

Abstract
The frontline tuberculosis drug isoniazid (INH) inhibits InhA, the NADH-dependent fatty acid biosynthesis (FAS-II) enoyl reductase from Mycobacterium tuberculosis (MTB), via formation of a covalent adduct with NAD+ (the INH-NAD adduct). Resistance to INH can be correlated with many mutations in MTB, some of which are localized in the InhA cofactor binding site. While the InhA mutations cause a substantial decrease in the affinity of InhA for NADH, surprisingly the same mutations result in only a small impact on binding of the INH-NAD adduct. Based on the knowledge that InhA interacts in vivo with other components of the FAS-II pathway, we have initiated experiments to determine whether enzyme inhibition results in structural changes that could affect protein–protein interactions involving InhA and how these ligand-induced conformational changes are modulated in the InhA mutants. Significantly, while NADH binding to wild-type InhA is hyperbolic, the InhA mutants bind the cofactor with positive cooperativity, suggesting that the mutations permit access to a second conformational state of the protein. While cross-linking studies indicate that enzyme inhibition causes dissociation of the InhA tetramer into dimers, analytical ultracentrifugation and size exclusion chromatography reveal that ligand binding causes a conformational change in the protein that prevents cross-linking across one of the dimer–dimer interfaces in the InhA tetramer. Interestingly, a similar ligand-induced conformational change is also observed for the InhA mutants, indicating that the mutations modulate communication between the subunits without affecting the two conformational states of the protein that are present.
Keywords: enoyl reductase, InhA, fatty acid synthesis, isoniazid, Mycobacterium tuberculosis
Mycobacterium tuberculosis (MTB), the etiological agent of the pulmonary disease tuberculosis (TB), is an intracellular human pathogen that has afflicted humankind for thousands of years. Currently, TB is the most common cause of death in patients with HIV, and it is estimated that one-third of the world's population has been infected with MTB. Drug-susceptible strains of MTB can be treated with a cocktail of inhibitors, including isoniazid (INH), rifampicin (RIF), ethambutol (ETH), and pyrazinamide (PZA). However, the escalating numbers of patients infected with multi-drug-resistant (MDR) mycobacterial strains poses a substantial public health risk (Heymann 2006).
Although INH has been the gold standard in anti-TB chemotherapy since its discovery in 1952, the CDC estimates that over 7% of new cases in the United States are resistant to this drug (Middlebrook 1952; World Health Organization 2003). INH resistance arises primarily from mutations in katG, which codes for a mycobacterial catalase-peroxidase enzyme that is responsible for drug activation (Zhang et al. 1992). While the ultimate target of activated INH has been the subject of much debate, it is clear that INH inhibits mycolic acid biosynthesis via effects on the fatty acid biosynthesis (FAS-II) pathway, leading to an accumulation of saturated hexacosanoic acid and cell lysis (Takayama et al. 1975; Vilcheze et al. 2000). Following activation, INH forms an adduct with NAD(H), and it is this adduct that inhibits InhA, the enoyl reductase in the FAS-II pathway (Zhang et al. 1992; Quemard et al. 1995). While mutations in inhA correlate with INH resistance, there is also evidence that INH forms a complex with KasA, the FAS-II β-ketoacyl synthase condensing enzyme (Banerjee et al. 1994; Mdluli et al. 1996, 1998; Marrakchi et al. 2000; Slayden et al. 2000; Vilcheze et al. 2000; Larsen et al. 2002; Kremer et al. 2003). In addition, it has now been shown that a second INH-NADP adduct can inhibit other mycobacterial enzymes such as dihydrofolate reductase (Argyrou et al. 2006). Thus, it is likely that the in vivo effects of INH are complex. Since INH resistance arises primarily from a dysfunction in INH activation, compounds that inhibit the target(s) of INH but do not require activation are likely to be active against drug-resistant MTB. Consequently, understanding the exact mechanism of INH action is of critical importance.
While the majority of INH-resistant clinical isolates have mutations in katG, polymorphisms are also observed in inhA and kasA, supporting the proposals that these enzymes are targets for INH (Zhang et al. 1992; Dessen et al. 1995; Basso et al. 1998). The InhA mutations, such as Ile21Val, Ile47Thr, and Ser94Ala, cluster in and around the cofactor binding site, resulting in a large (20–160-fold) reduction in affinity of NADH for the enzyme (Dessen et al. 1995; Basso et al. 1998; Vilcheze et al. 2006). However, inhibition experiments with the INH-NAD adduct, which is a slow onset inhibitor of wild-type InhA with an overall Ki value of 0.75 nM, demonstrate that the InhA mutations have only a small effect on adduct affinity, thus questioning the relevance of the mutations to INH resistance. Given that the inhibition assays were performed on the isolated enzyme, we speculated that interactions between InhA and other proteins in the Mycobacterium might be critical for modulating the activity and inhibition of the enzyme. Indeed, there is increasing evidence that components of the FAS-II pathway associate with each other in vivo. Bloch and coworkers isolated a high MW fraction from Mycobacterium smegmatis cell lysate with FAS-II activity (Odriozola et al. 1977) that was subsequently shown to contain the FAS-II components InhA and MabA (Marrakchi et al. 2000, 2002). More recently, two-hybrid experiments have provided evidence for the existence of both heterotypic and homotypic protein–protein interactions within the members of the FAS-II pathway (Veyron-Churlet et al. 2004, 2005).
In order to determine whether the clinical inhA mutations affect InhA inhibition within the context of a multienzyme complex, we need to identify the interactions between the FAS-II components and determine how these interactions are affected by mutagenesis and/or enzyme inhibition. Based on the knowledge that the interaction of MabA, the FAS-II ketoreductase, with other FAS-II enzymes was altered when tetrameric MabA was converted to a dimeric species, we set out to determine whether the oligomerization state of InhA was affected by enzyme inhibition (Veyron-Churlet et al. 2004). In addition, we have also probed the effect of mutagenesis on both the oligomerization state of the enzyme and, using bacterial-two-hybrid methods, on the ability of InhA to interact with KasA. While initial cross-linking experiments raised the tantalizing possibility that inhibition resulted in dissociation of the InhA tetramer into dimers, more detailed studies, using analytical ultracentrifugation and size exclusion chromatography, now reveal that inhibition causes a conformational change in the enzyme. The relevance of these observations to INH drug resistance is discussed.
Results
Bacteria 2-hybrid
Interactions between wild-type InhA and KasA
In order to probe interactions between members of the FAS-II pathway, the kasA gene was cloned into the bait vector, pBT (KasA-pBT), and the inhA gene was cloned into the target vector, pTRG (InhA-pTRG). Each of these vectors was cotransformed with either an empty target or bait vector, respectively, to ensure that the plasmids did not falsely activate transcription. When screened on selective media, none of the plasmids constructed autoactivated either the HIS3 or aadA reporter genes. A separate positive and negative control was performed utilizing the pBT-LGF2 and either the pTRG-Gal11P or an empty pTRG vector. All controls yielded the expected results. Following cotransformation into the Escherichia coli reporter strain, a derivative of XL-1 Blue cells, with KasA-pBT and InhA-pTRG, numerous colonies developed on selective and dual selective media, indicating that the fusion proteins produced by the two vectors interacted. While tetracycline and chloramphenicol resistance are encoded by the plasmids pTRG and pBT, respectively, streptomycin resistance, as well as the ability to utilize 3-AT, is indicative of activation of the reporter gene from a positive protein–protein interaction. Colonies from this interaction were confirmed by PCR and sequencing to check for the presence of both vectors.
Interactions between INH resistant InhA and KasA
The InhA-pTRG plasmids were mutated to encode Ile21Val, Ile47Thr, or Ser94Ala, in order to determine if the InhA mutations observed in INH-resistant clinical MTB isolates affected the ability of InhA to interact with KasA. Each of the mutants was screened with the KasA-pBT plasmid by cotransformation and evaluated as described above. All three mutant forms of InhA retained their interaction with KasA, with growth of colonies on selective and dual selective media comparable to that of wild-type InhA. The number and size of the colonies formed by the InhA mutants was similar to that of wild-type InhA, suggesting that the strength of the InhA-KasA interaction had not been affected by the mutations.
Cross-linking of InhA
Wild-type InhA
Using the chemical cross-linker bis(sulfosuccinimidyl)suberate (BS3), we cross-linked the wild-type InhA protein. Analysis by denaturing SDS-PAGE resulted in the detection of equal amounts of dimeric (61.34 kDa) and tetrameric (122.68 kDa) protein, with a less intense band of uncross-linked monomer (30.67 kDa) (Fig. 1). Our results are in agreement with previous reports that InhA is a tetramer (Rozwarski et al. 1999; Marrakchi et al. 2002). To evaluate the effect of INH on the oligomerization of InhA, the cross-linking experiment was repeated with INH-inhibited InhA. The inhibition was performed with the addition of NADH, MnCl2, and INH, to form the inhibitory INH-NAD adduct (Rawat et al. 2003). Controls lacking either INH or INH/MnCl2 were also performed. In contrast to the uncomplexed wild-type InhA, cross-linking of InhA inhibited with the INH-NAD adduct or bound to NADH cofactor with BS3 yielded only monomer and dimer bands on SDS-PAGE (Fig. 1).
Figure 1.

Cross-linking of wild-type and mutant InhA proteins. Each type of InhA is represented in four lanes, all in the presence of the BS3 cross-linker. (Lane 1) Purified recombinant proteins, all displaying the characteristic monomer, dimer, and tetramer bands. (Lane 2) Proteins in the presence of excess NADH. Wild-type InhA, which has a high affinity for NADH, shows a disappearance of the tetramer band in the presence of cofactor. In contrast all three mutants have a decreased affinity for NADH and retain the tetrameric form. (Lane 3) Proteins in the presence of excess NADH and MnCl2. The presence of MnCl2 does not alter the results found in lane 2. (Lane 4) Proteins under inhibiting conditions, containing NADH, MnCl2, and INH. MnCl2 is used in place of KatG to activate INH. All four proteins have similar affinity for the inhibitory adduct and upon binding, the tetrameric form of the protein can no longer be observed.
MALDI analysis of cross-linking
In an attempt to determine the location of the BS3 cross-links, unreacted wild-type InhA as well as dimeric and tetrameric forms of the cross-linked protein were isolated from an SDS-PAGE gel by digestion with trypsin and Glu-C and subjected to MALDI analysis. In general, the yield of the cross-linked fragments isolated from the SDS-PAGE was too low to enable cross-links across interface B in the tetramer (Fig. 2B) from being identified by this method (data not shown). However, we were able to obtain evidence for the formation of a cross-link across interface A between residues Lys8 and Lys240. These residues are within 11 Å of each other in the X-ray structure and thus potentially within the range that could be covalently cross-linked by BS3 (Fig. 2A). Analysis with the Automatic Spectrum Assignment Program (ASAP) revealed a peptide with a mass of 2081 in both the dimeric and tetrameric sample, representing the Lys8-BS3-Lys240 species (Fig. 3, inset). The mass spectrum in Figure 3 also contains intense peaks with m/z values of 1731, 1913, and 2187, consistent with the masses of non-lysine-containing peptides that were unaffected by the cross-linking.
Figure 2.
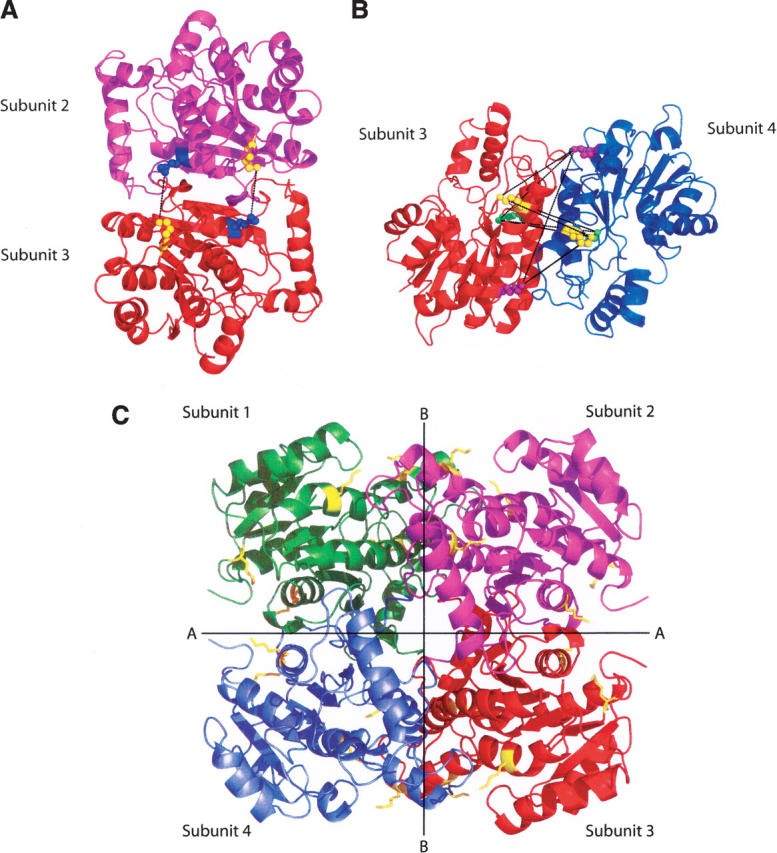
Interactions across the dimer–dimer interfaces in InhA. (A) Dimer formed by subunits 2 and 3 (1BVR.pdb), rotated 90° to clearly show the cross-links, in black, formed between Lys240 (blue spheres) and Lys8 (yellow spheres), across interface A. The two lysine pairs are separated by 9.86 Å and 9.89 Å, respectively. (B) Dimer formed by subunits 3 and 4 (1BVR.pdb), rotated to show all nine possible cross-links (in black) across interface B. The three lysines, 118, 132, and 165, in each monomer are highlighted in yellow, purple, and green. Distances between pairs of lysine residues from subunit 3 to subunit 4 range from 18.58 Å to 32.25 Å. In this ligand-bound structure, none of the lysine pairs are close enough to be cross-linked by BS3. (C) The InhA tetramer, taken from the structure of InhA bound to a C16 substrate and NAD+ (1BVR.pdb). The subunits are labeled 1–4, and the two distinct dimer–dimer interfaces, A and B, are shown. All lysine residues are shown in yellow.
Figure 3.

MALDI-MS of InhA. Representative MALDI spectra of the trypsin/Glu-C double digestion reaction of InhA in the absence (top) and in the presence of the BS3 cross-linker (bottom). Intermolecular cross-linked peptide masses were assigned using the Automatic Spectrum Assignment Program (ASAP) (http://roswell.ca.sandia.gov/∼mmyoung/asap.html). InhA peptide masses are designated above each peak; examples include trypsin fragments 1731 m/z (amino acids 28–43), 1913 m/z (amino acids 10–27), and 2186 m/z (amino acids 136–153). The cross-linked peptides [GK(8)R]-BS3-[DATPVAK(240)TVCALLSD] are designated with a circle (2081 m/z) and represent the cross-link that appears in both the dimer and tetramer (data not shown) band, but not in the uncross-linked sample (see insets).
INH resistant InhA mutants
Similar to wild-type InhA, cross-linking of the three INH resistant mutants, Ile21Val, Ile47Thr, or Ser94Ala, gave three bands on SDS-PAGE assigned to monomer, dimer, and tetrameric forms of the protein (Fig. 1). In each case, the inhibition of the enzyme with the INH-NAD adduct resulted in loss of the band assigned to tetramer. In contrast, cross-linking in the presence of saturating concentrations of NADH yielded a lower amount of the tetramer upon SDS-PAGE (Fig. 1). These data are consistent with the knowledge that the three mutants have a decreased affinity for the cofactor despite being able to bind the INH-NAD adduct with similar affinity to wild-type InhA (Basso et al. 1998; Rawat et al. 2003).
Lysine mutants
InhA contains nine lysine residues, three of which, Lys118, Lys132, and Lys165, can potentially be involved in cross-links across interface B in the InhA tetramer (Fig. 2B). The replacement of each of these lysines individually with arginine had no effect on cross-linking in the unliganded enzyme, which is seen as the retention of the tetramer (Fig. 4A). However, following reaction with BS3 the amount of tetramer was only substantially reduced for the Lys118Arg/Lys165Arg double mutant (Fig. 4B), providing evidence that multiple cross-links can form at interface B (Fig. 2B). This indicates that an 11 Å cross-link must be able to form between the Lys118–Lys165 or a combination of cross-links via Lys118–Lys132 and Lys165–132 residues across the B interface in the unliganded protein. Since these residues are separated by 26 Å or 23 Å and 19 Å, respectively, in crystal structures of InhA, ligand binding must cause a substantial movement of the two dimers with respect to each other.
Figure 4.
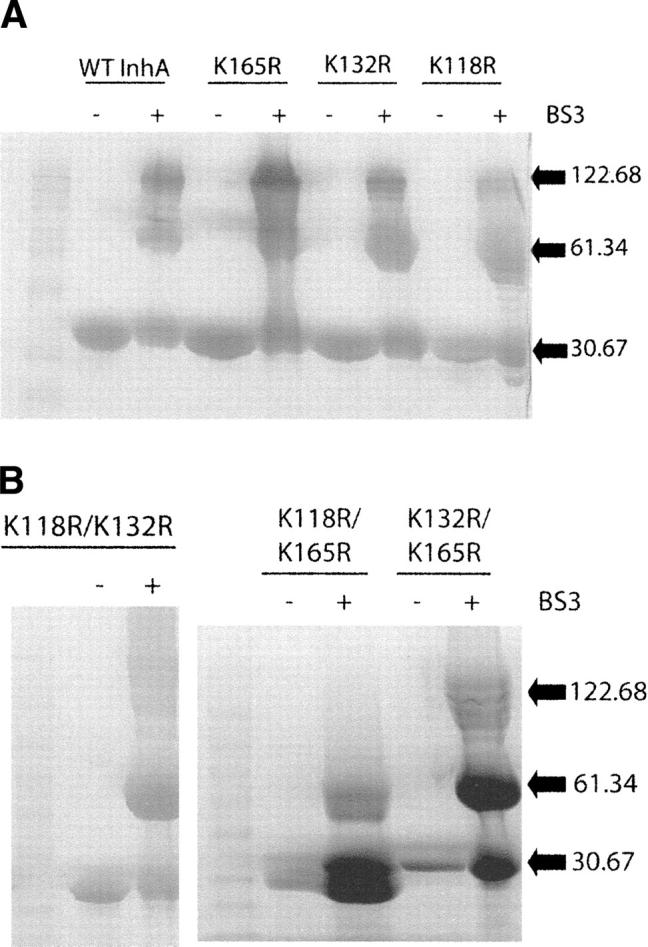
SDS-PAGE of cross-linked wild-type InhA and the interface B lysine mutants. (A) SDS-PAGE of wild-type InhA and three lysine mutants, Lys118Arg, Lys132Arg, and Lys165Arg, after cross-linking with BS3. The three lysines are thought to form cross-links over interface B. The wild-type InhA and all single mutants appear identical, giving monomers (30.7 kDa) in the absence of cross-linker (−), and monomers, dimers (61.3 kDa), and tetramers (122.7 kDa) in the presence of cross-linker (+). (B) Cross-linking of the Lys118Arg/Lys132Arg, Lys118Arg/Lys165Arg, and Lys132Arg/Lys165Arg double mutants. No tetramer can be observed for the Lys118Arg/Lys165Arg double mutant.
Analytical ultracentrifugation (AUC) and gel filtration
AUC–Sedimentation equilibrium (SE)
SE analysis was performed to determine the oligomeric state(s) of the wild-type InhA protein in the presence and absence of NADH or the INH-NAD adduct. In each case, data analysis gave a MW within 5% of that expected for tetramer at all three concentrations of protein studied (3 μM, 10 μM, and 30 μM) (Table 1, Figure 5A–C). This result conflicts with the cross-linking studies, which suggest that ligand binding causes the dissociation of InhA into dimers. To reconcile this apparent inconsistency, we used gel filtration chromatography and sedimentation velocity (SV) to study the effect of ligand biding on InhA.
Table 1.
Sedimentation equilibrium (SE), sedimentation velocity (SV), and gel filtration (GF) chromatography of free and ligand-bound InhA

Figure 5.
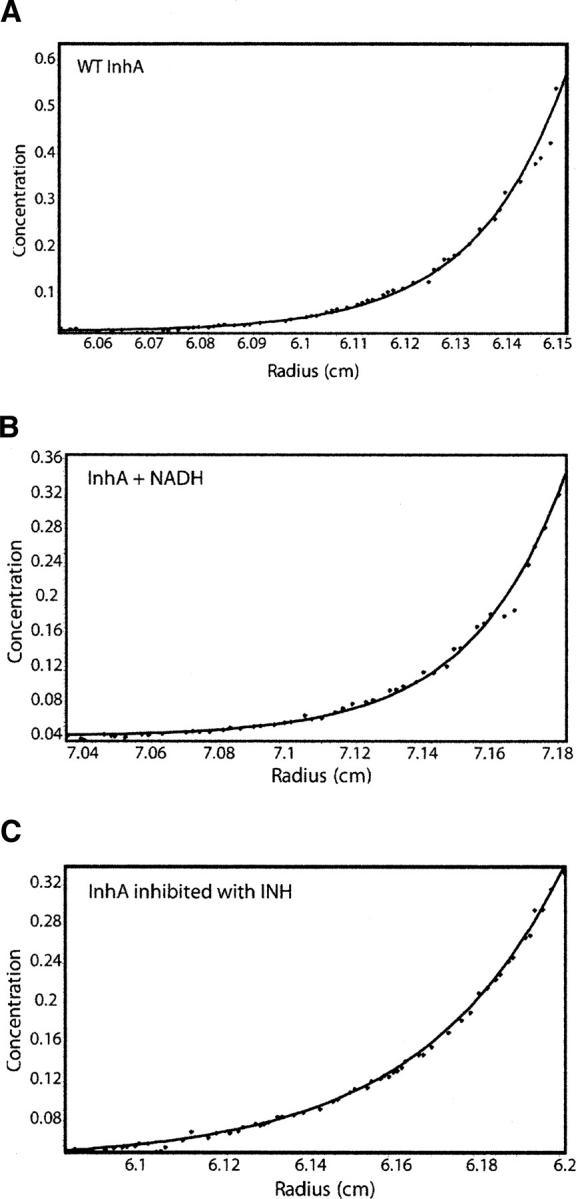
SE-AUC analysis shows unbound and bound forms of InhA are tetramers. Representative sedimentation equilibrium data for (A) unbound wild-type InhA, (B) InhA incubated with excess NADH, and (C) wild-type InhA inhibited by the INH-NAD adduct. Data analysis was performed with HeteroAnalysis software, and in all three cases InhA was found to be tetrameric.
Gel filtration
Unbound and inhibited wild-type InhA were eluted from the size exclusion column at elution volumes of 70.28 ± 0.16 mL and 71.15 ± 0.15 mL, respectively. These elution volumes correspond to a MW of 114.6 ± 1.7 kDa and 106.3 ± 2.3 kDa, respectively, close to the expected MW for the InhA tetramer, which is 122.7 kDa (Table 1). Since both SE and size exclusion chromatography indicate that inhibited InhA remains tetrameric, we hypothesized that ligand binding caused a conformational change that altered the ability of the protein tetramer to be cross-linked. This possibility was examined using sedimentation velocity experiments.
AUC–Sedimentation velocity (SV)
Using Sedfit, the MWs of free and ligand-bound InhA were all shown to be close to the expected MW for InhA tetramer (Table 1). However, in contrast to the SE analysis, the SV experiments revealed a clear change in the sedimentation coefficient(s) value for the enzyme upon ligand binding. Thus, while free InhA had an s-value of 5.82 ± 0.15 S, InhA bound to NADH or inhibited by the INH-NAD adduct had s-values of 6.32 ± 0.15 S and 6.33 ± 0.15 S, respectively (Table 1, Fig. 6).
Figure 6.

SV-AUC analysis of free and ligand-bound InhA sedimentation velocity data for wild-type InhA in the absence (a) and presence of NADH (b) and the INH-NAD adduct (c). The program SedFit was used for data analysis. There is a clear increase in s-value upon binding of either NADH or INH-NAD, corresponding to the generation of a more compact tetrameric structure.
Discussion
Despite intense investigation, the precise molecular basis for the mode of action of INH is still not completely understood. While it is clear that KatG, the mycobacterial catalase-peroxidase, is essential for INH activation, the drug may have multiple targets within the cell (Zhang et al. 1992). Both InhA and KasA, two components of the FAS-II pathway, have been proposed as INH targets while Blanchard and coworkers recently demonstrated that INH could also inhibit dihydrofolate reductase (Banerjee et al. 1994; Mdluli et al. 1996, 1998; Marrakchi et al. 2000; Slayden et al. 2000; Vilcheze et al. 2000; Larsen et al. 2002; Kremer et al. 2003; Argyrou et al. 2006; Vilcheze et al. 2006). The effect of INH on mycolic acid biosynthesis can be most clearly understood through inhibition of long chain fatty acid biosynthesis, and it has been shown that the INH-NAD adduct is a very effective in vitro inhibitor of InhA. In an attempt to unravel the complexities of INH action, we previously studied the inhibition of wild-type InhA by the INH-NAD adduct and three mutants, Ile21Val, Ile47Thr, or Ser94Ala, that correlate with resistance to INH. While the InhA mutations dramatically decrease the affinity of NADH for the enzyme (Dessen et al. 1995; Basso et al. 1998), we were surprised to find that these mutations had only a minimal effect on adduct affinity (Rawat et al. 2003). Based on these observations, we speculated that the full effect of the InhA mutations on enzyme reactivity and inhibition might only be fully realized in the context of the living cell and, specifically, when InhA is present in a noncovalent multienzyme complex with other FAS-II components (Odriozola et al. 1977; Marrakchi et al. 2000, 2002; Rawat et al. 2003).
Initially, therefore, we set out to evaluate whether the INH-resistant InhA mutations affected the ability of InhA to interact with other components of the FAS-II pathway. We chose to study the ability of InhA to interact with KasA, since InhA directly precedes KasA in the FAS-II pathway and since an interaction between InhA and KasA might explain how independent studies have suggested that both InhA and KasA are targets for INH. However, while the bacteria two-hybrid experiments showed the anticipated interaction between wild-type InhA and KasA (Veyron-Churlet et al. 2004), these studies failed to show any difference in the ability of the three InhA mutants to interact with KasA. In fact, the strength of the interaction appears unaltered by these mutations.
While the bacteria two-hybrid method provides qualitative information on the ability of two proteins to interact, the interacting partners are not necessarily present in their native oligomerization states, which could be important in the present context since InhA is a tetramer in solution while KasA is a dimer. Importantly, while our two-hydrid experiments were in progress, Veyron-Churlet et al. (2004, 2005) demonstrated that mutations at the dimer–dimer interface of MabA, the FAS-II β-ketoacyl-ACP reductase, altered the ability of this protein to interact with KasA and KasB. Consequently, we designed a second set of experiments to directly assess whether inhibition of InhA by the INH-NAD adduct altered the oligomerization state of InhA, since this would likely affect InhA-dependent interactions in the FAS-II complex, and also whether the Ile21Val, Ile47Thr, and Ser94Ala mutations then altered the response of InhA to inhibition. Blanchard and coworkers had previously shown that the InhA mutants bind NADH cooperatively, in contrast to wild-type InhA, indicating that not only had the mutations altered the affinity of InhA for NADH, they had also affected communication within the InhA subunits (Basso et al. 1998).
In agreement with previous studies (Rozwarski et al. 1999; Marrakchi et al. 2002), cross-linking wild-type InhA with the homobifunctional chemical cross-linking reagent BS3 yielded bands on SDS consistent with tetrameric InhA, in addition to dimeric and monomeric forms of the protein. This reagent contains two N-hydroxysuccinimide esters, which are reactive with the primary amines of lysine residues, of which there are nine in each of the InhA monomers (Fig. 2C). Interestingly, when the same experiment was repeated on InhA inhibited by the INH-NAD adduct, only bands associated with dimeric and monomeric InhA were observed. A similar result was obtained when InhA was incubated exclusively with the cofactor NADH, suggesting that binding of cofactor or inhibitor to the tetrameric InhA has caused the protein to dissociate into dimers (Fig. 1). When we extended these studies to the three mutant InhA proteins, similar data were obtained in the presence of the INH-NAD adduct. However, even at saturating concentrations of NADH, cross-linking experiments indicated significant amounts of tetrameric InhA were present. The similarity in response of the three mutant proteins is consistent with our inhibition studies, which show very similar affinities of wild-type and mutant enzyme for the INH-NAD adduct (Ki 0.75–5 nM). The observation that tetrameric InhA can still be observed for the mutant enzymes in the presence of NADH may result from incomplete binding of the cofactor and/or incomplete occupancy of the InhA subunits. Alternatively, the cross-linking data may indicate that NADH binding populates a second conformational state in the mutant InhA proteins that is fundamentally different from the ligand bound population observed for the wild-type enzyme.
In order to further probe the effect of ligand binding on the structure of InhA, we performed an additional set of experiments using AUC and size exclusion chromatography. In contrast to the cross-linking data, these methods indicated that inhibited and cofactor-bound enzyme was still tetrameric. However, sedimentation velocity experiments did reveal an alteration in sedimentation coefficient for wild-type and mutant InhA proteins upon ligand binding (Table 1). Thus, one simple conclusion we can draw from all these data is that ligand binding causes a conformational change in the InhA tetramer that prevents one or more lysine pairs across a dimer–dimer interface from being cross-linked.
While there are several crystal structures of InhA in complex with a variety of ligands, there are no structures of uncomplexed InhA and, thus, no high resolution information on the structure of InhA before ligand binding. However, by analogy to AUC studies on the hexameric Helicobacter pylori VirB11 protein, which undergoes a 5%–7% increase in s-value upon binding ATP, we can conclude that the change in s-value observed for InhA upon complexation indicates that the protein structure has become more compact (Savvides et al. 2003). In order to provide more information on the nature of the ligand-induced conformational change, we performed a series of cross-linking experiments on InhA mutants in which we replaced candidate lysine residues with arginines.
Analysis of the ligand-bound InhA structure indicates that the protein contains two dimer–dimer interfaces, A and B (Fig. 2C). In order to cross-link the four monomers within the tetramer, lysine residues at both dimer–dimer interfaces must be within 11.4 Å of each other. Across interface A, only Lys8 and Lys240 are close enough to be cross-linked by BS3, as they are 9.8 Å apart (Fig. 2A; Rozwarski et al. 1999). Comparative MALDI analysis of the trypsin/Glu-C digests of the uncross-linked monomeric InhA and the cross-linked dimer reveals the generation of a mass peak of 2081, confirming the expected m/z for the reacted BS3 plus the two peptides containing Lys8 and Lys240 (Fig. 3). In contrast, no lysine residues were sufficiently close to each other to allow cross-linking between subunits at interface B. These data suggest, therefore, that the conformational change resulting from ligand binding causes one or more lysine pairs at the B interface to move apart, since observation of tetrameric protein on SDS-PAGE also requires formation of cross-links over interface B for the unliganded protein. To provide more information on the ligand-induced conformational change, we consequently replaced Lys118, Lys132, and Lys165 with arginine residues. While the single-lysine mutants all formed tetramer for unliganded enzyme on reaction with BS3, no tetramer could be observed for the Lys118Arg/Lys165Arg double mutant. Since Lys165 is involved in cofactor binding, the interaction of this residue with the NAD ribose can explain why ligand binding prevents this residue from participation in a cross-link. However, the fact that a cross-link can form between Lys118 in two different monomers, which are over 21 Å apart, indicates that these residues must be within 11 Å of each other in the unliganded protein. Thus ligand binding must cause a substantial movement of the two dimers with respect to each other. In order to provide further information on the ligand-induced conformational change, we are currently using X-ray crystallography to study the cross-linked InhA protein tetramer.
In summary we have provided direct evidence for a ligand-induced conformational change upon interaction of InhA with the INH-NAD adduct. However, while the modulation in InhA structure caused by enzyme inhibition may have a critical impact on the ability of InhA to interact with other components of the FAS-II pathway, similar changes in structure are also observed for the Ile21Val, Ile47Thr, and Ser94Ala InhA mutants. Currently, the only difference we are able to discern between wild-type and mutant InhAs is the observation that some fraction of the mutant InhA proteins form tetramers upon reaction with BS3 even in the presence of saturating NADH. However, the mutations have little effect on the affinity of InhA for the INH-NAD adduct and do not affect the ability of InhA to undergo the observed ligand-induced conformational change (Rawat et al. 2003). If these mutations arise from drug pressure, then they must modulate the antibacterial activity of INH. Since there is evidence that InhA interacts with other components of the FAS-II pathway in vivo, our current hypothesis remains that the mutations affect InhA inhibition and/or the consequences of this inhibition within the context of the FAS-II multienzyme complex. Alterations in the affinity of the adduct for InhA modulated via protein–protein interactions could also then affect the partitioning of the adduct between different enzyme targets within the cell, including the proteins identified recently by affinity chromatography (Argyrou et al. 2006). Further studies on the inhibition of InhA in the presence of other enzymes from the FAS-II pathway are currently in progress.
Materials and Methods
Bacteria 2-hybrid
Bacterial strains and media
E. coli strain XL1-Blue MRF was grown at 37°C in Luria-Bertani (LB) broth or on LB agar plates, containing either 25 μg/mL chloramphenicol (dissolved in 100% ethanol) or 12.5 μg/mL tetracycline (dissolved in 50% ethanol). Validation of protein–protein interaction was completed in BacterioMatch II Validation Reporter competent E. coli cells. All interaction experiments were carried out on nonselective, selective, or dual selective screening media. Nonselective medium was prepared by first autoclaving 7.5 g Bacto agar in 380 mL H2O, followed by the addition of 50 mL of 10× M9 salts (Qbiogene), 67.5 mL M9 media additive, and 0.5 mL each of chloramphenicol and tetracycline, when the solution had cooled to 50°C. M9 media additive was prepared in two parts. Part one contained 10 mL of filter-sterilized 20% glucose, 5 mL of 5 mM filter-sterilized adenine HCl, and 50 mL of autoclaved 10× His-dropout supplement, while part two contained 0.5 mL of each of the following filter-sterilized components: 1 M MgSO4, 1 M thiamine HCl, 10 mM ZnSO4, 100 mM CaCl2, and 50 mM IPTG. Subsequently, the two parts were combined and then added to the autoclaved media. Selective screening medium was prepared by adding 2.5 mL of 1 M 3-amino-1,2,4-triazole (3-AT), dissolved in DMSO, to nonselective screening medium. Dual-selective screening medium was prepared by adding 0.5 mL of 12.5 mg/mL streptomycin to selective screening medium. Finally, M9 His-dropout broth was prepared by adding 50 mL 10× M9 salts and 67.5 mL of M9 media additive to 380 mL H2O. All solutions were stored at 4°C until required.
Plasmid construction
PCR was used to amplify the kasA and inhA genes from the genomic H37Rv DNA using the following primers: inhA 5′-GCTCCTAGGATGACAGGACTGCTGG-3′ (forward), 5′-ATAAGAATGCGGCCGCTTAGAGCAATTGGGTGT-3′ (reverse). kasA 5′-ATAAGAATGCGGCCGCAGATGATTCAGCCTT-3′ (forward), and 5′-ATGTCGAATTCCTCAGTAACGCCCGAAGGC-3′ (reverse). PCR products were digested with the appropriate restriction enzymes and cloned in frame into either the bait plasmid, pBT, to produce fusion proteins with the DNA-binding domain, the full-length bacteriophage λ repressor protein, λcI, or the target vector, pTRG, to produce hybrid proteins with the activation domain, the N-terminal of α-subunit of RNA polymerase. The three InhA mutants, Ile21Val, Ile47Thr, and Ser94Ala, were constructed as previously described. All cloning/mutagenesis steps were performed in E. coli XL1-blue MRF cells (Stratagene).
Transformations
All protocols were adapted from the Stratagene BacterioMatch kit. BacterioMatch II Validation reporter competent E. coli cells were used for all interaction experiments. Transformations were performed in Falcon 2059 polypropylene tubes using a mixture containing 100 μL of cells, 1.7 μL of β-mercaptoethanol, and 50–100 ng of plasmid (bait and target). Reactions were incubated on ice for 30 min (shaking gently every 10 min). Cotransformation reactions were then heat shocked at 42°C for 35 sec. After 2 min of incubation on ice, 900 μL of sterile SOC medium were added to each tube. SOC medium was prepared by autoclaving 20 g of tryptone, 5 g of yeast extract, and 0.5 g of NaCl to 950 mL of H2O, followed by the addition of 10 mL of 1 M MgCl2, 10 mL of 1 M MgSO4, and 2 mL of 20% (w/v) glucose, all filter sterilized. After 1.5 h of recovery incubation in a 37°C shaker bath, the cells were centrifuged at 700 g for 15 min at room temperature. Pellets were washed (twice) in 1 mL M9 His-dropout broth and centrifuged again for 15 min. Pellets were then resuspended in 600 mL of M9 His-dropout broth and incubated for an additional 2 h in a 37°C shaker bath. Cotransformants were plated on nonselective medium, for calculating the efficiency of the uptake of both bait and target plasmid, as well as selective and dual selective media for detection of positive interactions. Plates were incubated at 37°C for 1–3 d, to ensure the detection of both strong and weak interactions. All cotransformations were performed with both positive (pBT-LGF2/pTRG-Gal11P) and negative (pBT-LGF2/pTRG) controls. Positive control vectors pBT-LGF2 and pTRG-Gall1P produce the dimerization domain of the yeast Gal4 protein and a domain from a mutant Gal11 protein, respectively, which have been shown to interact in this strain of reporter E. coli.
Protein expression and purification
Wild-type and mutant InhA proteins were constructed, expressed, and purified as described previously (Parikh et al. 1999). Briefly, all InhA variant proteins were expressed with N-terminal hexa-histidine motifs in BL21 (DE3) pLysS cells (Novagen) and were subsequently purified by nickel affinity chromatography and immediately exchanged into 30 mM PIPES, 150 mM NaCl, and 1 mM EDTA (pH 6.8) by gel filtration using a Sephadex G-25 (Amersham Biosciences) column. Protein concentrations were determined using ɛ280 = 37.3 M−1 cm−1 for the wild-type, Ile21Val, Ile47Thr, and Ser94Ala proteins.
Cross-linking of the wild-type and mutant InhAs
The purified proteins were exchanged into the cross-linking reaction buffer (20 mM Na2HPO4, 0.15 M NaCl at pH 7.5) directly prior to the experiment. The cross-linking reaction contained varied concentrations (0.1–10 μM) of wild-type or mutant InhA and a 50-fold excess of bis(sulfosuccinimidyl)suberate (BS3) (Pierce) in 5 mM sodium acetate (pH 5.0). After allowing the cross-linking reaction to proceed for 30 min at room temperature, the reactions were quenched with 1 M Tris (pH 7.5) (final concentration 50 mM) and incubated for an additional 15 min. The cross-linked proteins were then concentrated and analyzed using 4%–20% gradient SDS-PAGE gels. Cross-linking reactions were also performed on inhibited InhA, prepared as described below, and in the presence of 150 μM NADH (Sigma).
Protein digestion and MALDI analysis
Three protein samples were extracted after SDS-PAGE analysis: wild-type InhA without BS3, wild-type dimer, and tetramer with BS3. Standard protein extraction and digestion with trypsin and Glu-C was performed. Briefly, protein bands were excised from the gel and washed overnight in 50% methanol (v/v) and 5% acetic acid (v/v). Unless otherwise noted, all steps were carried out at room temperature. Wash buffer was removed and a second wash was carried out for an additional 3 h. Gel pieces were then dehydrated with acetonitrile for 10 min and then dried thoroughly with a CentriVap. Samples were then reduced with 10 mM DTT for 30 min, followed by alkylation with 100 mM iodoacetamide for 30 min. Samples were then dehydrated with acetonitrile and rehydrated with 100 mM ammonium bicarbonate, twice. Trypsin (20 ng/μL) and endoproteinase Glu-C (200 ng/μL) were prepared in ice-cold 50 mM ammonium bicarbonate (pH 7.8). Dried gel fragments were rehydrated with trypsin and Glu-C for 10 min with vortexing, excess trypsin/Glu-C was removed, and digestion was carried out overnight at 37°C. Peptides were then extracted from the gel with 50% acetonitrile (v/v) and 5% formic acid (v/v). The volume of the extract was reduced and adjusted to 20 μL with 50% acetonitrile (v/v) and 1% TFA (v/v) in preparation for MALDI. Peptides were analyzed using a Bruker Daltronics AutoFlex II MALDI-TOF/TOF (Fig. 3). Peptide masses were calculated using the program MS-Digest (http://prospector.ucsf.edu/prospector/4.0.7/html/msdigest.htm). Intermolecular and intramolecular cross-linked peptide masses were assigned using the Automatic Spectrum Assignment Program (ASAP) (http://roswell.ca.sandia.gov/∼mmyoung/asap.html).
Inactivation of wild-type and mutant InhA proteins by INH
Inactivation reactions were performed as previously described (Rawat et al. 2003). Briefly, each reaction contained 0.3 μM InhA, 300 μM INH, 1 μM MnCl2, and 150 μM NADH in 100 mM Na2HPO4 buffer (pH 7.5). After incubating for 2 h at room temperature, residual enzyme activity was assessed by monitoring the ability of the samples to oxidize NADH upon the addition of trans-2-dodecenoyl-CoA (Parikh et al. 1999; Rawat et al. 2003). The inactivation reactions were then prepared for cross-linking with BS3, as described above. Inactivated InhA was further purified by chromatography on G-25 to remove excess NADH, and analyzed using analytical ultracentrifugation (AUC) and gel filtration chromatography.
Structure-based mutagenesis
Lys118, Lys132, and Lys165 were all mutated to Arg using a standard mutagenesis protocol. Primers for the Lys118Arg mutation were 5′-TACGCGGATGTGTCCAGGGGCATCCACATCTCG-3′ (forward) and 5′-CGAGATGTGGATGCCCCTGGACACATCCGCGTA-3′ (reverse), and primers for the Lys132Arg mutation were 5′-TATGCTTCGATGGCCAGGGCGCTGCTGCCGATC-3′ (forward) and 5′-GATCGGCAGCAGCGCCCTGGCCATCGAAGCATA-3′ (reverse). All mutated nucleotides are designated in underlined bold. The Lys165Arg InhA mutation was available from previous studies (Parikh et al. 1999). In addition, the double-mutants Lys118Arg/Lys132Arg, Lys118Arg/Lys165Arg, and Lys132Arg/Lys165Arg were also constructed. All six InhA mutants were expressed, purified, and assayed as previously described (Parikh et al. 1999).
Analytical ultracentrifugation (AUC)
All protein samples were dialyzed into 20 mM Na2HPO4, 150 mM NaCl (pH 7.5) immediately prior to analysis. AUC experiments were performed at 20°C using a Beckman Coulter Optima XL-I analytical ultracentrifuge equipped with a scanning UV/VIS spectrometer to monitor absorbance during radial scans. Experiments were performed using a four-cell, An-60 Ti analytical rotor and either a six-channel, charcoal-filled Epon centerpiece (for sedimentation equilibrium, SE) or a two-channel, aluminum centerpiece (for sedimentation velocity, SV). For SE, samples were analyzed at three concentrations, 3 μM, 10 μM, and 30 μM and included free InhA, InhA plus NADH, and inhibited InhA. SE was performed at 18k rpm and the data were collected for 24 h or until successive scans at A280 nm remained constant. Partial specific volumes (0.7406) and solvent density (1.00727) were calculated using SEDNTERP software (www.bbri.org/RASMB/rasmb.html). Each data set was analyzed using HeteroAnalysis Version 1.0.102. For SV, samples with an A280 nm and A230 nm ranging from 0.5 to 0.9 were utilized. Data sets were collected for a minimum of 150 scans at a running speed of either 40k or 60k rpm. Each data set was analyzed using the Sedfit program (www.analyticalultracentrifugation.com) (Schuck 1998; 2000; Dam and Schuck 2004).
Gel filtration chromatography
A prepacked HiLoad 16/60 Superdex 200 column was equilibrated with 20 mM Na2HPO4, 150 mM NaCl (pH 7.5). Calibration of the column was performed using blue dextran to measure the void volume in combination with a Gel Filtration Standard kit (Bio-Rad). The data were fitted to the equation y = −3.24x + 5.95. After calibration, the column was reequilibrated with the same buffer and wild-type InhA samples varying in concentration from 3 to 120 μM were applied to the column, as well as inhibited InhA samples. Column effluent was monitored at A280 nm and plotted against the elution volume.
Acknowledgments
This work was supported by National Institutes of Health Grant AI44639 to P.J.T. N.A.K. was supported by the National Research Service Award T32 AI007539 and GAANN fellowship. We thank Dr. Steven Berkowitz and Biogen Idec, Inc., for assistance obtaining and analyzing the AUC data. Genomic H37Rv MTB DNA was obtained from the “Tuberculosis Research Materials and Vaccine Testing” center at Colorado State University as part of NIH, NIAID Contract No. HHSN266200400091C.
Footnotes
Reprint requests to: Peter J. Tonge, Department of Chemistry, Stony Brook University, Stony Brook, NY 11794-3400, USA; e-mail: peter.tonge@sunysb.edu; fax: (631) 632-7934.
Abbreviations: MTB, Mycobacterium tuberculosis; FAS-II, fatty acid synthase type two; INH, isoniazid; INH-NAD, isoniazid-NAD adduct; SE, sedimentation equilibrium; SV, sedimentation velocity.
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.062749007.
References
- Argyrou A., Vetting, M.W., Aladegbami, B., and Blanchard, J.S. 2006. Mycobacterium tuberculosis dihydrofolate reductase is a target for isoniazid. Nat. Struct. Mol. Biol. 13: 408–413. [DOI] [PubMed] [Google Scholar]
- Banerjee A., Dubnau, E., Quemard, A., Balasubramanian, V., Um, K.S., Wilson, T., Collins, D., de Lisle, G., and Jacobs Jr, W.R. 1994. inhA, a gene encoding a target for isoniazid and ethionamide in Mycobacterium tuberculosis . Science 263: 227–230. [DOI] [PubMed] [Google Scholar]
- Basso L.A., Zheng, R., Musser, J.M., Jacobs Jr, W.R., and Blanchard, J.S. 1998. Mechanisms of isoniazid resistance in Mycobacterium tuberculosis: Enzymatic characterization of enoyl reductase mutants identified in isoniazid-resistant clinical isolates. J. Infect. Dis. 178: 769–775. [DOI] [PubMed] [Google Scholar]
- Dam J. and Schuck, P. 2004. Calculating sedimentation coefficient distributions by direct modeling of sedimentation velocity concentration profiles. Methods Enzymol. 384: 185–212. [DOI] [PubMed] [Google Scholar]
- Dessen A., Quemard, A., Blanchard, J.S., Jacobs Jr, W.R., and Sacchettini, J.C. 1995. Crystal structure and function of the isoniazid target of Mycobacterium tuberculosis . Science 267: 1638–1641. [DOI] [PubMed] [Google Scholar]
- Heymann D.L. 2006. Resistance to anti-infective drugs and the threat to public health. Cell 124: 671–675. [DOI] [PubMed] [Google Scholar]
- Kremer L., Dover, L.G., Morbidoni, H.R., Vilcheze, C., Maughan, W.N., Baulard, A., Tu, S.C., Honore, N., Deretic, V., Sacchettini, J.C., et al. 2003. Inhibition of InhA activity, but not KasA activity, induces formation of a KasA-containing complex in mycobacteria. J. Biol. Chem. 278: 20547–20554. [DOI] [PubMed] [Google Scholar]
- Larsen M.H., Vilcheze, C., Kremer, L., Besra, G.S., Parsons, L., Salfinger, M., Heifets, L., Hazbon, M.H., Alland, D., Sacchettini, J.C., et al. 2002. Overexpression of inhA, but not kasA, confers resistance to isoniazid and ethionamide in Mycobacterium smegmatis, M. bovis BCG, and M. tuberculosis . Mol. Microbiol. 46: 453–466. [DOI] [PubMed] [Google Scholar]
- Marrakchi H., Laneelle, G., and Quemard, A. 2000. InhA, a target of the antituberculous drug isoniazid, is involved in a mycobacterial fatty acid elongation system, FAS-II. Microbiol. 146: 289–296. [DOI] [PubMed] [Google Scholar]
- Marrakchi H., Ducasse, S., Labesse, G., Montrozier, H., Margeat, E., Emorine, L., Charpentier, X., Daffe, M., and Quemard, A. 2002. MabA (FabG1), a Mycobacterium tuberculosis protein involved in the long-chain fatty acid elongation system FAS-II. Microbiol. 148: 951–960. [DOI] [PubMed] [Google Scholar]
- Mdluli K., Sherman, D.R., Hickey, M.J., Kreiswirth, B.N., Morris, S., Stover, C.K., and Barry 3rd, C.E. 1996. Biochemical and genetic data suggest that InhA is not the primary target for activated isoniazid in Mycobacterium tuberculosis . J. Infect. Dis. 174: 1085–1090. [DOI] [PubMed] [Google Scholar]
- Mdluli K., Slayden, R.A., Zhu, Y., Ramaswamy, S., Pan, X., Mead, D., Crane, D.D., Musser, J.M., and Barry 3rd, C.E. 1998. Inhibition of a Mycobacterium tuberculosis β-ketoacyl ACP synthase by isoniazid. Science 280: 1607–1610. [DOI] [PubMed] [Google Scholar]
- Middlebrook G. 1952. Sterilization of tubercle bacilli by isonicotinic acid hydrazide and the incidence of variants resistant to the drug in vitro. Am. Rev. Tuberc. 65: 765–767. [PubMed] [Google Scholar]
- Odriozola J.M., Ramos, J.A., and Bloch, K. 1977. Fatty acid synthetase activity in Mycobacterium smegmatis. Characterization of the acyl carrier protein-dependent elongating system. Biochim. Biophys. Acta 488: 207–217. [DOI] [PubMed] [Google Scholar]
- Parikh S., Moynihan, D.P., Xiao, G., and Tonge, P.J. 1999. Roles of tyrosine 158 and lysine 165 in the catalytic mechanism of InhA, the enoyl-ACP reductase from Mycobacterium tuberculosis . Biochemistry 38: 13623–13634. [DOI] [PubMed] [Google Scholar]
- Quemard A., Sacchettini, J.C., Dessen, A., Vilcheze, C., Bittman, R., Jacobs Jr, W.R., and Blanchard, J.S. 1995. Enzymatic characterization of the target for isoniazid in Mycobacterium tuberculosis . Biochemistry 34: 8235–8241. [DOI] [PubMed] [Google Scholar]
- Rawat R., Whitty, A., and Tonge, P.J. 2003. The isoniazid-NAD adduct is a slow, tight-binding inhibitor of InhA, the Mycobacterium tuberculosis enoyl reductase: Adduct affinity and drug resistance. Proc. Natl. Acad. Sci. 100: 13881–13886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozwarski D.A., Vilcheze, C., Sugantino, M., Bittman, R., and Sacchettini, J.C. 1999. Crystal structure of the Mycobacterium tuberculosis enoyl-ACP reductase, InhA, in complex with NAD+ and a C16 fatty acyl substrate. J. Biol. Chem. 274: 15582–15589. [DOI] [PubMed] [Google Scholar]
- Savvides S.N., Yeo, H.J., Beck, M.R., Blaesing, F., Lurz, R., Lanka, E., Buhrdorf, R., Fischer, W., Haas, R., and Waksman, G. 2003. VirB11 ATPases are dynamic hexameric assemblies: New insights into bacterial type IV secretion. EMBO J. 22: 1969–1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuck P. 1998. Sedimentation analysis of noninteracting and self-associating solutes using numerical solutions to the Lamm equation. Biophys. J. 75: 1503–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuck P. 2000. Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and Lamm equation modeling. Biophys. J. 78: 1606–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slayden R.A., Lee, R.E., and Barry 3rd, C.E. 2000. Isoniazid affects multiple components of the type II fatty acid synthase system of Mycobacterium tuberculosis . Mol. Microbiol. 38: 514–525. [DOI] [PubMed] [Google Scholar]
- Takayama K., Schnoes, H.K., Armstrong, E.L., and Boyle, R.W. 1975. Site of inhibitory action of isoniazid in the synthesis of mycolic acids in Mycobacterium tuberculosis . J. Lipid Res. 16: 308–317. [PubMed] [Google Scholar]
- Veyron-Churlet R., Guerrini, O., Mourey, L., Daffe, M., and Zerbib, D. 2004. Protein–protein interactions within the fatty acid synthase-II system of Mycobacterium tuberculosis are essential for mycobacterial viability. Mol. Microbiol. 54: 1161–1172. [DOI] [PubMed] [Google Scholar]
- Veyron-Churlet R., Bigot, S., Guerrini, O., Verdoux, S., Malaga, W., Daffe, M., and Zerbib, D. 2005. The biosynthesis of mycolic acids in Mycobacterium tuberculosis relies on multiple specialized elongation complexes interconnected by specific protein–protein interactions. J. Mol. Biol. 353: 847–858. [DOI] [PubMed] [Google Scholar]
- Vilcheze C., Morbidoni, H.R., Weisbrod, T.R., Iwamoto, H., Kuo, M., Sacchettini, J.C., and Jacobs Jr, W.R. 2000. Inactivation of the inhA-encoded fatty acid synthase II (FASII) enoyl-acyl carrier protein reductase induces accumulation of the FASI end products and cell lysis of Mycobacterium smegmatis . J. Bacteriol. 182: 4059–4067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilcheze C., Wang, F., Arai, M., Hazbon, M.H., Colangeli, R., Kremer, L., Weisbrod, T.R., Alland, D., Sacchettini, J.C., and Jacobs Jr, W.R. 2006. Transfer of a point mutation in Mycobacterium tuberculosis inhA resolves the target of isoniazid. Nat. Med. 12: 1027–1029. [DOI] [PubMed] [Google Scholar]
- World Health Organization. 2003. Global TB Control Report. World Health Organization, Geneva, Switzerland.
- Zhang Y., Heym, B., Allen, B., Young, D., and Cole, S. 1992. The catalase-peroxidase gene and isoniazid resistance of Mycobacterium tuberculosis . Nature 358: 591–593. [DOI] [PubMed] [Google Scholar]