

Abstract
Adenylosuccinate lyase (ASL) of Bacillus subtilis contains three conserved histidines, His68, His89, and His141, identified by affinity labeling and site-directed mutagenesis as critical to the intersubunit catalytic site. The pH-Vmax profile for wild-type ASL is bell-shaped (pK 1 = 6.74 and pK 2 = 8.28). Only the alkaline side changes with temperature, characteristic of histidine pKs. To identify determinants of pK 2 in the enzyme-substrate complex, we replaced residues at two positions close to His68 (but not to His89 or His141) in the structure. Compared with the specific activity of 1.75 μmol adenylosuccinate reacting/min/mg of wild-type enzyme at pH 7.0, mutant enzymes D69E, D69N, R310Q, and R310K exhibit specific activities of 0.40, 0.04, 0.00083, and 0.10, respectively. While D69E has a K m for adenylosuccinate similar to that of wild-type ASL, D69N and R310K exhibit modest increases in K m, and R310Q has an 11-fold increase in K m. The mutant enzymes show no significant change in molecular weight or secondary structure. The major change is in the pH-Vmax profile: pK 2 is 8.48 for the D69E mutant and is decreased to 7.83 in D69N, suggesting a proximal negative charge is needed to maintain the high pK of 8.28 observed for wild-type enzyme and attributed to His68. Similarly, R310Q exhibits a decrease in its pK 2 (7.33), whereas R310K shows little change in pK 2 (8.24). These results suggest that Asp69 interacts with His68, that Arg310 interacts with and orients the β-carboxylate of Asp69, and that His68 must be protonated for ASL to be active.
Keywords: adenylosuccinate lyase, pH-Vmax profile, site-directed mutagenesis
Adenylosuccinate lyase (ASL) plays a critical role in both cellular replication and metabolism via its involvement in the de novo purine biosynthetic pathway, the end products of which serve as precursors for DNA and RNA synthesis, as intermediates in biosynthetic reactions, as energy storage depots, and as metabolic regulators (Ratner 1972). The importance of ASL is indicated by the existence of ASL deficiency, a human genetic disease resulting in autism, mental retardation, muscle wasting, and/or epilepsy (Jaeken and Van den Berghe 1984; Van den Berghe and Jaeken 2001).
ASL catalyzes the cleavage of adenylosuccinate (SAMP) to AMP and fumarate, the second step in the conversion of inosine monophosphate (IMP) to adenosine monophosphate (AMP) (Ratner 1972). Steady-state kinetics and inhibition studies suggest a uni–bi mechanism with a strong preference for product release in which fumarate leaves the enzyme before AMP (Bridger and Cohen 1968). It has been proposed that the cleavage proceeds by a β-elimination mechanism, involving the attack on the β-H of adenylosuccinate by an amino acid of the enzyme functioning as the general base, and the protonation of either the N1 or N6 position of adenylosuccinate by an enzymic amino acid functioning as a general acid (Hanson and Havir 1972).
ASL of Bacillus subtilis is a homotetramer, with a molecular weight of ∼200,000, composed of subunits with 431 amino acids. Studies on the B. subtilis ASL have shown that the enzyme has four active sites, with each active site formed by three subunits each contributing one or more amino acids (Brosius and Colman 2002). Previous affinity labeling and site-directed mutagenesis studies of the B. subtilis ASL enzyme have identified three histidines as critical residues of the intersubunit catalytic site: His68, His141, and His89 (Lee et al. 1997, 1998, 1999; Brosius and Colman 2000). It has been postulated that one of these histidines acts as the general base and one histidine functions as the general acid to protonate the AMP leaving group; His141 and His68 have been suggested to act as the general base–general acid (Lee et al. 1999).
Kinetic studies over the pH range 6–9 reveal a bell-shaped pH-Vmax profile for the wild-type ASL enzyme with pK 1 and pK 2 values of about 6.7 and 8.3, respectively. These pKs presumably represent those of ionizable groups of the enzyme–substrate complex. Studies of the temperature dependence of the pH-Vmax profile of wild-type ASL have shown that the pK 1 value does not change with temperature (excluding histidine as that ionizable group), while the pK 2 value decreases with increasing temperature, with a ΔHi value of 9.9 kcal/mol, characteristic of histidines (Brosius and Colman 2000). We have recently shown that mutation of Ser94, which is close to His89 but not to the other two important histidines, results in a decrease in pK2 (Segall et al. 2007). To test whether the ionization of another amino acid is also reflected in the pK 2 of the wild-type pH-Vmax profile, we now focus on Asp69, which is 3.63 Å from His68, 9.31 Å from His89, and 10.74 Å from His141, based on the structure of ASL (Toth and Yeates 2000; Brosius and Colman 2002; Segall and Colman 2004). Replacement of Asp69 would be expected to perturb the ionization of His68 but not of the other two histidines. Thus, if substitution for Asp69 results in a change in pK2, His68 would be implicated as an ionizable group reflected in pK2. This paper reports the results of replacing, by site-directed mutagenesis, amino acids close to or interacting with His68. A preliminary version of this paper has been presented (Sivendran et al. 2005).
Results
Expression and purification of wild-type and mutant enzymes
His68 is one of the three conserved histidines that are critical for the intersubunit catalytic site (Lee et al. 1998, 1999). Because the negatively charged Asp69 is close to the critical His68 (but not to His89 or His141) in the crystal structure of ASL, mutations were made at position 69. Since the positively charged Arg310 is close to Asp69 in the ASL crystal structure, mutations were also made at position 310. Asp69 and Arg310 are conserved in ASLs from bacteria to humans, as indicated in the representative sequences shown in Figure 1. To study the effect of these conserved charged residues on the pK of histidine 68, the negatively charged aspartate at 69 was mutated to the uncharged asparagine and the negatively charged glutamate. The arginine at 310 was replaced by the neutral glutamine and the positively charged lysine. The wild-type and mutant pBHis plasmid encoding B. subtilis ASL were expressed in Escherichia coli, and the enzymes were purified as described in Materials and Methods. The wild-type and all mutant enzymes were homogeneous as indicated by the single band for each on SDS-PAGE (Supplemental Fig. S1).
Figure 1.

Amino acid sequence alignments of adenylosuccinate lyase from B. subtilis, T. maritima, Gallus gallus, Mus musculus, and Homo sapiens according to CLUSTALW. The asterisks indicate that the amino acids at the position are identical.
Circular dichroism spectroscopy of wild-type and mutant enzymes
Circular dichroism (CD) spectra of wild-type and mutant enzymes were measured to evaluate any changes in the secondary structure of the enzymes. The CD spectra of all mutant and wild-type enzymes are shown in Supplemental Figure S2. The spectra of all mutant enzymes are very similar to that of wild type, indicating that the mutations do not cause any detectable changes in the secondary structure of the enzymes.
Molecular weight determination
The oligomeric state of the wild-type and mutant enzymes was analyzed by light-scattering photometry. ASL has been shown to exist in solution as an equilibrium mixture of dimer and tetramer with their relative proportions dependent on the protein concentration (Palenchar and Colman 2003). Thus, the molecular weights of wild-type and mutant enzymes were measured at the same protein concentration (0.25 mg/mL); the results are shown in Table 1. The M r values of wild-type and mutant enzymes are similar; these are consistent with an equilibrium mixture of dimer and tetramer, with the tetramer predominating. Light scattering could not be used on the R310Q mutant enzyme because of its tendency to aggregate. Therefore, native polyacrylamide gel electrophoresis was used to compare the sizes of wild-type and R310Q enzymes. Figure 2 shows a representative gel (9%). The wild-type and R310Q mutant enzymes migrate to similar positions, yielding molecular weights of 147 and 148 kDa, respectively; both enzymes were added to the gels at a lower protein concentration, accounting for the lower molecular weight observed by this method as compared with light scattering.
Table 1.
Molecular weight determined by light scattering

Figure 2.

Native polyacrylamide gel electrophoresis. A representative 9% gel is shown: lane 1, R310Q; lane 2, wild type (WT ASL); lane 3, yeast alcohol dehydrogenase (ADH) (141,000); lane 4, bovine serum albumin (BSA) (67,000); lane 5, horse spleen ferritin (450,000); lane 6, chicken egg ovalbumin (43,000).
Kinetics of mutant enzymes
The kinetic properties at pH 7.0 for wild-type and mutant enzymes are summarized in Table 2. Compared with the wild-type Vmax of 1.87 μmol/min/mg, the mutant enzymes D69E, D69N, R310K, and R310Q display markedly decreased Vmax values. Of the Asp69 mutants, and of the Arg310 mutants, those in which the original charge was preserved (D69E and R310K) exhibit the smallest changes in Vmax, and the K m values for SAMP of these mutant enzymes are not very different from the wild-type value of 2 μM. In contrast, the K m for SAMP is elevated about 11-fold in R310Q. Table 2 also reports values of k cat/K m. The mutant enzymes, in which charged amino acids were replaced with neutral residues, as in D69N and R310Q, exhibit the most striking decreases in these values.
Table 2.
Kinetics of wild-type and mutant B. subtilis adenylosuccinate lyases

pH Dependence on Vmax for wild-type and mutant ASL enzymes
To evaluate the effect of the mutations on the kinetically detectable ionizable groups, the velocity at 300 μM SAMP was measured as a function of pH (from pH 6.0 to pH 9.3) for all the enzymes in the AMP and fumarate formation reaction. The Km for SAMP increases with pH over the pH range used. For example, the Km varies from 1.9 to 33 μM for wild type, from 1.3 to 14 μM for the D69E mutant, and from 9 to 43 μM for the D69N mutants. In most cases, SAMP concentration is high relative to the Km over the pH range used; therefore, the velocity at 300 μM SAMP measures the Vmax at each pH. For the R310Q mutant, however, the 300 μM SAMP was only ∼6 times the Km in the high pH range; therefore, for this mutant the rates measured at 300 μM SAMP were used to calculate Vmax from the equation: Vmax = vobs(1 + Km/[SAMP]). The wild-type and all mutant enzymes show a bell-shaped pH-V max curve. Representative graphs for wild type, D69E, and D69N are shown in Figure 3. The pK values were determined by using the equation V max (at each pH) = V max intrinsic/[1 + 10(pK1 − pH) + 10(pH − pK2)], where pK 1 and pK 2 are the pK values for the ionizable groups of the enzyme–substrate complex responsible for the left and right limbs, respectively, of the pH-Vmax curve; V max (at each pH) is the value at saturating SAMP concentration at each pH; and V max intrinsic is the pH-independent maximum velocity for the enzyme. The pK 1, pK 2, and V max intrinsic values are summarized in Table 3. The ionization of His68 has been postulated to be reflected in pK 2 (Lee et al. 1999; Brosius and Colman 2000), even though this pK 2 is higher than the value of 6.0 given for a typical histidine (Garrett and Grisham 1999). The wild-type enzyme has pK 1 and pK 2 values of 6.74 and 8.28, respectively. In contrast, the D69N and R310Q mutant enzymes have pK 2 values of 7.83 and 7.33, respectively. Thus, removing the charge at position 69 and 310 lowers the pK 2 value, whereas changing the amino acid but maintaining the same charge does not alter pK 2 appreciably (e.g., for D69E enzyme pK 2 is 8.48, while for R310K enzyme, pK 2 is 8.24, similar to that of a wild-type enzyme). Since the amino acid at position 69 is close to His68 in the homology model, pK 2 must reflect the effect on the ionization of His68 resulting from the charge at position 69.
Figure 3.

pH-Vmax profiles of wild-type (WT) and mutant adenylosuccinate lyase enzymes. The pH dependence of Vmax of WT (black), D69E (red), and D69N (green). Note the different scales for Vmax for the three enzyme samples.
Table 3.
pK values determined from pH-Vmax profiles of wild-type and mutant adenylosuccinate lyase enzymes

Discussion
This study seeks to identify an amino acid whose ionization is reflected in pK 2 of the wild-type pH-Vmax profile. We constructed, expressed, purified, and characterized enzymes with site-directed mutations of residues Asp69 and Arg310 of the B. subtilis ASL to test their effect on pK2. Sequence alignment revealed that amino acid residues Asp69 and Arg310 are conserved in all species and are close to or interact with His68, according to the homology model.
The Thermatoga maritima enzyme was the first ASL crystal structure to be published (Toth and Yeates 2000). The B. subtilis and T. maritima enzymes share 50% identity plus 23% similarity at the amino acid sequence level. Based on the strong resemblance of the amino acid sequences of the two enzymes, the homology model of B. subtilis ASL (which is based on the crystal structure of T. maritima enzyme) is likely to be a reasonable representation of the actual structure (Segall and Colman 2004). We have previously postulated that the protonated form of His68 acts as a general acid in the catalytic reaction and/or is involved in an electrostatic interaction with a carboxylate of SAMP to facilitate the binding of substrate (Segall and Colman 2004). Recently, Tsai et al. have determined the structure of an inactive mutant of E. coli ASL with bound SAMP and they found that the histidine equivalent to His68 in the B. subtilis enzyme is actually closer to the carboxylate of SAMP (Tsai et al., in press).
In the homology model of the B. subtilis ASL in Figure 4, His68 is 3.6 Å from the carboxylate of Asp69, and a proximal negative charge would raise the pK of the protonated form of histidine. A similar strategy was used for acetoacetate decarboxylase; in that case, the pK of the active site Lys115 was markedly changed by mutating the amino acid at position 116 (Highbarger et al. 1996). Also shown in Figure 4 for ASL, Asp69 is 3.6 Å from the guanidino group of Arg310. An electrostatic interaction between the negatively charged carboxylate of Asp and the positively charged Arg could properly orient Asp69 to interact with His68.
Figure 4.
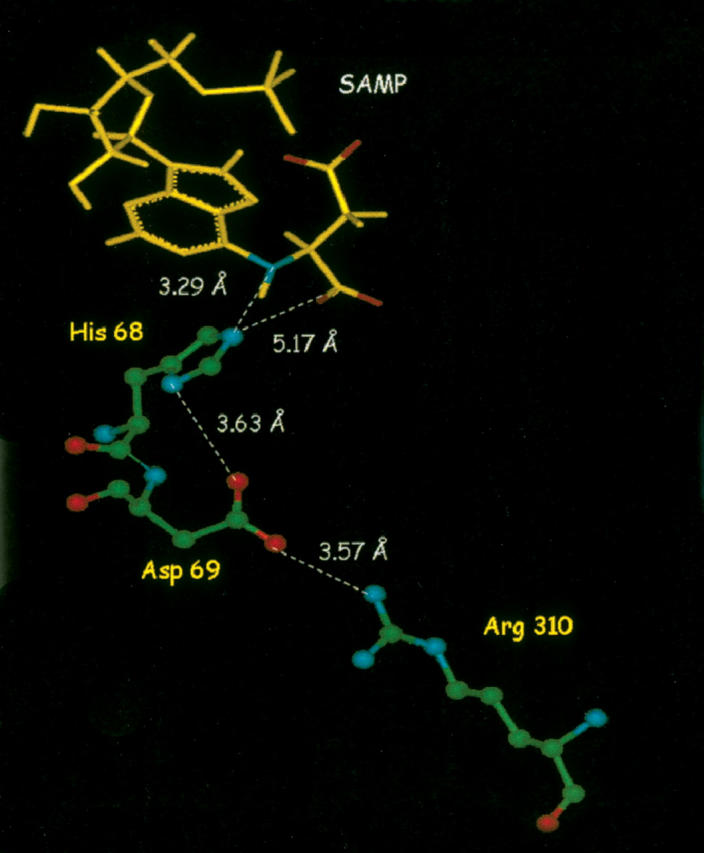
Homology model of B. subtilis adenylosuccinate lyase SAMP (yellow). Relevant active-site amino acids are colored according to the atom (green = carbon; red = oxygen; blue = nitrogen) and shown in ball and stick format.
The kinetic data show that all mutant enzymes display greatly decreased specific activities, especially in the cases where charged amino acids were replaced with neutral residues, suggesting that Asp69 and Arg310 are important for catalytic function. The biophysical characteristics of these mutant enzymes are not appreciably different from those of wild-type enzyme in their secondary or quaternary structure. The Asp69-to-Asn mutant enzyme has a 50-fold lower activity and Arg310-to-Gln has a 2500-fold lower activity. Removing the charge from Asp69 by replacing it with Asn also causes a fourfold increase in K m; and replacing positively charged Arg310 by neutral Gln causes an 11-fold increase in the K m, indicating that there is weakened affinity between the enzyme and substrate.
The pH-Vmax profiles of the Asp69 mutant enzymes show that when the same negative charge is maintained (as in D69E), pK 2 is similar to that of wild-type enzyme, with a pK 2 value of 8.48, and His68 is maintained in a positively charged form. The small increase in pK 2 in D69E may be due to the negatively charged COO− of Glu69 being closer than that of Asp69 to His68. When the negative charge is removed (D69N), the positively charged form of His68 is not stabilized and the pK 2 decreases, as seen by the pK 2 value of 7.83. In the pH-Vmax profile of the Arg310 mutant enzyme, in which the positive charge is maintained (R310K), the electrostatic interaction with Asp69 remains, and the pK 2 (8.24) is high, as in wild-type enzyme. In contrast, when the positive charge is replaced by a neutral amino acid, as in R310Q, there is no attraction between Asp69 and Gln310; Asp69 may not be positioned to interact with His68 and the pK 2, representing the ionization of His68, decreases to 7.33. These data indicate that His68 must be maintained as a positively charged species for the enzyme to be fully active. In D69N and R310Q, the pK 2 is likely decreased because of their effects on His68 ionization.
The right-hand limb of the pH-Vmax profile has been suggested to reflect the ionization of both His68 and His89 (Brosius and Colman 2000). When His68 is replaced by a neutral amino acid, as in H68Q, the pH-Vmax profile is determined by the ionization of His89 (Lee et al. 1999). For H68Q, although the specific activity is <1% that of wild-type enzyme (Lee et al. 1998), the shape of the pH-Vmax curve is similar to that of the normal enzyme (Lee et al. 1999); this result can be interpreted as indicating that the ionization of His89 is unperturbed in the His68 mutant. Reciprocally, when His89 is replaced by a neutral acid, as in H89Q, the pH-Vmax curve reflects the ionization of only His68 (Brosius and Colman 2000). For H89Q, the specific activity is also only 1% that of wild-type enzyme; but, (in contrast to H68Q) pK2 of H89Q is decreased appreciably (Brosius and Colman 2000). His89 is considered to be involved in binding the AMP moiety of the substrate and in orienting the substrate for catalysis. If the binding of substrate is altered within the active site, the ionization of His68 in the enzyme–substrate complex may be modified indirectly.
The wild-type ASL enzyme pH-rate profile in the reverse reaction, using AMP and fumarate as substrates, gives pK 1 and pK 2 values of 6.53 ± 0.05 and 9.19 ± 0.07, respectively (Palenchar 2003). Heats of ionization studies of the wild-type ASL enzyme show that the pK 1 value reflects the ionization in the enzyme–substrate complex of a neutral acid such as a carboxyl group or phosphoric acid, due to the ▵Hi value of 0 kcal/mol (Brosius and Colman 2000). Therefore, to determine whether the pK 1 is reflecting the ionization of the phosphate group of the substrate, adenosine 5′-O-thiomonophosphate (AMPS) with a pK of ∼5.4 was used as substrate (Frey 1989). Since the pK value of a typical phosphate group is ∼6.8, if the ionization of a phosphoryl group is represented in pK 1, a substantial shift in pK 1 will be observed when using AMPS as substrate. The pK 1 and pK 2 values with AMPS and fumarate are 6.58 ± 0.07 and 8.91 ± 0.08, respectively (Palenchar 2003). Since there was no significant difference in the pK 1 value when using AMP or AMPS as substrates, the pK 1 of the pH-rate profile cannot be reflective of the dissociation of a proton from the substrate's phosphoryl moiety. Instead, the pK 1 value may represent the deprotonation of COO− groups from the succinyl portion of enzyme-bound SAMP and the increase in pK 1 in D69E and R310Q may reflect changes in the orientation of SAMP as a result of the mutations introduced.
Recently we reported that in the S94A mutant of ASL, perturbation of the interaction of Ser94 with His89 causes a decrease in pK2 of the pH-Vmax profile (Segall et al. 2007). In addition, we have preliminary evidence that replacement of another amino acid close to His89 results in a decrease in pK2. Glu27 is only 4.1 Å from the side chain of His89, while it is 12.1 Å and 16.3 Å from His68 and His141, respectively. The E27Q mutant, in which the neutral Gln replaces the negatively charged Glu, exhibits a pK2 of the pH-Vmax curve that is 0.4 lower than in the wild-type enzyme. In the present study we focused on Asp69, which is 3.6 Å from His68, 9.3 Å from His89, and 10.7 Å from His141. Thus, the changes seen in the pH-Vmax profile due to the mutations introduced at Asp69 and Arg310 provide the first experimental demonstration that His68 is also a residue reflected in pK2 of the pH-Vmax profile.
In conclusion, this study indicates that mutations introduced at positions 69 and 310 of the B. subtilis ASL enzyme affect the catalytic activity and affinity for substrate, especially when the charge is changed. Also, the shift seen in the pK 2 of the pH-Vmax profile with these mutations indicates that pK2 reflects the ionization of His68, since Asp69 is close to His68 (but not to His89 or His141) in the structure. His68 has been proposed to act as the general acid in the catalytic reaction and it may also be involved in an electrostatic interaction with a carboxylate of the adenylosuccinate to facilitate the binding of substrate.
Materials and Methods
Materials
SAMP, HEPES, 2-(N-morpholino)ethanesulfonic acid (MES), N-tris-(hydroxymethyl)methyl-3-aminopropanesulfonic acid (TAPS), and imidazole were purchased from Sigma. Ni-NTA resin was purchased from Qiagen. Oligonucleotides for both sequencing and site-directed mutagenesis were obtained from Biosynthesis. All other chemicals were of reagent grade.
Site-directed mutagenesis
Mutations to the pBHis plasmid (a gift from Dr. Jack E. Dixon, University of California, San Diego) that encodes ASL of B. subtilis were constructed using the Stratagene QuikChange mutagenesis kit. The following oligonucleotides and their complements were used to generate the Asp69 mutant enzymes: CACGCGCCATAACGTTGTCGCTTTTAC (D69N) and CACGCGCCATGAAGTTGTCGCTTTTAC (D69E). The Arg310 mutant enzymes were constructed using the following oligonucleotides and their complements: CTTCAGCAGAAAAAATTATTCTTCCGGATG (R310K) and CTTCAGCAGAACAGATTATTCTTCCGG (R310Q). In each case, the mutated codon is underlined. The mutations were confirmed by DNA sequencing carried out at the University of Delaware Center for Agricultural Biotechnology using an ABI Prism model 377 DNA sequencer (PE Biosystems).
The pBHis plasmid, which encodes a His6 tag on the N terminus of ASL, was expressed in E. coli strain BL21 (DE3), and the enzymes were purified to homogeneity using Qiagen nickel nitrilotriacetic acid–agarose (Redinbo et al. 1996; Lee et al. 1997). The purity of the wild-type and mutant enzymes was evaluated by 12% polyacrylamide gels containing 0.1% sodium dodecyl sulfate (Laemmli 1970), as well as by N-terminal sequencing. The protein concentrations were determined by the absorbance at 280 nm using E 1% 280 nm = 10.6 (Lee et al. 1997). The purified enzymes were stored at −80°C in 20 mM potassium phosphate containing 20 mM sodium chloride at pH 7.
Kinetics of B. subtilis ASL
The wild-type and mutant enzyme activity toward SAMP was measured by the time-dependent decrease in absorbance at 282 nm using the difference extinction coefficient of 10,000/M/cm between SAMP and AMP. The enzymes were incubated at a minimum concentration of 0.4 mg/mL in 20 mM potassium phosphate containing 20 mM sodium chloride at pH 7 for 30 min at 25°C before activity measurement (Palenchar and Colman 2003). Standard assay conditions of 50 mM HEPES at pH 7.0 at 25°C with 60 μM SAMP were used for the determination of specific activity, expressed as μmol of SAMP converted/min/mg of enzyme used. The K m for the wild-type and mutant enzymes was determined by varying the substrate concentration under the same conditions. The data were analyzed by ν versus [S] with standard error estimates obtained from the Sigma Plot software (SPSS Inc.).
The pH dependence of Vmax was determined for wild-type and mutant enzymes using 300 μM SAMP, from pH 6.0–pH 9.3, at 25°C using the buffers MES (pH 6.0–6.9), HEPES (pH 6.8 – pH 8.0), and TAPS (pH 7.9–9.3) with a constant ionic strength of 0.03 M in all buffers. The rates were measured at 290 nm for these experiments, using a difference extinction coefficient of 4050/M/cm to calculate the rates. The pH of each assay mixture was measured after the rate determination and the data were analyzed by Sigma Plot software. The spectra of SAMP and AMP do not change over this pH range (Hampton 1962) so the same difference extinction coefficient could be used to calculate the rates.
The pH dependence of Vmax was also determined in the direction of SAMP formation for wild-type ASL enzyme using 1 mM AMP and 10 mM fumarate as substrates. The same MES, HEPES, and TAPS buffers were used from pH 6.0–pH 8.5 at 25°C, and the rates were measured at 290 nm because of the high absorbance at 282 nm. The difference extinction coefficient of 4050/M/cm was used to calculate the rates. The wild-type ASL enzyme pH-Vmax profile in the direction of SAMP formation was also determined using 1 mM AMPS, under the same conditions as for AMP.
CD spectroscopy
A Jasco J-710 spectropolarimeter was used to measure ellipticity as a function of wavelength from 250 to 200 nm in 0.2-nm increments using a 0.1-cm cylindrical quartz cuvette. The enzymes were incubated at 0.4 mg/mL for 30 min at 25°C before measuring the spectra. The samples were scanned five times and averaged, and the spectrum of the buffer, containing 20 mM potassium phosphate and 20 mM sodium chloride at pH 7, was subtracted. The final protein concentration was determined by a dye-binding assay (Bio-Rad protein assay), based on the method of Bradford (1976), using a Bio-Rad 2550 RIA plate reader with a 600-nm filter. Wild-type ASL was used as the protein standard. The mean residue ellipticity [θ] (deg cm2/dmol) was calculated from the equation [θ] = θ/10 nCl, where θ is the measured ellipticity in millidegrees, C is the molar concentration of the enzyme subunits, l is the path length in centimeters, and n is the number of residues per subunit (437, including the His6 tag).
Molecular weight determination by light scattering
A minDAWN laser photometer (Wyatt Technology Corp.) set at a laser wavelength of 690 nm was used to determine the wild-type and mutant enzyme molecular weights. Molecular weight was measured at a protein concentration range of 0.1–0.4 mg/mL. The enzyme was preincubated at 25°C in 20 mM potassium phosphate containing 20 mM sodium chloride at pH 7, and filtered through a 0.20-μm filter before being used. The data were collected and analyzed using ASTRA software for windows (Wyatt 1993). The protein concentration was determined using the Bio-Rad protein assay (Bradford 1976).
Molecular weight determination of R310Q by native polyacrylamide gel electrophoresis
For wild-type and R310Q mutant enzyme, the molecular weight was determined by native polyacrylamide gel electrophoresis, by the method of Hedrick and Smith (1968). Gels were prepared consisting of 20 mM potassium phosphate at pH 7, 0.12% (v/v) N,N,N′,N′-tetramethylethylenediamine, 0.2 mg/mL ammonium persulfate, and appropriate volumes of distilled water and 37.5:1 acrylamide/bisacrylamide mixture to yield final acrylamide concentrations of 5%–9%. All samples and standards were prepared in potassium phosphate at pH 7 and were run at 25°C and 25 mA; the running buffer was also 20 mM potassium phosphate at pH 7.0.
Homology model of B. subtilis ASL
A homology model of the B. subtilis ASL, based on the crystal structure of the T. maritima enzyme (Toth and Yeates 2000), has been constructed (Brosius and Colman 2002; Segall and Colman 2004). The homology model was used to study the amino acid substitutions with energy minimization computational results obtained using software programs from Biosym Technologies (dynamics calculations from Discover and graphical display using Insight II) on an Indigo 2 workstation from Silicon Graphics.
Electronic supplemental material
The Electronic supplemental material contains two figures. Supplemental Figure S1 illustrates the single band of representative purified enzymes on SDS-PAGE. Supplemental Figure S2 shows the circular dichroism spectra of wild-type and mutant enzymes.
Acknowledgment
This work was supported by NIH grant RO1-DK60504.
Footnotes
Supplemental material: see www.proteinscience.org
Reprint requests to: Roberta F. Colman, Department of Chemistry and Biochemistry, University of Delaware, Newark, DE 19716, USA; e-mail: rfcolman@chem.udel.edu; fax: (302) 831-6335.
Abbreviations: ASL, adenylosuccinate lyase; SAMP, adenylosuccinate; AMP, adenosine monophosphate; AMPS, adenosine 5′-O-thiomonophosphate; HEPES, N-(2-hydroxyethyl)piperazine-N’-2-ethanesulfonic acid; MES, 2-(N-morpholino)ethanesulfonic acid; TAPS, N-tris-(hydroxymethyl)methyl-3-aminopropanesulfonic acid; CD, circular dichroism; B. subtilis, Bacillus subtilis; T. maritima, Thermatoga maritima
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.072927207.
References
- Bradford M.M. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72: 248–254. [DOI] [PubMed] [Google Scholar]
- Bridger W.A. and Cohen, L.H. 1968. The kinetics of adenylosuccinate lyase. J. Biol. Chem. 243: 644–650. [PubMed] [Google Scholar]
- Brosius J.L. and Colman, R.F. 2000. A key role in catalysis for His89 of adenylosuccinate lyase of Bacillus subtilis . Biochemistry 39: 13336–13343. [DOI] [PubMed] [Google Scholar]
- Brosius J.L. and Colman, R.F. 2002. Three subunits contribute amino acids to the active site of tetrameric adenylosuccinate lyase: Lys268 and Glu275 are required. Biochemistry 41: 2217–2226. [DOI] [PubMed] [Google Scholar]
- Frey P.A. 1989. Chiral phosphorothioates: Stereochemical analysis of enzymatic substitution at phosphorous. Adv. Enzymol. Relat. Areas Mol. Biol. 62: 119–201. [DOI] [PubMed] [Google Scholar]
- Garrett H.R. and Grisham, C.M. 1999. Amino acids. In Biochemistry, 2nd ed, pp. 81–105. Saunders College Publishing, Philadelphia, PA.
- Hampton A. 1962. Studies of the action of adenylosuccinase with 6-thio analogues of adenylosuccinic acid. J. Biol. Chem. 237: 529–535. [PubMed] [Google Scholar]
- Hanson K.R. and Havir, E.A. 1972. The enzymatic elimination of ammonia. In The enzymes, 3rd ed. (ed. P.D. Boyer). Vol. Vol. 7, pp. 75–166. Academic Press, New York. [Google Scholar]
- Hedrick J.L. and Smith, A.J. 1968. Size and charge isomer separation and estimation of molecular weights of proteins by disc gel electrophoresis. Arch. Biochem. Biophys. 126: 155–164. [DOI] [PubMed] [Google Scholar]
- Highbarger L.A., Gerlt, J.A., and Kenyon, G.L. 1996. Mechanism of the reaction catalyzed by acetoacetate decarboxylase. Importance of lysine 116 in determining the pK a of active-site lysine 115. Biochemistry 35: 41–46. [DOI] [PubMed] [Google Scholar]
- Jaeken J. and Van den Berghe, G. 1984. An infantile autistic syndrome characterized by the presence of succinylpurines in body fluids. Lancet 2: 1058–1061. [PubMed] [Google Scholar]
- Laemmli U.K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227: 680–685. [DOI] [PubMed] [Google Scholar]
- Lee T.T., Worby, C., Dixon, J.E., and Colman, R.F. 1997. Identification of His141 in the active site of Bacillus subtilis adenylosuccinate lyase by affinity labeling with 6-(4-bromo-2,3-dioxobutyl)thioadenosine 5′-monophosphate. J. Biol. Chem. 272: 458–465. [PubMed] [Google Scholar]
- Lee T.T., Worby, C., Bao, Z.-Q., Dixon, J.E., and Colman, R.F. 1998. Implication of His68 in the substrate site of Bacillus subtilis adenylosuccinate lyase by mutagenesis and affinity labeling with 2-[(4-bromo-2,3-dioxobutyl)thiol]adenosine 5′-monophosphate. Biochemistry 37: 8481–8489. [DOI] [PubMed] [Google Scholar]
- Lee T.T., Worby, C., Bao, Z.Q., Dixon, J.E., and Colman, R.F. 1999. His68 and His141 are critical contributors to the intersubunit catalytic site of adenylosuccinate lyase of Bacillus subtilis . Biochemistry 38: 22–32. [DOI] [PubMed] [Google Scholar]
- Palenchar J.B. 2003, University of Delaware, Newark.
- Palenchar J.B. and Colman, R.F. 2003. Characterization of a mutant Bacillus subtilis adenylosuccinate lyase equivalent to a mutant enzyme found in human adenylosuccinate lyase deficiency: Asparagine 276 plays an important structural role. Biochemistry 42: 1831–1841. [DOI] [PubMed] [Google Scholar]
- Ratner S. 1972. Argininosuccinases and adenylosuccinases. In The enzymes, 3rd ed. (ed. P.D. Boyer). Vol. Vol. 7, pp. 167–197. Academic Press, New York. [Google Scholar]
- Redinbo M.R., Eide, S.M., Stone, R.L., Dixon, J.E., and Yates, T.O. 1996. Crystallization and preliminary analysis of Bacillus subtilis adenylosuccinate lyase, an enzyme implicated in infantile autism. Protein Sci. 5: 786–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segall M. and Colman, R.F. 2004. Gln212, Asn270, and Arg301 are critical for catalysis by adenylosuccinate lyase from Bacillus subtilis . Biochemistry 43: 7391–7402. [DOI] [PubMed] [Google Scholar]
- Segall M.L., Cashman, M.A., and Colman, R.F. 2007. Important roles of hydroxylic amino acid residues in the function of Bacillus subtilis adenylosuccinate lyase. Protein Sci. 16: 441–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivendran S., Segall, M.L., Rancy, P.C., and Colman, R.F. 2005. Effect of Asp69 and R310 on the pK of His68, a key catalytic residue of adenylosuccinate lyase (ASL). FASEB J. 39: A307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toth E.A. and Yeates, T.O. 2000. The structure of adenylosuccinate lyase, an enzyme with dual activity in the de novo purine biosynthetic pathway. Structure 8: 163–174. [DOI] [PubMed] [Google Scholar]
- Tsai M., Koo, J., Yip, P., Colman, R.F., Segall, M.L., and Howell, P.L. 2007. Substrate and product complexes of E. coli adenylosuccinate lyase provide new insights into the enzymatic mechanism. J. Mol. Biol. (in press) doi: 10.1016/j.jmb.2007.04.052. [DOI] [PMC free article] [PubMed]
- Van den Berghe G. and Jaeken, J. 2001. Adenylosuccinate lyase deficiency. In The metabolic and molecular basis of inherited diseases, 8th ed. (eds. C.R. Scriver et al.). Vol. Vol. II. McGraw-Hill, New York.
- Wyatt P.J. 1993. Light scattering and the absolute characterization of macromolecules. Anal. Chim. Acta 272: 1–40. [Google Scholar]