

Abstract
Globular proteins are assemblies of α-helices and β-strands, interconnected by reverse turns and longer loops. Most short turns can be classified readily into a limited repertoire of discrete backbone conformations, but the physical–chemical determinants of these distinct conformational basins remain an open question. We investigated this question by exhaustive analysis of all backbone conformations accessible to short chain segments bracketed by either an α-helix or a β-strand (i.e., α-segment-α, β-segment-β, α-segment-β, and β-segment-α) in a nine-state model. We find that each of these four secondary structure environments imposes its own unique steric and hydrogen-bonding constraints on the intervening segment, resulting in a limited repertoire of conformations. In greater detail, an exhaustive set of conformations was generated for short backbone segments having reverse-turn chain topology and bracketed between elements of secondary structure. This set was filtered, and only clash-free, hydrogen-bond–satisfied conformers having reverse-turn topology were retained. The filtered set includes authentic turn conformations, observed in proteins of known structure, but little else. In particular, over 99% of the alternative conformations failed to satisfy at least one criterion and were excluded from the filtered set. Furthermore, almost all of the remaining alternative conformations have close tolerances that would be too tight to accommodate side chains longer than a single β-carbon. These results provide a molecular explanation for the observation that reverse turns between elements of regular secondary can be classified into a small number of discrete conformations.
Keywords: protein structure/folding, computational analysis of protein structure, reverse turns
Globular proteins are built on scaffolds of repetitive secondary-structure α-helices and β-strands (Levitt and Chothia 1976). These iso-directional elements are interconnected by short turns and longer loops (Rose and Seltzer 1977) that usually reverse the overall chain direction (Rose et al. 1985). The backbone torsion angles (ϕ,ψ angles) of most turn conformations are found to lie within a limited set of discrete basins, and accordingly work in this area has focused largely on ϕ,ψ-based turn classification (Chou and Fasman 1977; Richardson 1981; Rose et al. 1985; Sibanda and Thornton 1991; Ring et al. 1992; Efimov 1993; Donate et al. 1996; Wojcik et al. 1999; Engel and DeGrado 2005; Lahr et al. 2005). The very fact that turns are compatible with such classification prompted us to investigate the physical-chemical origin of this phenomenon.
Our analysis focuses on short turns bracketed between α-helices or β-strands, resulting in four distinct microenvironments: α-turn-α, β-turn-β, α-turn-β, and β-turn-α. Classic β-turns are four residues long (i to i + 3), but only the central residues (i + 1, i + 2) affect turn conformation (Venkatachalam 1968). Upon analyzing the central residues in three-, four-, and five-residue turns, we find that their backbone conformations are particular to each of the four microenvironments. This realization suggests that the limited repertoire of backbone conformations observed in turns is conditioned by local restrictions that are secondary-structure specific.
Earlier studies emphasized the paramount importance of sterics and hydrogen bonding in restricting accessible conformational space (Srinivasan and Rose 1999; Fitzkee and Rose 2004, 2005). If these same factors are the primary determinants of turn conformations, then authentic turns should be clash-free and hydrogen-bond–satisfied; conversely, other conceivable backbone conformations should fail at least one of these criteria.
We tested this proposition using simulations: Conformations of short-chain segments embedded in the four microenvironments were sampled exhaustively and then filtered for clash-free, hydrogen-bond–satisfied structures having reverse-turn chain topology. Survivors include authentic turns observed in solved protein structures, but almost all other conformers (>99%) are filtered away, and most remainders can be rationalized. These results confirm the hypothesis that the conformational biases of short turns between elements of repetitive secondary structure are established via local restrictions imposed by sterics and hydrogen bonding.
Results
Turns used in this study were defined as chain segments from one to three residues long, bracketed between elements of repetitive secondary structure (α-helix or β-strand) having crossing angles between 110° and 180° (Fig. 1). All segments satisfying these criteria were culled from the Protein Coil Library (http://www.roselab.jhu.edu/) (Fitzkee et al. 2005b), a repository of contiguous protein fragments that are neither α-helix nor β-strand, excised from a large data set of solved, nonredundant structures (Wang and Dunbrack Jr. 2003). Coil-library fragments are typically short (Fig. 1), and a substantial fraction of the one-, two-, and three-residue fragments correspond to the central residues in turns.
Figure 1.

Fragments that link elements of secondary structure. Two- and three-residue fragments were extracted from the Protein Coil Library (Fitzkee et al. 2005b), a repository of all non-α-helix and non-β-sheet residues culled from the PISCES server (Wang and Dunbrack Jr. 2003). This fragment data set was supplemented by all one-residue links. (A) Histogram of all non-α-helix and non-β-sheet fragment lengths. One-, two-, and three-residue fragments are the most abundant (shaded bars). (B) Histogram of observed crossing angles between secondary-structure elements. Crossing angles between elements linked by one-, two-, and three-residue fragments (shaded bars) have angles in the range of 110° to 180°, reversing the overall chain direction.
Turns were partitioned by bracketing secondary-structure types into four classes (α-turn-α, β-turn-β, α-turn-β, and β-turn-α), and each class was further subdivided into turns having one, two, or three central residues. For each of these 12 categories, the observed backbone torsion angles (ϕ,ψ angles) were plotted as contour maps (Fig. 2).
It is apparent that the turn conformations fall into a small number of discrete clusters (Fig. 2). Further, these conformations are unique with respect to the surrounding secondary structure (for example, Fig. 2, cf. A, D, G, and J). In all, 28 turn conformations were identified by visual examination, including both canonical β-turns (e.g., between β-strands; Fig. 2K) and many noncanonical turns. Representative ϕ,ψ values for all 28 turn types are listed in Table 1. While some observed structures are not included within the distinct conformational clusters highlighted in Figure 2, these outliers are sparsely distributed across the Ramachandran map (Supplemental Fig. 1).
Figure 2.

Turn conformations populate a limited set of discrete conformations. Backbone ϕ,ψ angles of fragments are contoured, revealing discrete conformational basins. The four rows correspond to the four microenvironments, and panels in each row are plots of the observed conformations of one-, two-, and three-residue fragments: (row 1, A–C) helix–helix; (row 2, D–F) helix–strand; (row 3, G–I) strand–helix; and (row 4, J–L) strand–strand. Every fragment length (one-, two-, or three-residue) in each microenvironment (α–α, α–β, β–α, and β–β) is identified by an arbitrarily assigned color (red, blue, or yellow) that is maintained in successive panels of that turn type. Contour levels are measured by the vertical bars associated with these 12 (3 fragment lengths × 4 microenvironments) possible categories. It is apparent that only a small number of discrete conformations (28 in all, listed in Table 1) are observed in the 24 backbone ϕ,ψ pairs (one for each panel in the figure) for these 12 categories, and their conformations are particular to each secondary-structure microenvironment.
Table 1.
Conformations of observed turns between elements of secondary structure

We confirmed that the observed turn conformations are uniquely compatible with their bracketing secondary-structure types by interchanging turns and microenvironments. Specifically, representative ϕ,ψ values for turns within their observed secondary-structure microenvironment (Table 1) were tested against each of the other three microenvironments. Each cross-matched case gave rise to violations of (1) turn-like chain topology (i.e., crossing angles between the bracketing secondary structure elements between 110° and 180°), (2) clash-free sterics, and/or (3) hydrogen-bond satisfaction. Illustrative examples of these three types of cross-matched violations are given next.
Example 1—A one-residue fragment between a helix–helix pair (Fig. 2A); when situated between two helices, this conformation reverses the direction of the protein chain (Fig. 3A, left), but when situated between two strands, the same conformation leaves the chain direction unchanged (Fig. 3A, right).
Example 2—A two-residue fragment between a strand–helix pair (Fig. 2H); when situated between a strand–helix pair, this conformation is clash-free (Fig. 3B, left), but when situated between a helix–helix pair, the same conformation results in steric overlap between two Cβ atoms (Fig. 3B, right).
Example 3—A three-residue fragment between a helix–helix pair (Fig. 2C, blue contour); when situated between a helix–helix pair, this conformation is fully hydrogen-bond–satisfied, with the amide nitrogen in the first turn residue serving as a helix cap (Fig. 3C, left). However, when situated between a strand–helix pair, the same amide nitrogen is occluded by a Cβ atom and cannot form a hydrogen bond to either the protein or the solvent (Fig. 3C, right).
Figure 3.

Three examples illustrating topological, steric, and hydrogen-bonding restrictions imposed by bracketing elements of secondary structure. Atoms that violate steric or hydrogen-bond restrictions are depicted as space-filling spheres, overlaid on a ball-and-stick representation of the structure, with ribbon diagrams marking helices and strands. (A) In a one-residue fragment, the same backbone conformation (ϕ,ψ = −120°, 90°) reverses the chain direction when situated between two α-helices (left) but not when situated between two β-strands (right). (B) In a two-residue fragment, the same backbone conformation (ϕ1,ψ1 = −90°, −30°; ϕ2,ψ2 = –80°, 130°) is sterically allowed in a strand–helix pair (left) but causes a steric clash between two β-carbons in a helix–helix pair (right). (C) In a three-residue fragment, the same backbone conformation (ϕ1,ψ1 = −75°, 5°; ϕ2,ψ2 = 65°, 40°; ϕ3,ψ3 = –100°, 80°) is fully hydrogen-bonded when situated between a helix–helix pair (left) but not when situated between a strand–helix pair (right).
Extending the analysis beyond these selected examples, we now report on exhaustive tests showing that, indeed, authentic turn types (in Table 1) do satisfy topological, steric, and hydrogen-bonding restrictions while, conversely, alternative conformers fail to do so.
Authentic turn conformations satisfy steric, hydrogen-bonding, and topological restrictions
All 28 authentic turn types were assessed in simulations by varying the backbone torsion angles of turn residues by 10° from their representative ϕ,ψ values (Table 1), with bracketing secondary structures held fixed as rigid units. Chain topology was assessed by the mean crossing angle, averaged over the ensemble of simulated structures. Steric restrictions and hydrogen-bond satisfaction were evaluated using the acceptance ratio from these simulations. An acceptance ratio of 1.0 indicates that all simulated structures are clash-free and fully hydrogen bonded. Indeed, all simulated structures were found to have high acceptance ratios (>0.8) with crossing angles that effectively reversed the overall chain direction (>120°; Fig. 4A).
Figure 4.
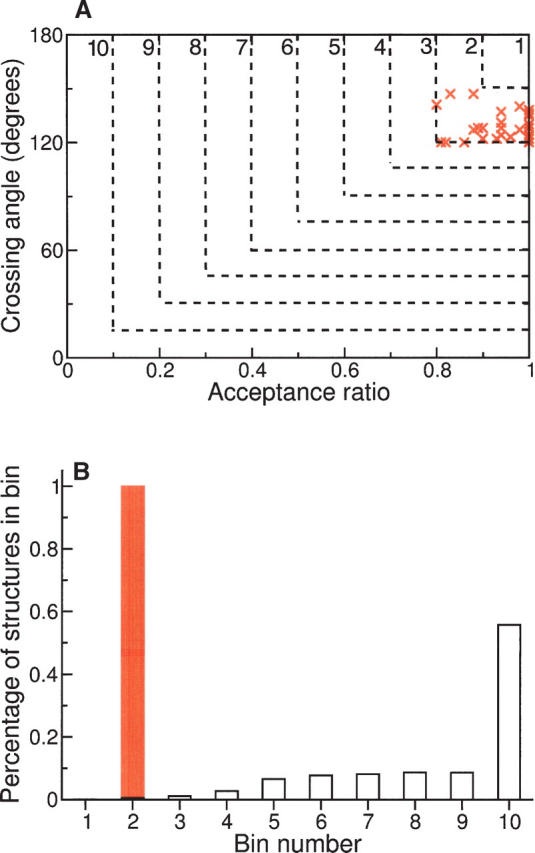
Simulated turns binned by crossing angle and acceptance ratio. (A) Crossing angles and acceptance ratios were subdivided into 10 bins (dashed lines). Effective turns reverse the chain direction (i.e., high crossing angles) and have backbone conformations that engender clash-free, hydrogen-bond–satisfied structures (i.e., high acceptance ratios); such conformers fall into low-numbered bins. Values from simulations of authentic turns (red ×'s) fall exclusively into bin 2, indicating that these structures satisfy topological, steric, and hydrogen-bonding restrictions. (B) A population histogram showing the fraction of simulated conformations in each bin. Simulations of alternative conformations (unfilled bars) are distributed across multiple, higher-numbered bins; most fall into bin 10, indicating either crossing angles that fail to reverse the chain direction or structures with steric violations and/or unsatisfied hydrogen bonds. In contrast, simulations using representative backbone torsion angles gleaned from proteins of known structure (Fig. 2) fall exclusively into bin 2 (red bar).
Most alternative backbone conformations are disallowed
Alternative backbone conformers were generated from a nine-state model (Scheme 1). These nine states cover the populated regions of the ϕ,ψ map (Hovmøller et al. 2002); each state is a circular region of radius 10°. Identical ranges (i.e., a 10° radius around a central value) were used for both alternative and authentic turns to normalize comparison of their resulting acceptance ratios. Using these nine states, conformations between pairs of secondary-structure elements were sampled exhaustively for one-, two-, and three-residue fragments (with 9, 92, and 93 states, respectively). Crossing angles and acceptance ratios were then calculated from the simulated ensembles after first eliminating any authentic turns (see details in Materials and Methods).
Scheme 1.

To assess the extent to which alternative conformers can mimic the observed turns shown in Figure 2, simulated populations were binned by acceptance ratio and crossing angle. Effective turns—those conformers having high acceptance ratios and high crossing angles—will fall into low-numbered bins in Figure 4, whereas non-turns—those conformers having low acceptance ratios and/or low crossing angles—will fall into higher-numbered bins. When classified in this way, all authentic turns do, in fact, lie within bin 2 (Fig. 4). Consequently, alternative conformers can be evaluated by whether or not they fall into low-numbered bins, like effective turns.
It is apparent that almost all alternative conformations have high bin numbers, corresponding to low crossing angles and/or low acceptance ratios (shown as unfilled histogram bars in Fig. 4B). Only 14 of the 2215 total alternative conformations fall into bin 1 or 2, i.e., more than 99% are outside the observed variation of authentic turns. Most of these 14 false positives have Cβ atoms that are too close to other atoms for all but an alanine or a glycine residue (Fig. 5; Table 2), and their acceptance ratios would have been reduced significantly had bulky side chains been included.
Figure 5.

False positives have tight tolerances. Alternative conformers simulated using the nine-state model result in 14 false positives that satisfy authentic turn criteria (Table 2), but most of these cases are disfavored owing to Cβ atoms with close tolerances, too tight to accommodate residues other than glycine or alanine. Accordingly, sparsely populated conformations in Fig. 2 can be rationalized by an incompatibility with bulky side chains. Cβ atoms in question are depicted as space-filling spheres, overlaid on a ball-and-stick representation of the structure, with ribbon diagrams marking helices and strands. (A) A three-residue fragment (ϕ1,ψ1 = 60°, 30°; ϕ2,ψ2 = 60°, 30°; ϕ3,ψ3 = −80°, 80°) that brings Cβ atoms into van der Waals contact. (B) A three-residue fragment (ϕ1,ψ1 = 60°, 30°; ϕ2,ψ2 = −80°, 160°; ϕ3,ψ3 = −80°, 160°) that brings Cβ atoms into close proximity.
Table 2.
False-positive turn conformations

Discussion
Pauling's paradigmatic models of repetitive secondary structure—α-helix (Pauling et al. 1951) and β-sheet (Pauling and Corey 1951)—were based solely on local hydrogen-bond satisfaction, sterics, and peptide bond geometry. Later, Venkatachalam predicted the existence of β-turn structures using identical principles (Venkatachalam 1968). Together, these several categories account for approximately three-quarters of protein structure, on average (Fitzkee et al. 2005a,b). The remaining quarter includes other recognizable categories, such as the one-, two-, and three-residue turns described in Figure 2. These turns have greater conformational heterogeneity than helix and sheet and might therefore be expected to involve additional determinants, more than just local hydrogen-bond satisfaction, sterics, and chain geometry. But our results suggest otherwise.
Reverse turns include both strict β-turns (Venkatachalam 1968) and more moderate bends (Rose et al. 1985) and loops (Leszczynski and Rose 1986). The peptide main chain changes its overall direction at these reverse-turn sites, and their abundance is the reason why globular proteins are, in fact, globular. Much, but not all, of the earlier literature has focused on strict β-turns that link strands of β-sheet (Richardson 1985; Wilmot and Thornton 1988; Sibanda and Thornton 1991; Gunasekaran et al. 1997; Ramirez-Alvarado et al. 1997; Chung et al. 1998). Efimov's modeling studies (Efimov 1993, 1994) encompass a more general definition of turns, including those at helix ends (Brunet et al. 1993). Helices often terminate in recognizable capping motifs (Aurora and Rose 1998) that both stabilize the helix and launch the main chain in a different direction, and consequently helix caps (Presta and Rose 1988; Richardson and Richardson 1988) can be an integral component of helix-adjacent turns (Lahr et al. 2005). Recently, Engel and DeGrado (2005) undertook a comprehensive analysis of links in helical hairpins and their populations (Fig. 6 and Table 4 in Engel and DeGrado 2005) resemble ours (Fig. 2; Table 1) (their analysis extends to nine-residue links, whereas ours is limited to three residues). Shortle has pointed out that the densely populated regions in ϕ,ψ space can be subdivided into discrete bins (Shortle 2003), consistent with subpopulations that are favored by the local structural environment. However, unlike most of these earlier studies, our present focus is on the molecular origin of turn conformations and on why only a few discrete populations are observed.
The physical-chemical determinants analyzed here are completely general in that they are properties of the protein backbone and therefore applicable to all residues (Rose et al. 2006). These determinants are also extensive. Even for a dipeptide, steric considerations limit accessible conformational space (Ramachandran and Sasisekharan 1968), and additional limitations are imposed in these congested turn conformations, where the chain folds back on itself (Fitzkee and Rose 2004). Further, polar groups must find either intra- or intermolecular hydrogen-bond partners because the energetic cost of an unsatisfied backbone polar group is high (Baldwin 2003). Accordingly, almost all backbone hydrogen bonds in X-ray structures of folded proteins are either satisfied (Fleming and Rose 2005) or can be minimized into satisfied conformations without substantial structural changes (Panasik et al. 2005).
Contouring the data, as in Figure 2, reveals the major populations by suppressing outliers. It is possible that such outliers are instances of rare but authentic turns, e.g., strained conformations (Herzberg and Moult 1991; Hodel et al. 1994). Upon excluding those false positives with obvious structural anomalies, four conformations with crossing angles and acceptance ratios resembling those of authentic turns remain in Table 2; these conformers are not highly populated in proteins. Another possibility is that some or all of the outliers are spurious conformations that legitimately belong to one of the major populations listed in Table 1, akin to the near-turns described in Panasik Jr. et al. (2005). Clearly, further work is needed to distinguish between these possibilities. Other interesting details are also beyond the scope of this current study, such as the differences in basin sizes and populations in Figure 2, and here, too, further analysis is needed, taking into account factors that were neglected initially, such as secondary-structure packing (Chothia et al. 1981) and electrostatic effects (Dasgupta et al. 2004). These issues notwithstanding, it is compelling that Pauling's three simple criteria—steric exclusion, hydrogen-bond satisfaction, and chain geometry—can largely account for the conformational diversity observed in short turns that interconnect elements of protein secondary structure.
Materials and Methods
Protein fragments that are neither α-helix nor β-strand were extracted from the Protein Coil Library (Fitzkee et al. 2005b) and sorted by length. These fragments were derived from the PISCES list (Wang and Dunbrack Jr. 2003) of structures with resolution of 2.0 Å or better and refinement factors of 0.25 or better. The list was supplemented by one-residue fragments, which are not included in the Protein Coil Library (http://www.roselab.jhu.edu/coil). Care was taken to avoid double-counting longer fragments as multiple shorter fragments.
In the context of their host proteins, the one-, two-, and three-residue fragments used in this study are flanked by secondary-structure elements of differing lengths. To control for length variability when computing crossing angles, all secondary-structure elements were set to be eight residues in length. Specifically, the two adjoining residues at either end of a given fragment were extended by an additional six residues; canonical backbone torsion angles were assigned to these six-residue extensions (ϕ,ψ = −62°, −42° for α-helix or ϕ,ψ = −120°, 135° for β-strand). Crossing angles were computed by taking the principal moments of inertia of each secondary structure element, calculated from its Cα coordinates; the moment with the smallest eigenvalue is the vector of least-squares best-fit to the long axis of that element (Rose and Seltzer 1977). The crossing angle between two elements was then computed as the scalar angle between their two best-fit vectors, each pointed in the N- to C-terminal direction to avoid sign ambiguities.
In simulations, the backbone torsions of turn residues were varied at random within 10° of their origin, with bracketing secondary-structure elements held rigid. Exceptions were made in the case of some one-residue fragments where clear conformational preferences observed in authentic turns affect the fragment-adjacent residue, as noted in Table 1. All residues were modeled as alanine except those with ϕ > 0°, which were modeled as glycine.
Average crossing angles and acceptance ratios were based on 100 simulated structures. Standard bond lengths, bond angles, atomic radii, and hydrogen-bond criteria were used, as described elsewhere (Fitzkee and Rose 2004, 2005). Atomic radii were scaled to 90% of their standard values to compensate for any simulation artifacts caused by using rigid models of secondary structure (Creamer and Rose 1994).
The ϕ,ψ origins of the nine states in Scheme 1 are (−160°,160°), (−80°,160°), (−160°,80°), (−80°,80°), (60°,30°), (−90°,0°), (90°,0°), (−60°,−30°), and (60°,−150°). Each state is a circular region of the Ramachandran map within 10° of these central values. Conformations were sampled exhaustively by varying the backbone through all possible combinations of states within every secondary-structure microenvironment (helix–helix, helix–strand, strand–helix, and strand–strand). In each of these four microenvironments, one-, two-, and three-residue fragments can visit 9, 81, and 729 possible states, respectively. However, conformers within 30° of any authentic turn in Figure 2 were disallowed, eliminating 64 conformers. In addition, conformers that simply extend an adjacent secondary-structure element were also disallowed. Specifically, states 1 and 3 were disallowed when immediately adjacent to a β-strand, and state 8 was disallowed when immediately adjacent to an α-helix; 997 additional conformations were eliminated by these criteria.
Acknowledgments
We thank Pat Fleming and Haipeng Gong for useful discussions. Support from a Wellcome Interfaces fellowship (T.O.S.) and the Mathers Foundation (G.D.R.) is gratefully acknowledged.
Footnotes
Supplemental material: see www.proteinscience.org
Reprint requests to: George D. Rose, T.C. Jenkins Department of Biophysics, Johns Hopkins University, 3400 N. Charles Street, Baltimore, MD 21218, USA; e-mail: grose@jhu.edu; fax: (410) 516-4118.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.072898507.
References
- Aurora R. and Rose, G.D. 1998. Helix capping. Protein Sci. 7: 21–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldwin R.L. 2003. In search of the energetic role of peptide hydrogen bonds. J. Biol. Chem. 278: 17581–17588. [DOI] [PubMed] [Google Scholar]
- Brunet A.P., Huang, E.S., Huffine, M.E., Loeb, J.E., Weltman, R.J., and Hecht, M.H. 1993. The role of turns in the structure of an α-helical protein. Nature 364: 355–358. [DOI] [PubMed] [Google Scholar]
- Chothia C., Levitt, M., and Richardson, D. 1981. Helix to helix packing in proteins. J. Mol. Biol. 145: 215–250. [DOI] [PubMed] [Google Scholar]
- Chou P.Y. and Fasman, G.D. 1977. β-Turns in proteins. J. Mol. Biol. 115: 135–175. [DOI] [PubMed] [Google Scholar]
- Chung Y.J., Christianson, L.A., Stanger, H.E., Powell, D.R., and Gellman, S.H. 1998. A β-peptide reverse turn that promotes hairpin formation. J. Am. Chem. Soc. 120: 10555–10556. [Google Scholar]
- Creamer T.P. and Rose, G.D. 1994. α-Helix-forming propensities in peptides and proteins. Proteins 19: 85–97. [DOI] [PubMed] [Google Scholar]
- Dasgupta B., Pal, L., Basu, G., and Chakrabarti, P. 2004. Expanded turn conformations: Characterization and sequence–structure correspondence in α-turns with implications in helix folding. Proteins 55: 305–315. [DOI] [PubMed] [Google Scholar]
- Donate L.E., Rufino, S.D., Canard, L.H., and Blundell, T.L. 1996. Conformational analysis and clustering of short and medium size loops connecting regular secondary structures: A database for modeling and prediction. Protein Sci. 5: 2600–2616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efimov A.V. 1993. Standard structures in proteins. Prog. Biophys. Mol. Biol. 60: 201–239. [DOI] [PubMed] [Google Scholar]
- Efimov A.V. 1994. Common structural motifs in small proteins and domains. FEBS Lett. 355: 213–219. [DOI] [PubMed] [Google Scholar]
- Engel D.E. and DeGrado, W.F. 2005. α–α Linking motifs and interhelical orientations. Proteins 61: 325–337. [DOI] [PubMed] [Google Scholar]
- Fitzkee N.C. and Rose, G.D. 2004. Steric restrictions in protein folding: An α-helix cannot be followed by a contiguous β-strand. Protein Sci. 13: 633–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzkee N.C. and Rose, G.D. 2005. Sterics and solvation winnow accessible conformational space for unfolded proteins. J. Mol. Biol. 353: 873–887. [DOI] [PubMed] [Google Scholar]
- Fitzkee N.C., Fleming, P.J., Gong, H., Panasik Jr, N., Street, T.O., and Rose, G.D. 2005a. Are proteins made from a limited parts list? Trends Biochem. Sci. 30: 73–80. [DOI] [PubMed] [Google Scholar]
- Fitzkee N.C., Fleming, P.J., and Rose, G.D. 2005b. The Protein Coil Library: A structural database of nonhelix, nonstrand fragments derived from the PDB. Proteins 58: 852–854. [DOI] [PubMed] [Google Scholar]
- Fleming P.J. and Rose, G.D. 2005. Do all backbone polar groups in proteins form hydrogen bonds? Protein Sci. 14: 1911–1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunasekaran K., Ramakrishnan, C., and Balaram, P. 1997. β-Hairpins in proteins revisited: Lessons for de novo design. Protein Eng. 10: 1131–1141. [DOI] [PubMed] [Google Scholar]
- Herzberg O. and Moult, J. 1991. Analysis of the steric strain in the polypeptide backbone of protein molecules. Proteins 11: 223–229. [DOI] [PubMed] [Google Scholar]
- Hodel A., Kautz, R.A., Adelman, D.M., and Fox, R.O. 1994. The importance of anchorage in determining a strained protein loop conformation. Protein Sci. 3: 549–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hovmøller S., Zhou, T., and Ohlson, T. 2002. Conformations of amino acids in proteins. Acta Crystallogr. D Biol. Crystallogr. 58: 768–776. [DOI] [PubMed] [Google Scholar]
- Lahr S.J., Engel, D.E., Stayrook, S.E., Maglio, O., North, B., Geremia, S., Lombardi, A., and DeGrado, W.F. 2005. Analysis and design of turns in α-helical hairpins. J. Mol. Biol. 346: 1441–1454. [DOI] [PubMed] [Google Scholar]
- Leszczynski J.F. and Rose, G.D. 1986. Loops in globular proteins: A novel category of secondary structure. Science 234: 849–855. [DOI] [PubMed] [Google Scholar]
- Levitt M. and Chothia, C. 1976. Structural patterns in globular proteins. Nature 261: 552–558. [DOI] [PubMed] [Google Scholar]
- Panasik N., Fleming, P.J., and Rose, G.D. 2005. Hydrogen-bonded turns in proteins: The case for a recount. Protein Sci. 14: 2910–2914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauling L. and Corey, R.B. 1951. The pleated sheet, a new layer configuration of polypeptide chains. Proc. Natl. Acad. Sci. 37: 251–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauling L., Corey, R.B., and Branson, H.R. 1951. The structure of proteins; two hydrogen-bonded helical configurations of the polypeptide chain. Proc. Natl. Acad. Sci. 37: 205–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Presta L.G. and Rose, G.D. 1988. Helix signals in proteins. Science 240: 1632–1641. [DOI] [PubMed] [Google Scholar]
- Ramachandran G.N. and Sasisekharan, V. 1968. Conformation of polypeptides and proteins. Adv. Protein Chem. 23: 283–437. [DOI] [PubMed] [Google Scholar]
- Ramirez-Alvarado M., Blanco, F.J., Niemann, H., and Serrano, L. 1997. Role of β-turn residues in β-hairpin formation and stability in designed peptides. J. Mol. Biol. 273: 898–912. [DOI] [PubMed] [Google Scholar]
- Richardson J.S. 1981. The anatomy and taxonomy of protein structure. Adv. Protein Chem. 34: 167–339. [DOI] [PubMed] [Google Scholar]
- Richardson J.S. 1985. A new twist for hairpin turns. Nature 316: 102–103. [Google Scholar]
- Richardson J.S. and Richardson, D.C. 1988. Amino acid preferences for specific locations at the ends of α-helices. Science 240: 1648–1652. [DOI] [PubMed] [Google Scholar]
- Ring C.S., Kneller, D.G., Langridge, R., and Cohen, F.E. 1992. Taxonomy and conformational analysis of loops in proteins. J. Mol. Biol. 224: 685–699. [DOI] [PubMed] [Google Scholar]
- Rose G.D. and Seltzer, J.P. 1977. A new algorithm for finding the peptide chain turns in a globular protein. J. Mol. Biol. 113: 153–164. [DOI] [PubMed] [Google Scholar]
- Rose G.D., Gierasch, L.M., and Smith, J.A. 1985. Turns in peptides and proteins. Adv. Protein Chem. 37: 1–109. [DOI] [PubMed] [Google Scholar]
- Rose G.D., Fleming, P.J., Banavar, J.R., and Maritan, A. 2006. A backbone-based theory of protein folding. Proc. Natl. Acad. Sci. 103: 16623–16633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shortle D. 2003. Propensities, probabilities, and the Boltzmann hypothesis. Protein Sci. 12: 1298–1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sibanda B.L. and Thornton, J.M. 1991. Conformation of β hairpins in protein structures: Classification and diversity in homologous structures. Methods Enzymol. 202: 59–82. [DOI] [PubMed] [Google Scholar]
- Srinivasan R. and Rose, G.D. 1999. A physical basis for protein secondary structure. Proc. Natl. Acad. Sci. 96: 14258–14263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatachalam C.M. 1968. Stereochemical criteria for polypeptides and proteins. V. Conformation of a system of three linked peptide units. Biopolymers 6: 1425–1436. [DOI] [PubMed] [Google Scholar]
- Wang G. and Dunbrack Jr, R.L. 2003. PISCES: A protein sequence culling server. Bioinformatics 19: 1589–1591. [DOI] [PubMed] [Google Scholar]
- Wilmot C.M. and Thornton, J.M. 1988. Analysis and prediction of the different types of β-turn in proteins. J. Mol. Biol. 203: 221–232. [DOI] [PubMed] [Google Scholar]
- Wojcik J., Mornon, J.P., and Chomilier, J. 1999. New efficient statistical sequence-dependent structure prediction of short- to medium-sized protein loops based on an exhaustive loop classification. J. Mol. Biol. 289: 1469–1490. [DOI] [PubMed] [Google Scholar]