYALDD). For fluorescence assays, plasmids bearing a GFP-based reporter were mobilized into E. coli strain BW7786 (Khlebnikov et al. 2001). The ΔclpX strain was constructed from BW7786 using the Wanner method (Datsenko and Wanner 2000).
For overproduction of GFP-tag, GFP, and GFP-tagDD, the genes were excised from pTrc99a, ligated into pQE8 (QIAGEN) to produce an N-terminal His6-fusion under control of the T7 promoter, and mobilized into E. coli BL21(DE3) (Novagen). E. coli clpP was cloned into pQE70 (QIAGEN), resulting in a C-terminally His6-tagged protein. E. coli clpX, sspB, and the gene encoding λ O were cloned into pET28a (Novagen) to produce N-terminally His6-tagged proteins. All constructs were transformed into E. coli BL21(DE3). Unless otherwise noted, E. coli strains were grown at 37°C in LB broth, with the addition of 100 μg/mL ampicillin, 30 μg/mL chloramphenicol, or 30 μg/mL kanamycin where appropriate. C. crescentus strain CB15N (Evinger and Agabian 1977) was grown in PYE medium (Ely 1991).
SICLOPPS libraries were constructed as previously described (Abel-Santos et al. 2003). For the SGWX5 library, the initial PCR reaction combined degenerate oligonucleotide SGW+5 (5′-ggaattcgccaatggggcgatcgcccacaattccggctggnnsnnsnnsnnsnnstgcttaagttttggc-3′) and CBDRev (ggaattcaagctttcattgaagctgccacaagg). For the second PCR reaction, CBDRev was combined with a forward primer named zipper (ggaattcgccaatggggcgatcgcc). Production of cyclic peptides from the SGX5PL library in E. coli was confirmed by butanol extraction and reversed-phase chromatography followed by mass spectrometric analysis of the purified cyclic peptides as described (Scott et al. 2001).
Proteins and peptides
Histidine-tagged versions of ClpP, ClpX, SspB, GFP, GFP-tag, GFP-tagDD, and λ O protein were purified from overproducing strains by metal-chelate chromatography followed by ion exchange chromatography, gel filtration, or both. In all cases, cells were grown at 30°C in LB broth with the appropriate antibiotics to OD600 = 0.6, 1 mM IPTG was added to induce protein production for 3.5 h, and cells were harvested by centrifugation. Cell pellets were resuspended in Wash buffer (50 mM NaH2PO4 at pH 8.0, 300 mM NaCl, 20 mM imidazole), lysed by sonication, and cleared by centrifugation at 26,000g for 15 min. The cleared lysate was added to 0.1% (v/v) Ni-NTA resin (QIAGEN) for 1 h, loaded into a column, and washed with 100 bed volumes of wash buffer. Bound protein was eluted with 5 bed volumes of wash buffer containing 500 mM imidazole, and fractions containing purified protein were identified by SDS-PAGE.
Fractions containing purified ClpX were combined and applied to a Superose 6 (GE Healthcare) gel filtration column equilibrated in buffer containing 50 mM Tris-HCl (pH 7.5), 300 mM NaCl, 2 mM DTT, and 10% glycerol; and fractions containing purified ClpX protein were identified by SDS-PAGE.
For purification of ClpP and λ O protein, fractions from metal-chelate chromatography were combined, dialyzed against buffer A1 (50 mM Tris-HCl at pH 8, 10 mM MgCl2, 5 mM DTT, 10 mM KCl), and applied to a MonoQ HR5/5 column (GE Healthcare). The column was washed in buffer A1, and bound protein was eluted with a linear gradient from 10 to 1000 mM KCl. Fractions containing purified protein were combined and dialyzed against buffer A2 (50 mM Tris-HCl at pH 7.5, 100 mM NaCl, 2 mM DTT, and 10% glycerol).
GFP, GFP-tag, and GFP-tagDD were purified as described for ClpP, except that fractions from the MonoQ column containing purified protein were applied to a Superdex 75 (GE Healthcare) gel filtration column equilibrated in buffer A2, and fractions containing the GFP variant were combined.
SspB was purified as described for ClpP, except that buffer A3 (50 mM MES at pH 6, 10 mM MgCl2, 1 mM DTT, 100 mM KCl) was used in place of buffer A1. For all proteins, concentrations were determined by UV absorbance at 280 nm.
Linear peptides were synthesized by the PSU Huck Institutes of the Life Sciences Macromolecular Core Facility (Hershey, PA). Linear peptides were cyclized by incubating peptide with excess 1-ethyl-3-(3′-dimethylaminopropyl)carbodiimeide (EDC) and 1-hydroxy-7-azabenzotriazole (HOAt) in 50 mL DMF. After 24 h of incubation, an aliquot of each reaction was analyzed by RP-HPLC to confirm cyclization. Successful reactions were assumed based on the increased retention time of peptide relative to the retention time of linear starting products. Reactions were evaporated and peptides partially purified by precipitation with diethyl ether. Final purification of cyclic peptides was accomplished by RP-HPLC. Mass was confirmed by use of electrospray-ionization mass spectrometry.
Screen for inhibitors of the tmRNA pathway
E. coli BW7786 cells containing the GFP-tag reporter and the SICLOPPS library were grown in LB broth with 30 μg/mL chloramphenicol, 100 μg/mL ampicillin, and 0.0002% arabinose at 37°C to OD600 = 0.3. IPTG was added to a final concentration of 1 mM, and the culture was grown for 3 h. Cells were sorted by fluorescence activated cell sorting (FACS) using a Beckman Coulter Elite cell sorter with Autoclone to isolate cells with GFP fluorescence, and selected cells were deposited on agar plates for clonal growth. Cells from each colony were grown in liquid culture as described above and examined by epifluorescence microscopy. The fluorescence intensity and the number of cells with fluorescence above background were scored using ImagePro software (MediaCybernetics). SICLOPPS plasmid DNA was prepared from selected clones, and the region encoding the cyclic peptide was sequenced. Peptide sequences were obtained from conceptual translation of the DNA sequences.
In vitro proteolysis assays
Proteolysis of GFP-tag was performed using a continuous fluorescence assay essentially as previously described (Levchenko et al. 2000). Briefly, loss of GFP fluorescence was monitored at 507 nm after excitation at 395 nm at 30°C using a Hitachi F-2000 Fluorescence Spectrophotometer in buffer R (25 mM HEPES-KOH at pH 7.6, 5 mM MgCl2, 50 mM KCl, 0.032% NP-40, 10% glycerol) and an ATP regeneration system (containing 4 mM ATP at pH 7, 16 mM creatine phosphate, and 0.32 mg/mL creatine kinase). Typically, reactions contained 0.1 μM ClpX6, 0.3 μM ClpP14, and equimolar concentrations (0.2–2.0 μM) of GFP-tag and SspB. Degradation of GFP-tag protein was confirmed by SDS-PAGE assays. To examine inhibition of GFP-tag proteolysis, peptides were incubated with ClpXP in reaction buffer for 5 min prior to the addition of GFP-tag. Plots of fluorescence versus time were fit with a single exponential function to determine the initial rate of proteolysis. Kinetic parameters were estimated using Eadie-Hofstee plots. Curve fitting for competitive inhibitors was performed using the Scientist program (MicroMath Scientific Software, Inc.).
Peptidase activity against IXP1 was assayed by incubating 200 μM IXP1 with 0.1 μM ClpX6, 0.3 μM ClpP14, and the ATP regeneration system in buffer R at 37°C. Samples were taken after 0, 5, and 60 min, and separated by reverse-phase HPLC using a Varian Microsorb-MV C18 column developed in a gradient from 0.1% trifluoroacetic acid in water to 0.1% trifluoroacetic acid in acetonitrile. UV absorbance at 280 nm was monitored, and the peak corresponding to the IXP1 cyclic peptide was determined by comparison to reactions containing only cyclic IXP1 or linear IXP1. The loss of cyclic IXP1 was determined by integrating the area under the cyclic IXP1 peak.
Proteolysis of λ O protein was assayed by incubating 1 μM λ O with 0.1 μM ClpX6, 0.3 μM ClpP14 in buffer R at 30°C. At various times the reaction was sampled, and the reaction was terminated by boiling in SDS-PAGE loading buffer and analyzed using SDS-polyacrylamide gels stained with Coomassie blue. The intensity of the band corresponding to intact λ O protein was measured using ImageQuant software (Molecular Dynamics), and plots of the intensity versus time were fit with single exponential functions to determine the half-life of λ O protein.
ATPase and peptidase assays
ClpX ATPase activity was measured by monitoring the increase in phosphate using a ternary hetero polyacid assay (Chen et al. 2003). One micromolar ClpX was incubated with or without 6 μM GFP-tag and varying concentrations of IXP1 in buffer P (4 mM ATP, 50 mM Tris-HCl at pH 7.5, 100 mM NaCl, 100 mM KCl, 10 mM MgCl2, 2 mM DTT, and 10% glycerol) at 37°C. At each time point, 10 μL were removed from the reaction and added to 265 μL of 0.88 M nitric acid for 2 min, 225 μL of color developing solution (44.4 mM bismuth nitrate, 31.1 mM ammonium molybdate, 0.11% ascorbic acid) were added, and the absorbance at 700 nm was determined. The rate of ATP hydrolysis was determined from plots of phosphate accumulation versus time.
ClpP peptidase activity was measured using the fluorogenic peptide Suc-Leu-Tyr-AMC: 0.1 μM ClpP was incubated with 0.5–1.0 mM Suc-Leu-Tyr-AMC and varying concentrations of IXP1 in buffer P at 37°C, and the fluorescence of AMC (excitation at 353 nm, emission at 442 nm) was monitored.
Antibacterial activity assays
To measure the effects of peptides on bacterial growth, cultures of C. crescentus were diluted to OD660 = 0.001, peptide was added at various concentrations, the cultures were incubated for 14 h at 30°C, and the MIC was determined as the lowest concentration of peptide that prevented growth. To determine the MBC, each culture was diluted 1:100 in PYE, 10 μL of the diluted culture were spread onto PYE agar plates and incubated overnight at 30°C, and the number of colonies on each plate was counted. The MBC was assigned as the concentration of peptide that reduced the number of colonies by 99.9% compared to cultures with no inhibitor.
Acknowledgments
This work was supported by NIH grant GM68720 to K.C.K. and DTRA JSTO CBD grant W911NF-06-1-0144 to K.C.K. and S.J.B.
Footnotes
Reprint requests to: Kenneth C. Keiler, Department of Biochemistry and Molecular Biology, Penn State University, 401 Althouse Laboratory, University Park, PA 16802, USA; e-mail: kkeiler@psu.edu; fax; (814) 863-7024.
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.072933007.
References
- Abel-Santos E., Scott, C.P., and Benkovic, S.J. 2003. Use of inteins for the in vivo production of stable cyclic peptide libraries in E. coli . Methods Mol. Biol. 205: 281–294. [DOI] [PubMed] [Google Scholar]
- Brotz-Oesterhelt H., Beyer, D., Kroll, H.P., Endermann, R., Ladel, C., Schroeder, W., Hinzen, B., Raddatz, S., Paulsen, H., Henninger, K., et al. 2005. Dysregulation of bacterial proteolytic machinery by a new class of antibiotics. Nat. Med. 11: 1082–1087. [DOI] [PubMed] [Google Scholar]
- Burton R.E., Baker, T.A., and Sauer, R.T. 2003. Energy-dependent degradation: Linkage between ClpX-catalyzed nucleotide hydrolysis and protein-substrate processing. Protein Sci. 12: 893–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B., Guo, Q., Guo, Z., and Wang, X. 2003. An improved activity assay method for arginine kinase based on a ternary heteropolyacid system. Tsinghua Sci. Technol. 8: 422–427. [Google Scholar]
- Datsenko K.A. and Wanner, B.L. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. 97: 6640–6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ely B. 1991. Genetics of Caulobacter crescentus . Methods Enzymol. 204: 372–384. [DOI] [PubMed] [Google Scholar]
- Evinger M. and Agabian, N. 1977. Envelope-associated nucleoid from Caulobacter crescentus stalked and swarmer cells. J. Bacteriol. 132: 294–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flynn J.M., Neher, S.B., Kim, Y.I., Sauer, R.T., and Baker, T.A. 2003. Proteomic discovery of cellular substrates of the ClpXP protease reveals five classes of ClpX-recognition signals. Mol. Cell 11: 671–683. [DOI] [PubMed] [Google Scholar]
- Gonciarz-Swiatek M., Wawrzynow, A., Um, S.J., Learn, B.A., McMacken, R., Kelley, W.L., Georgopoulos, C., Sliekers, O., and Zylicz, M. 1999. Recognition, targeting, and hydrolysis of the λ O replication protein by the ClpP/ClpX protease. J. Biol. Chem. 274: 13999–14005. [DOI] [PubMed] [Google Scholar]
- Gottesman S., Maurizi, M.R., and Wickner, S. 1997. Regulatory subunits of energy-dependent proteases. Cell 91: 435–438. [DOI] [PubMed] [Google Scholar]
- Horswill A.R., Savinov, S.N., and Benkovic, S.J. 2004. A systematic method for identifying small-molecule modulators of protein–protein interactions. Proc. Natl. Acad. Sci. 101: 15591–15596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C., Wolfgang, M.C., Withey, J., Koomey, M., and Friedman, D.I. 2000. Charged tmRNA but not tmRNA-mediated proteolysis is essential for Neisseria gonorrhoeae viability. EMBO J. 19: 1098–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenal U. and Fuchs, T. 1998. An essential protease involved in bacterial cell-cycle control. EMBO J. 17: 5658–5669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Julio S.M., Heithoff, D.M., and Mahan, M.J. 2000. ssrA (tmRNA) plays a role in Salmonella enterica serovar Typhimurium pathogenesis. J. Bacteriol. 182: 1558–1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keiler K.C., Waller, P.R., and Sauer, R.T. 1996. Role of a peptide tagging system in degradation of proteins synthesized from damaged messenger RNA. Science 271: 990–993. [DOI] [PubMed] [Google Scholar]
- Khlebnikov A., Datsenko, K.A., Skaug, T., Wanner, B.L., and Keasling, J.D. 2001. Homogeneous expression of the P(BAD) promoter in Escherichia coli by constitutive expression of the low-affinity high-capacity AraE transporter. Microbiology 147: 3241–3247. [DOI] [PubMed] [Google Scholar]
- Lessner F.H., Venters, B.J., and Keiler, K.C. 2007. Proteolytic adaptor for transfer-messenger RNA-tagged proteins from α-proteobacteria. J. Bacteriol. 189: 272–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levchenko I., Seidel, M., Sauer, R.T., and Baker, T.A. 2000. A specificity-enhancing factor for the ClpXP degradation machine. Science 289: 2354–2356. [DOI] [PubMed] [Google Scholar]
- Maurizi M.R., Thompson, M.W., Singh, S.K., and Kim, S.H. 1994. Endopeptidase Clp: ATP-dependent Clp protease from Escherichia coli . Methods Enzymol. 244: 314–331. [DOI] [PubMed] [Google Scholar]
- Moore S.D. and Sauer, R.T. 2005. Ribosome rescue: tmRNA tagging activity and capacity in Escherichia coli . Mol. Microbiol. 58: 456–466. [DOI] [PubMed] [Google Scholar]
- Okan N.A., Bliska, J.B., and Karzai, A.W. 2006. A role for the SmpB-SsrA system in Yersinia pseudotuberculosis pathogenesis. PLoS Pathog. 2: e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park E.Y., Lee, B.G., Hong, S.B., Kim, H.W., Jeon, H., and Song, H.K. 2007. Structural basis of SspB-tail recognition by the zinc binding domain of ClpX. J. Mol. Biol. 367: 514–526. [DOI] [PubMed] [Google Scholar]
- Scott C.P., Abel-Santos, E., Wall, M., Wahnon, D.C., and Benkovic, S.J. 1999. Production of cyclic peptides and proteins in vivo. Proc. Natl. Acad. Sci. 96: 13638–13643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott C.P., Abel-Santos, E., Jones, A.D., and Benkovic, S.J. 2001. Structural requirements for the biosynthesis of backbone cyclic peptide libraries. Chem. Biol. 8: 801–815. [DOI] [PubMed] [Google Scholar]
- Wah D.A., Levchenko, I., Rieckhof, G.E., Bolon, D.N., Baker, T.A., and Sauer, R.T. 2003. Flexible linkers leash the substrate binding domain of SspB to a peptide module that stabilizes delivery complexes with the AAA+ ClpXP protease. Mol. Cell 12: 355–363. [DOI] [PubMed] [Google Scholar]