

Abstract
The zinc finger HIT domain is a sequence motif found in many proteins, including thyroid hormone receptor interacting protein 3 (TRIP-3), which is possibly involved in maturity-onset diabetes of the young (MODY). Novel zinc finger motifs are suggested to play important roles in gene regulation and chromatin remodeling. Here, we determined the high-resolution solution structure of the zinc finger HIT domain in ZNHIT2 (protein FON) from Homo sapiens, by an NMR method based on 567 upper distance limits derived from NOE intensities measured in three-dimensional NOESY spectra. The structure yielded a backbone RMSD to the mean coordinates of 0.19 Å for the structured residues 12–48. The fold consists of two consecutive antiparallel β-sheets and two short C-terminal helices packed against the second β-sheet, and binds two zinc ions. Both zinc ions are coordinated tetrahedrally via a CCCC-CCHC motif to the ligand residues of the zf-HIT domain in an interleaved manner. The tertiary structure of the zinc finger HIT domain closely resembles the folds of the B-box, RING finger, and PHD domains with a cross-brace zinc coordination mode, but is distinct from them. The unique three-dimensional structure of the zinc finger HIT domain revealed a novel zinc-binding fold, as a new member of the treble clef domain family. On the basis of the structural data, we discuss the possible functional roles of the zinc finger HIT domain.
Keywords: NMR; structural genomics; solution structure; zinc finger HIT (zf-HIT); ZNHIT2 (protein FON); thyroid hormone receptor interacting protein 3 (TRIP-3, ZNHIT3)
Zinc-binding proteins play crucial roles in protein–protein and protein–nucleic acid interactions. Therefore, comprehensive structural information about zinc-binding proteins will facilitate a fundamental understanding of the zinc-binding proteins.
The zinc finger HIT (zf-HIT) is a novel zinc-binding domain with ∼50 amino acids and is one of the interesting targets for the RIKEN Structural Genomics/Proteomics Initiative (RSGI) project (Yokoyama et al. 2000). An amino acid sequence alignment revealed that these domains contain conserved cysteine (Cys) and histidine (His) residues that can potentially coordinate zinc atoms (Pfam database) (Fig. 1). Zf-HIT was named after the first protein that originally defined the domain: the yeast HIT1 protein (Kawakami et al. 1992). The function of this domain is unknown, but this domain is mainly found in nuclear proteins involved in gene regulation and chromatin remodeling. For example, the human thyroid hormone receptor interacting protein 3 (TRIP-3, ZNHIT3), composed of 155 amino acids, is characterized by the N-terminal zf-HIT domain spanning residues 1–52 (Iwahashi et al. 2002). Intriguingly, TRIP-3 is also a co-activator associated with hepatocyte nuclear factor-4α (HNF-4α), which is a transcription factor and a member of the steroid hormone receptor superfamily. Maturity-onset diabetes of the young (MODY) is a monogenic form of diabetes caused by autosomal dominant inheritance, with an early-onset and pancreatic β-cell dysfunction. Mutations of the HNF-4α gene are associated with a subtype of MODY characterized by impaired insulin secretion in response to a glucose load. Therefore, TRIP-3 is considered to be possibly involved in MODY.
Figure 1.
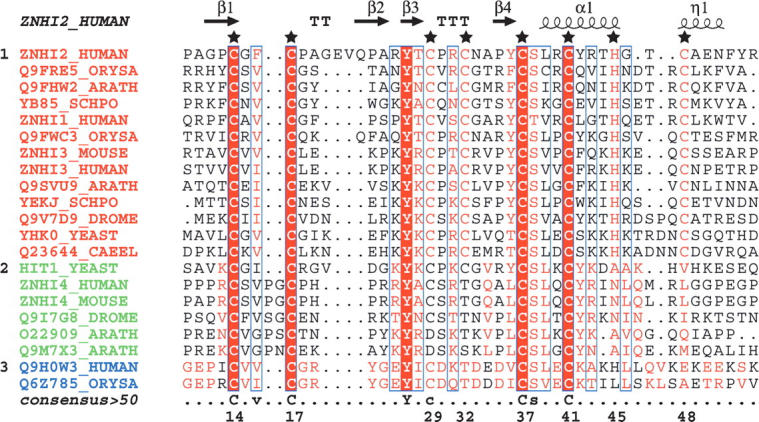
Structural-based multiple-sequence alignment of the zf-HIT domain, based on seed sequences from Pfam. (★) The seven Cys and one His residues for coordination with zinc atoms are numbered at the bottom of the sequence. Labels and elements of secondary structures are reported above the aligned sequences. The three subgroups are shown with different colors for the title of the sequence. (Red) The first subgroup; (green) second subgroup; (blue) third subgroup. The sequence alignment output was made with the ESPript program (Gouet et al. 1999).
Despite the medical importance of the zf-HIT-containing proteins described above, conventional sequence alignments did not reveal any significant homology with proteins of known structure or function. Thus, the structure and function of the zf-HIT domain remain to be elucidated. Here we report the first solution structure of a zf-HIT domain, comprising residues 1–46 from ZNHIT2 (protein FON), encoded by the C11orf5 gene on Chromosome 11q13–q22 from Homo sapiens (Lemmens et al. 2000; O'Brien et al. 2000). Our structural analysis revealed that the zf-HIT domain, with a compact, globular fold, contains two consecutive antiparallel β-sheets followed by a C-terminal short α-helix and 310-helix, which pack against the second β-sheet. The domain binds two zinc ions with a Cys4 and Cys2-His-Cys coordination mode in a cross-brace zinc finger architecture. We compared this structure with those of other zinc-binding motifs, such as the B-box, RING finger, and PHD domains from the same treble cleft domain family with an interleaved zinc binding mode, and now propose that zf-HIT may function as a protein–protein interaction domain.
Results
Resonance assignments
The 2D 1H–15N HSQC spectrum (Fig. 2) showed dispersed, sharp NH signals with similar intensities for non-proline residues, characteristic of a folded protein. The polypeptide backbone resonance assignments are complete, except for the amide proton of Leu39 (see Materials and Methods for the numbering of residues) in the nonnative expression tail regions. The side-chain assignments of the non-labile protons are complete, with the exception of Hζ of Phe52 and Hɛ of Phe16. Among the labile side-chain protons, the imide groups of all of the asparagine and glutamine residues were assigned using intraresidual NOEs (Wüthrich 1986). For all of the Xxx-Pro bonds, the trans conformation was confirmed independently by intense Xxx (Hα)-Pro (Hδ) sequential NOESY cross peaks (Wüthrich 1986), and by the 13Cβ and 13Cγ chemical shift differences (Schubert et al. 2002). The assignments were confirmed by tracing the d NN, d αN, and d βN connectivities on NOESY experiments, following the original methodology of Wüthrich (1986). Secondary structural elements were identified during this process by using the middle- and long-range NOE connectivities. The overall secondary structure of zf-HIT of ZNHIT2 comprises the following elements: β1 (Gly12–Cys14), β2 (Gln23–Arg26), β3 (Tyr27–Cys29), β4 (Ala34–Tyr36), αA (Leu39–His45), and αB (Cys48–Phe52).
Figure 2.

Two-dimensional 1H–15N HSQC spectrum of uniformly 13C/15N-labeled zf-HIT domain of ZNHIT2 (600 MHz, 298K, protein concentration 1.1 mM in 20 mM phosphate buffer at pH 6.0, 100 mM NaCl). Amino acid assignments of each peak, according to the correlation of the 1H–15N atoms of an amino acid, are shown. Horizontal lines identify side-chain NH2 groups of asparagine and glutamine residues.
Zinc coordination
To determine the Zn2+:protein stoichiometry, zf-HIT was chemically modified at the cysteine residues by p-hydroxy-mercuribenzoic acid (PHMB), and the released Zn2+ was quantified using a metallochromic indicator, 4-(2-pyridylazo) resorcinol (PAR) (Hunt et al. 1984, 1985). This modification released an amount of Zn2+ corresponding to ∼1.8-fold of the zf-HIT molecule (data not shown). Thus, we concluded that zf-HIT binds two Zn2+ ions tightly with its Cys residues. In addition, zinc coordination by the seven Cys residues was confirmed by the chemical shifts of their Cβ atoms, which were between 30.6 and 34.4 ppm, consistent with the sulfur groups coordinating Zn2+ (Lee et al. 1992). Furthermore, the 2D 1H–15N long-range HMQC spectrum (Pelton et al. 1993) of the 15N-labeled zf-HIT domain in the presence of the Zn2+ ions clearly showed two of the 15N resonances, corresponding to Nɛ2 of His45 shifted downfield as a result of zinc coordination, indicating that this His residue is in the ɛ tautomeric state, with the Nδ1 protonated.
The experiment described above showed that eight amino acids, seven Cys and one His, are used in the Zn2+ coordination, indicating that the zf-HIT domain contains two Zn2+ ions. Correspondingly, the structural calculation based on the NOE constraints (Supplemental Table S1) revealed that Cys14, Cys17, Cys37, and Cys41 are clustered and oriented to coordinate one Zn2+ ion. Similarly, Cys29, Cys32, Cys48, and His45 are in proximity for another Zn2+ ion. Therefore, we decided to determine the structure of this domain on the basis of the information about the coordination of the two Zn2+ ions.
Structure calculation
The statistics regarding the quality and precision of the final 20 energy-minimized conformers (Fig. 3A) that represent the solution structure of the zf-HIT domain of ZNHIT2 are summarized in Table 1. The structures are well defined and show excellent agreement with the experimental data. Among the 1355 cross-peaks that had been identified in the 15N- and 13C-edited 3D NOESY spectra, 99% were assigned by the program CYANA2.1 (Güntert 2004). Almost 12.6 NOE distance constraints per residue, including 171 long-range distance constraints as well as the restraints for the coordination with zinc ions (described in Materials and Methods), were used in the final structure calculations with CYANA2.1 without hydrogen-bond constraints. The final structures were further refined with AMBER9 (http://amber.scripps.edu). The precision of the structure is characterized by RMSD values to the mean coordinates of 0.19 Å for the backbone and 0.76 Å for all heavy atoms, excluding the unstructured regions at the chain termini of residues 1–11 and 49–59 (see Fig. 2A). Finally, the quality of the structure is also reflected by the fact that 73.4% of the (ϕ,ψ) backbone torsion angle pairs were found in the most favored regions, and 26.6% were within the additionally allowed regions of the Ramachandran plot, according to the program PROCHECK_NMR (Laskowski et al. 1996).
Figure 3.

Solution structure of the zf-HIT domain from ZNHIT2. (A) Stereoview of the ensemble of the 20 final energy-minimized structures of the zf-HIT domain (residues 8–53). Secondary elements are colored in cyan, red, and magenta for β-strands, α-helix, and 310-helix, respectively. (B) Ribbon presentation of the zf-HIT domain (residues 8–53). The side chains of the residues coordinating the zinc atoms and the zinc ions are shown in blue and yellow, respectively.
Table 1.
Statistics of the 20 final solution structures of the zf-HIT1 domain in ZNHIT2 from Homo sapiens

Tertiary structure
The structure of zf-HIT comprises two consecutive β-sheets structures followed by one short α-helix and a 310-helix packed against the second β-sheet, and this ββα310 structure forms the core of the zf-HIT fold (Fig. 3B). There is no other regular secondary structure. The zf-HIT domain of ZNHIT2 binds two zinc ions, using its seven Cys and one His residues, in a cross-brace manner to form a compact, globular fold, which resembles those of the B-box, RING, and PHD domains. As mentioned above, the first zinc-binding site is formed by Cys14, Cys17, Cys37, and Cys41, and the second one is formd by Cys29, Cys32, Cys48, and His45, respectively. These two zinc ions are located ∼12.5 Å apart in a cross-brace manner. Cys14 and Cys17 reside at the tip of the first β-sheet, while Cys37 and Cys41 are located at the end of the fourth β-strand and at the N-terminal end of the short α-helix, respectively. Cys29 and Cys32 are located at the tip of the second β-sheet structure, while His45 and Cys48 are on the kinked α-helix (Fig. 3B). The tertiary structure of zf-HIT contains the common structural features of the treble clef domain including B-box, RING, and PHD domains. We thus conclude that zf-HIT is a new member of the treble clef domain family of zinc-binding domains.
The zf-HIT of protein FON is not a DNA-binding domain
To examine whether the zf-HIT domain is a DNA-binding domain, we carried out an in vitro binding assay by the surface plasmon resonance method, using double-stranded cytomegalovirus promoter (CMV-TATA) oligonucleotides (Fig. 4). The SANT and SWIRM domains of MYSM1 were tested as positive and negative controls for this DNA-binding experiment (Yoneyama et al. 2007). The zf-HIT domain did not show DNA-binding activity (Fig. 4). We also tested its DNA-binding activity using two different (i.e., AT-rich and GC-rich) sequences of double-stranded oligonucleotides, but we could not detect any DNA-binding activity on the zf-HIT domain (data not shown). Although the first helix of the zf-HIT domain protrudes two arginine residues toward the outside, our results suggest that the zf-HIT domain in protein FON is not a DNA-binding domain.
Figure 4.

DNA-binding assay with the zf-HIT domain. Binding of the indicated protein to the cytomegalovirus promoter (CMV-TATA) DNA was monitored by a Biacore 3000 instrument. Each injection started at 60 and ended at 120 sec. The binding activity of the proteins and the CMV-TATA DNA was indicated as the difference in the resonance units (RU) between the DNA-bound and -unbound flow cells.
Discussion
Sequence analysis of zf-HIT domain
A search of the Pfam database (Bateman et al. 2004) revealed that the zf-HIT-containing proteins (total 121) are mainly divided into three groups: 76.0% of the zf-HIT proteins contain a single zf-HIT domain without any other annotated domains; 12.4% have an additional PAPA-1 homology sequence; and 11.6% have an additional DEAD-box helicase domain (hereafter denoted as the first, second, and third subgroups, respectively). ZNHIT2 and TRIP-3 belong to the first subgroup. A sequence alignment of the zf-HIT domains (Fig. 1) and a phylogenic analysis (from the Pfam database) showed that they are also classified into three subgroups. Intriguingly, these subgroups from the phylogenic analysis also correspond to the classification of zf-HIT-containing proteins according to the domain organization pattern described above.
A sequence alignment revealed the characteristic amino acids in these subgroups. Seven Cys and one His residues are well conserved, which facilitated the identification of the CCCC-CCHC motif in the first subgroup. However, the Cys and His residues corresponding to the fourth, seventh, and eighth positions are not conserved in the second and third subgroups (Fig. 1). Therefore, it is still unclear whether all of the Cys and His residues in the first subgroup are involved in the coordination with zinc ions. In the present study, the zf-HIT domain from ZNHIT2, belonging to the first subgroup, binds two zinc ions, and the region including the CCCC-CCHC motif forms a compact fold and adopts the structure of a cross-brace-type zinc finger domain. Whether the second and third subgroups of zf-HIT domains bind one zinc ion will be determined by further experiments. The results obtained here indicated that, despite the previous general annotation about the zf-HIT domain, the domains from the second and third subgroups are distinct from that from the first subgroup.
Besides the eight zinc-coordinating residues, several other amino acids are either highly conserved or somewhat conserved in the zf-HIT domain. Tyr27 and Ser38 are highly conserved residues in zf-HIT domains. Hydrophobic amino acids corresponding to Phe16, Tyr27, Tyr36, Leu39, and Tyr42 are well conserved (Fig. 5A). Among them, Phe16 and Tyr36 constitute the structural core of the fold; and Tyr27, Leu39, and Tyr42 are involved in the formation of the hydrophobic patch on the back surface (Fig. 5B). In addition, the types of amino acids located at the positions corresponding to Arg26 and Tyr42 are characteristic of the subgroups in zf-HIT: mainly a positively charged amino acid for Arg26 and an aromatic one for Tyr42 in the first and second subgroups, and a negatively charged amino acid and a positively charged one for these positions in the third subgroup. The highly conserved Ser38, with its small size, may be necessary to support the turn between the second β-sheet and the central α-helix.
Figure 5.
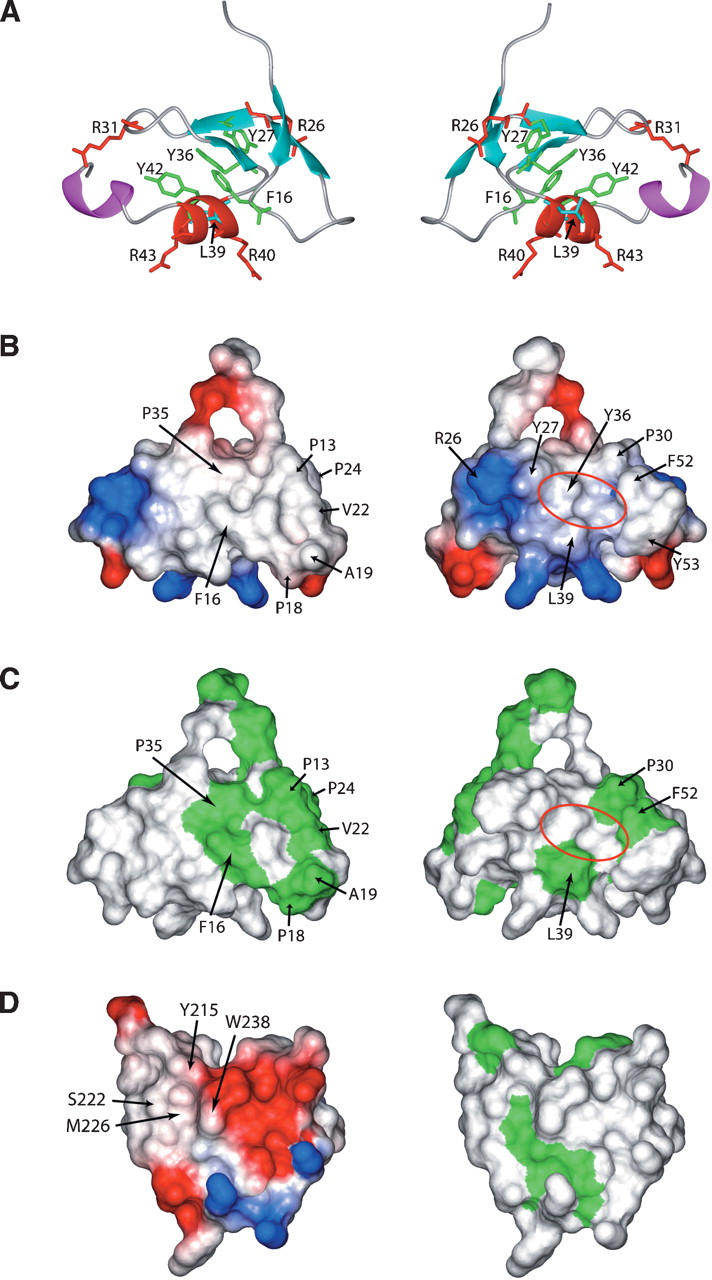
Presentation of ribbon diagrams, electrostatic surfaces, and hydrophobic surfaces of zf-HIT, and potential and hydrophobic surfaces of the PHD finger from ING2. (A) Side chains of the conserved hydrophobic residues are shown in green (for aromatic residues F16, Y27, Y36, and Y42) and cyan (for aliphatic residue L39). Side chains of all positively charged residues (R26, R31, R40, and R43) are shown in red. (B) Electrostatic surface presentation of the zf-HIT domain. The blue and red colors represent positive and negative electrostatic surface potential, respectively. (C) Hydrophobic surface presentation of the zf-HIT domain. The green color represents the hydrophobic surface. (D) Electrostatic and hydrophobic surfaces of the PHD finger of ING2. (A–C) The back surface (right panel) is the 180° rotation of the front surface (left panel). (D) Both the electrostatic potential and hydrophobic surfaces have a similar orientation on the back surfaces in A, B, and C.
Consensus sequence analysis of the zf-HIT, MYND, B-box, RING, and PHD domains
Zf-HIT belongs to the cross-brace zinc finger protein family, including the B-box, RING, and PHD domains, with the same interleaved zinc-binding mode. However, the arrangements of the Cys and His ligand residues in the two zinc-binding sites differ from each other. The MYND domain is a conserved zinc-binding domain with two zinc ions (Gross and McGinnis 1996; Spadaccini et al. 2006). The alignment of the consensus sequences of the zf-HIT, MYND, B-box, RING, and PHD fingers is shown in Figure 6A. zf-HIT and MYND have the same CCCC (for ZN1) and CCHC (for ZN2) motif; however, there are CCHC-CCCC in the PHD finger, CCCH-CCCC in the RING finger, and a rather different CCCC-CCHH type in the B-box domain. It is worthwhile to point out that there is an alternative Asp (D) for the fourth residue in the B-box and His (H) for the fifth residue in the RING finger for coordination with the zinc atom.
Figure 6.

Comparison of the cross-brace zinc-binding folds. (A) Alignment of the consensus sequences for the zf-HIT, MYND, B-box, RING, and PHD domains. (B) Schematic presentation of the cross-braced zinc-binding pattern and secondary structure elements in the zf-HIT, B-box, RING, and PHD domains. (C) Ribbon presentation of the zf-HIT (this study), B-box (2FFW), RING (1CHC), and PHD (2G6Q) domains. The α-helix is colored red and the 310 helix is magenta. The β-sheet is colored cyan. The side chains of the coordinating residues are shown as blue stick models and the zinc atoms are yellow spheres.
Comparison with other zinc-binding domains
The zf-HIT domain adopts a unique zinc-binding protein fold resembling that of the members of the treble clef domain family, such as the B-box, RING, and PHD fingers. Dali searches (Holm and Sander 1995) failed to yield any significant structural homologs. Although MYND possesses the same CCCC-CCHC motif as zf-HIT, a recent structural report of the MYND finger revealed that the tertiary structure has two consecutive zinc-binding sites resembling the LIM domain, but not the cross-brace-type zinc finger (Spadaccini et al. 2006). Therefore, we will focus on a comparison of the three-dimensional structures of the representative B-box (2FFW) (Massiah et al. 2006), RING (1CHC) (Barlow et al. 1994), and PHD (2G6Q) (Pena et al. 2006) domains with that of the zf-HIT domain (this study) (Figs. 6B,C and 7A–C).
Figure 7.

Superposition of the ZNHIT2 zf-HIT domain with the known domain structures of (A) the B-box (2FFW), (B) RING (1CHC), (C) PHD (2G6Q), (D) C2H2 (1UN6, finger5 of TFIIIA), and (F) cys2 domain of PKCδ (1PTQ). In each case, the zf-HIT domain is colored as in its ribbon diagram in Figure 3; the B-box, RING, PHD, C2H2 and cys2 domains are colored green, yellow, blue, orange, and purple, respectively. (E) Ribbon diagram of the cys2 domain of PKCδ. The side chains of the coordinating residues are shown in stick models (blue). Zinc ions are presented by yellow spheres.
These fingers all share some structural characteristics within the ββα core. However, the members from the treble clef domain family are distinct from each other, with unique three-dimensional characteristics. The most obvious difference between the zf-HIT, B-box, RING, and PHD fingers exists in the structure of the central α-helices and the following region. Among them, the RING finger usually has a longer α-helix than that in the zf-HIT and B-box structures. However, the PHD finger has only one turn for its central α-helix. In addition, the orientation of the central α-helix against the central β-sheet in the ββα core is different in these fingers. RING domains contain two highly conserved hydrophobic residues just before the fifth and after the sixth zinc ligand residues that construct the hydrophobic core of the structure. On the other hand, PHD domains contain the absolutely conserved aromatic residue at the two residues proceeding to the seventh zinc ligand, which contacts within the hydrophobic core. Correspondingly, the C-terminal chains approach the second zinc-binding site from the opposite direction between PHD and RING domains (Dodd et al. 2004). In the case of the zf-HIT domain of the protein FON, the highly conserved residues Phe16 and Tyr36, which are located on the N-terminal zinc knuckle of the β-hairpin and one residue preceding the fifth zinc ligand residue, make the hydrophobic core hold the architecture of the ββα core, comprising the β3 (Tyr27–Cys29), β4 (Pro35–Tyr36), αA (Leu39–His45), and αB (Thr47–Glu50) elements, and with the sandwiched second zinc ion, resembles that of the canonical C2H2-type zinc finger exemplified by the fifth finger of TFIIIA (Fig. 7D).
Other than the aforementioned domains from the treble clef domain family, the Cys2 domain of PKCδ (from the pre-SCOP database, PDB ID: 1PTQ) (Fig. 7E; Zhang et al. 1995) is similar to the zf-HIT domain with the structural elements. However, in the Cys2 domain, the zinc ligand corresponding to the seventh one in the zf-HIT domain is missing. One zinc ligand from the N-terminal before the second one, which corresponds to the first zinc ligand in zf-HIT domain, takes the role of the seventh one in the zf-HIT domain (Fig. 7E,F).
Features of the surface of the zf-HIT domain
The zf-HIT domain has several highly conserved residues in addition to the zinc ligands. As mentioned in the sequence analysis of zf-HIT, the conserved residues Tyr27, Leu39, and Tyr42 form the hydrophobic patch on the back surface, implying the interaction with another molecule. The front surface also has several large hydrophobic patches; however, the hydrophobic residues Pro13, Pro18, Ala19, Val22, and Pro24, which contribute to them, are not conserved in zf-HIT (Fig. 1). Thus, the hydrophobic patches on the front surface are not a general feature of the zf-HIT domain.
The electrostatic surface of the zf-HIT domain contains a few small, positively charged patches (Fig. 5B). Generally, it is hard for a small, positively charged patch to interact with nucleic acids. Based on the surface analysis, we hypothesize that the zf-HIT domain may function in protein–protein interactions through its hydrophobic patch.
Implications for the function of the zf-HIT domain
The present study revealed a novel zinc finger HIT domain as a new member of the treble clef domain family. Noting a certain similarity to the zf-C2H2 domain (Fig. 7D), which has multiple modes of DNA and RNA recognition (Lu et al. 2003; Hall 2005), we tested whether the zf-HIT domain could bind to nucleic acids. Our experiment demonstrated that the zf-HIT domain of protein FON is not a DNA-binding domain (Fig. 4). Also the preliminary 1H–15N chemical shift perturbation experiments did not detect any interaction with single-stranded poly-uridine and poly-adenine RNA sequences (data not shown). These NMR titration experiments suggest that the zf-HIT domain may not be an RNA-binding domain.
Our studies suggest that the zf-HIT domain presents a hydrophobic patch formed by the conserved residues Tyr27, Leu39, and Tyr42 (Fig. 5B,C). In the case of the PHD finger of ING2, the aromatic cage on the surface recognizes the trimethylated Lys4 residue in the histone 3 tail specifically (Fig. 5B–D; Pena et al. 2006). Thus, we tested whether the zf-HIT domain also has a similar histone-tail-binding activity like that of the PHD domain. We performed the binding experiment of the first 20 amino acids with trimethylated K4 of the histone H3 on the zf-HIT domain with a Biacore 3000 instrument. Our results (data not shown) show that the zf-HIT domain of protein FON does not bind the histone H3 tail.
In summary, the present study revealed that the zf-HIT domain is a new treble clef domain and binds two zinc atoms in a compact ββα core, which shares some structural similarities with the B-box, RING, and PHD domain structures. At the present time, the function of the novel zf-HIT domain is still unclear, and more functional analyses are necessary to elucidate the role of the zf-HIT domain. This structural study will provide guidelines for the functional analysis of the zf-HIT domain.
Materials and Methods
Protein sample
For the structure determination, a single 1.1 mM uniformly 13C- and 15N-labeled sample was prepared in 20 mM phosphate buffer at pH 6.0, 100 mM NaCl, 1 mM dithiothreitol, and 0.02% NaN3, with the addition of D2O to 10% (v/v), using the cell-free protein synthesis system (Kigawa et al. 2004). The engineered protein sample used for the NMR measurements comprised 46 amino acid residues from zf-HIT and the first seven and last six residues of tags, and is numbered 1–59 throughout this paper. The domain is surrounded by nonnative flanking sequences (N-terminal GSSGSSG and C-terminal PSGSSG) that are related to the expression and purification system.
Determination of the number of zinc ions
Experiments were performed in 20 mM Tris-HCl (pH 8.0), 200 mM NaCl, 5 μM ZnCl2, and 1 mM iminodiacetic acid (IDA). The protein concentration was determined from the peak volume of gel filtration chromatography (λ = 215 nm), using a bovine serum albumin solution as a standard. The free zinc concentration was determined from the absorbance of 50 μM 4-(2-pyridylazo) resorcinol (PAR) at 500 nm. The cysteine residues were modified by 250 μM p-hydroxy-mercuribenzoic acid (PHMB) in a 2.2 μM sample, followed by a 30-min incubation at room temperature. The amount of zinc bound to the sample was determined by measuring the increase of free zinc released by the PHMB modification. The zinc:protein stoichiometry was estimated from the ratio between the amount of released zinc and the protein.
NMR spectroscopy and resonance assignments
Standard triple resonance NMR experiments were recorded with the uniformly 13C-/15N-labeled sample, for the determination of the sequence-specific backbone and side-chain chemical shift assignments. For the collection of conformational constraints, 3D 15N- and 13C-edited NOESY-HSQC spectra were recorded with an 80-msec mixing time. NMR experiments were performed at 25°C on Bruker DRX 600 and Bruker AVANCE 800 spectrometers equipped with an xyz-pulse field gradient. The 1H, 15N, and 13C chemical shifts were referenced relative to the frequency of the 2H lock resonance of water, which was referenced through the magnetogyric ratios by external calibration on DSS.
Water suppression was achieved either by selective presaturation or by including a WATERGATE module (Piotto et al. 1992) in the original pulse sequences prior to the acquisition. The tautomeric state of the histidine ring was determined with a 2D heteronuclear multiple quantum coherence (1H–15N HMQC) experiment optimized for histidine side chains (Pelton et al. 1993). The raw NMR data were processed with the NMRPipe program (Delaglio et al. 1995). The NMRView program (Johnson and Blevins 1994) was used for interactive spectrum analysis.
Structure calculations
Peak lists for the NOESY spectra were generated by interactive peak picking, and peak volumes were determined by the automatic integration function of NMRView (Johnson and Blevins 1994). The three-dimensional structure was determined by combined automated NOESY cross-peak assignment (Herrmann et al. 2002) and structure calculation with torsion angle dynamics (Güntert et al. 1997) implemented in the CYANA program (Güntert 2004). The standard CYANA protocol of seven iterative cycles of NOE assignment and structure calculation, followed by a final structure calculation, was applied. In each cycle, the structure calculation started from 100 randomized conformers, and the standard CYANA simulated annealing schedule (Güntert et al. 1997) was used with 30,000 torsion angle dynamics steps per conformer. Initial rounds of calculations with only NOE restraints defined the general fold of the domain and revealed the zinc coordination. In the final refinement stage, distance restraints were added for Zn-Sγ (2.25–2.35 Å) and Zn-Nɛ2 (1.95–2.05 Å) (Summers et al. 1992; Wang et al. 2003) and for the bonds between the four zinc coordinating atoms, to ensure tetrahedral zinc coordination geometry. The 20 structures from CYANA calculation were further refined with AMBER9 (http://amber.scripps.edu) using the Generalized Born (GB) model (Xia et al. 2002) to get the final structures. During AMBER calculations, distance and dihedral angles were applied with force constants of 32 kcal/mol−1Å and 250 kcal/mol−1 rad2, respectively. The zinc coordination was restrained to be tetrahedral with lower and upper limits with force constants of 500 kcal/mol−1Å−1. PROCHECK_NMR (Laskowski et al. 1996) was used to validate the final structures. Structural figures were prepared with the MOLMOL program (Koradi et al. 1996).
DNA-binding assay
The DNA-binding assay was carried out on a Biacore 3000 instrument using streptavidin sensor chips (Biacore). Biotin-labeled oligonucleotides (CMV-TATA: Biotin-5′-TGGGAGGTCTATATAAGCAGAGCTCG-3′) were annealed with the complementary unlabeled oligonucleotides in a buffer containing 100 mM sodium chloride. Approximately 500 resonance units of the double-stranded oligonucleotides were immobilized to the sensor chip, using HBS-N buffer (10 mM HEPES buffer at pH 7.4 containing 150 mM sodium chloride). The indicated proteins were loaded for 1 min at a 20 μL/min flow rate onto the oligonucleotide-immobilized sensor chip, and the flow was continued for 10 min. Control surfaces with no oligonucleotide attached were used to correct for refractive index differences between the protein solution and the buffer. The results were analyzed using the BIAevaluation 4.1 software.
Protein Data Bank (PDB) accession number
The 20 conformers energy-minimized by AMBER9 of ZNHIT2 (residues 1–59) have been deposited in the Protein Data Bank (PDB entry 1X4S).
Acknowledgments
We thank Dr. Li Hua for critical reading and comments on the manuscript. We are grateful to Satoru Watanabe, Natsuko Matsuda, Yoko Motoda, Atsuo Kobayashi, Masaru Hanada, Masaomi Ikari, Fumiko Hiroyasu, Miyuki Sato, Satoko Yasuda, Yuri Tsuboi, Emi Nunokawa, Yasuko Tomo, Yukako Miyata, Yukiko Fujikura, Takushi Harada, Yuri Tomabechi, and Yuki Kamewari-Hayami for the sample preparation. This work was supported by the RIKEN Structural Genomics/Proteomics Initiative (RSGI), the National Project on Protein Structural and Functional Analyses of the Ministry of Education, Culture, Sports, Science and Technology of Japan (MEXT).
Footnotes
Supplemental material: see www.proteinscience.org
Reprint requests to: Shigeyuki Yokoyama, RIKEN Genomic Sciences Center, Protein Research Group, 1-7-22, Suehiro, Tsurumi, Yokohama 230-0045, Japan; e-mail: yokoyama@biochem.s.u-tokyo.ac.jp; fax: 81-45-503-9195; or Yutaka Muto, RIKEN Genomic Sciences Center, Protein Research Group, 1-7-22, Suehiro, Tsurumi, Yokohama 230-0045, Japan; e-mail: ymuto@gsc.riken.jp; fax: 81-45-503-9195.
Abbreviations: NOE, nuclear Overhauser effect; NOESY, NOE spectroscopy; HSQC, heteronuclear single-quantum coherence.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.062635107.
References
- Barlow P.N., Luisi, B., Milner, A., Elliott, M., and Everett, R. 1994. Structure of the C3HC4 domain by 1H-nuclear magnetic resonance spectroscopy. A new structural class of zinc-finger. J. Mol. Biol. 237: 201–211. [DOI] [PubMed] [Google Scholar]
- Bateman A., Coin, L., Durbin, R., Finn, R.D., Hollich, V., Griffiths-Jones, S., Khanna, A., Marshall, M., Moxon, S., Sonnhammer, E.L.L., et al. 2004. The Pfam protein families database. Nucleic Acids Res. 28: D138–D141, doi: 10.1093/nar/gkh121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaglio F., Grzesiek, S., Vuister, G.W., Zhu, G., Pfeifer, J., and Bax, A. 1995. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6: 277–293. [DOI] [PubMed] [Google Scholar]
- Dodd R.B., Allen, M.D., Brown, S.E., Sanderson, C.M., Duncan, L.M., Lehner, P.J., Bycroft, M., and Read, R.J. 2004. Solution structure of the Kaposi's sarcoma-associated herpesvirus K3 N-terminal domain reveals a novel E2-binding C4HC3-type RING domain. J. Biol. Chem. 279: 53840–53847. [DOI] [PubMed] [Google Scholar]
- Gouet P., Courcelle, E., Stuart, D.I., and Metoz, F. 1999. ESPript: Analysis of multiple sequence alignments in PostScript. Bioinformatics 15: 305–308. [DOI] [PubMed] [Google Scholar]
- Gross C.T. and McGinnis, W. 1996. DEAF-1, a novel protein that binds an essential region in a deformed response element. EMBO J. 15: 1961–1970. [PMC free article] [PubMed] [Google Scholar]
- Güntert P. 2004. Automated NMR structure calculation with CYANA. Methods Mol. Biol. 278: 353–378. [DOI] [PubMed] [Google Scholar]
- Güntert P., Mumenthaler, C., and Wuthrich, K. 1997. Torsion angle dynamics for NMR structure calculation with the new program DYANA. J. Mol. Biol. 273: 283–298. [DOI] [PubMed] [Google Scholar]
- Hall T.M. 2005. Multiple modes of RNA recognition by zinc finger proteins. Curr. Opin. Struct. Biol. 15: 367–373. [DOI] [PubMed] [Google Scholar]
- Herrmann T., Guntert, P., and Wuthrich, K. 2002. Protein NMR structure determination with automated NOE-identification in the NOESY spectra using the new software ATNOS. J. Biomol. NMR 24: 171–189. [DOI] [PubMed] [Google Scholar]
- Holm L. and Sander, C. 1995. Dali: A network tool for protein structure comparison. Trends Biochem. Sci. 20: 478–480. [DOI] [PubMed] [Google Scholar]
- Hunt J.B., Neece, S.H., Schachman, H.K., and Ginsburg, A. 1984. Mercurial-promoted Zn2+ release from Escherichia coli aspartate transcarbamoylase. J. Biol. Chem. 259: 14793–14803. [PubMed] [Google Scholar]
- Hunt J.B., Neece, S.H., and Ginsburg, A. 1985. The use of 4-(2-pyridylazo)resorcinol in studies of zinc release from Escherichia coli aspartate transcarbamoylase. Anal. Biochem. 146: 150–157. [DOI] [PubMed] [Google Scholar]
- Iwahashi H., Yamagata, K., Yoshiuchi, I., Terasaki, J., Yang, Q., Fukui, K., Ihara, A., Zhu, Q., Asakura, T., Cao, Y., et al. 2002. Thyroid hormone receptor interacting protein 3 (trip3) is a novel coactivator of hepatocyte nuclear factor-4α. Diabetes 51: 910–914. [DOI] [PubMed] [Google Scholar]
- Johnson B.A. and Blevins, R.A. 1994. NMR View: A computer program for the visualization and analysis of NMR data. J. Biomol. NMR 4: 603–614. [DOI] [PubMed] [Google Scholar]
- Kawakami K., Shafer, B.K., Garfinkel, D.J., Strathern, J.N., and Nakamura, Y. 1992. Ty element-induced temperature-sensitive mutations of Saccharomyces cerevisiae . Genetics 131: 821–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kigawa T., Yabuki, T., Matsuda, N., Matsuda, T., Nakajima, R., Tanaka, A., and Yokoyama, S. 2004. Preparation of Escherichia coli cell extract for highly productive cell-free protein expression. J. Struct. Funct. Genomics 5: 63–68. [DOI] [PubMed] [Google Scholar]
- Koradi R., Billeter, M., and Wuthrich, K. 1996. MOLMOL: A program for display and analysis of macromolecular structures. J. Mol. Graph. 14: 29–32. [DOI] [PubMed] [Google Scholar]
- Laskowski R.A., Rullmannn, J.A., MacArthur, M.W., Kaptein, R., and Thornton, J.M. 1996. AQUA and PROCHECK-NMR: Programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR 8: 477–486. [DOI] [PubMed] [Google Scholar]
- Lee M.S., Palmer III, A.G., and Wright, P.E. 1992. Relationship between 1H and 13C NMR chemical shifts and the secondary and tertiary structure of a zinc finger peptide. J. Biomol. NMR 2: 307–322. [DOI] [PubMed] [Google Scholar]
- Lemmens I.H., Farnebo, F., Piehl, F., Merregaert, J., Van de Ven, W.J., Larsson, C., and Kas, K. 2000. Molecular characterization of human and murine C11orf5, a new member of the FAUNA gene cluster. Mamm. Genome 11: 78–80. [DOI] [PubMed] [Google Scholar]
- Lu D., Searles, M.A., and Klug, A. 2003. Crystal structure of a zinc-finger–RNA complex reveals two modes of molecular recognition. Nature 426: 96–100. [DOI] [PubMed] [Google Scholar]
- Massiah M.A., Simmons, B.N., Short, K.M., and Cox, T.C. 2006. Solution structure of the RBCC/TRIM B-box1 domain of human MID1: B-box with a RING. J. Mol. Biol. 358: 532–545. [DOI] [PubMed] [Google Scholar]
- O'Brien K.P., Tapia-Paez, I., Stahle-Backdahl, M., Kedra, D., and Dumanski, J.P. 2000. Characterization of five novel human genes in the 11q13–q22 region. Biochem. Biophys. Res. Commun. 273: 90–94. [DOI] [PubMed] [Google Scholar]
- Pelton J.G., Torchia, D.A., Meadow, N.D., and Roseman, S. 1993. Tautomeric states of the active-site histidines of phosphorylated and unphosphorylated IIIGlc, a signal-transducing protein from Escherichia coli, using two-dimensional heteronuclear NMR techniques. Protein Sci. 2: 543–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pena P.V., Davrazou, F., Shi, X., Walter, K.L., Verkhusha, V.V., Gozani, O., Zhao, R., and Kutateladze, T.G. 2006. Molecular mechanism of histone H3K4me3 recognition by plant homeodomain of ING2. Nature 442: 100–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piotto M., Saudek, V., and Sklenar, V. 1992. Gradient-tailored excitation for single-quantum NMR spectroscopy of aqueous solutions. J. Biomol. NMR 2: 661–665. [DOI] [PubMed] [Google Scholar]
- Schubert M., Labudde, D., Oschkinat, H., and Schmieder, P. 2002. A software tool for the prediction of Xaa-Pro peptide bond conformations in proteins based on 13C chemical shift statistics. J. Biomol. NMR 24: 149–154. [DOI] [PubMed] [Google Scholar]
- Spadaccini R., Perrin, H., Bottomley, M.J., Ansieau, S., and Sattler, M. 2006. Structure and functional analysis of the MYND domain. J. Mol. Biol. 358: 498–508.16527309 [Google Scholar]
- Summers M.F., Henderson, L.E., Chance, M.R., Bess Jr, J.W., South, T.L., Blake, P.R., Sagi, I., Perez-Alvarado, G., Sowder III, R.C., Hare, D.R., et al. 1992. Nucleocapsid zinc fingers detected in retroviruses: EXAFS studies of intact viruses and the solution-state structure of the nucleocapsid protein from HIV-1. Protein Sci. 1: 563–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B., Alam, S.L., Meyer, H.H., Payne, M., Stemmler, T.L., Davis, D.R., and Sundquist, W.I. 2003. Structure and ubiquitin interactions of the conserved zinc finger domain of Npl4. J. Biol. Chem. 278: 20225–20234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wüthrich K. 1986. NMR of proteins and nucleic acids. Wiley, New York.
- Xia B., Tsui, V., Case, D.A., Dyson, H.J., and Wright, P.E. 2002. Comparison of protein solution structures refined by molecular dynamics simulation in vacuum, with a generalized Born model, and with explicit water. J. Biomol. NMR 22: 317–331. [DOI] [PubMed] [Google Scholar]
- Yokoyama S., Matsuo, Y., Hirota, H., Kigawa, T., Shirouzu, M., Kuroda, Y., Kurumizaka, H., Kawaguchi, S., Ito, Y., Shibata, T., et al. 2000. Structural genomics projects in Japan. Prog. Biophys. Mol. Biol. 73: 363–376. [DOI] [PubMed] [Google Scholar]
- Yoneyama M., Tochio, N., Umehara, T., Koshiba, S., Inoue, M., Yabuki, T., Aoki, M., Seki, E., Matsuda, T., Watanabe, S., et al. 2007. Structural and functional differences of SWIRM domain subtypes. J. Mol. Biol. 369: 222–238. [DOI] [PubMed] [Google Scholar]
- Zhang G., Kazanietz, M.G., Blumberg, P.M., and Hurley, J.H. 1995. Crystal structure of the Cys2 activator-binding domain of protein kinase Cδ in complex with phorbol ester. Cell 81: 917–924. [DOI] [PubMed] [Google Scholar]