

Abstract
The mitochondrial serine protease HtrA2/Omi helps to maintain mitochondrial function by handling misfolded proteins in the intermembrane space. In addition, HtrA2/Omi has been implicated as a proapoptotic factor upon release into the cytoplasm during the cell death cascade. The protein contains a C-terminal PDZ domain that packs against the protease active site and inhibits proteolytic activity. Engagement of the PDZ domain by peptide ligands has been shown to activate the protease and also has been proposed to mediate substrate recognition. We report a detailed structural and functional analysis of the human HtrA2/Omi PDZ domain using peptide libraries and affinity assays to define specificity, X-ray crystallography to view molecular details of PDZ–ligand interactions, and alanine-scanning mutagenesis to probe the peptide-binding groove. We show that the HtrA2/Omi PDZ domain recognizes both C-terminal and internal stretches of extended, hydrophobic polypeptides. High-affinity ligand recognition requires contacts with up to five hydrophobic side chains by distinct sites on the PDZ domain. However, no particular residue type is absolutely required at any position, and thus, the HtrA2/Omi PDZ domain appears to be a promiscuous module adapted to recognize unstructured, hydrophobic polypeptides. This type of specificity is consistent with the biological role of HtrA2/Omi in mitochondria, which requires the recognition of diverse, exposed stretches of hydrophobic sequences in misfolded proteins. The findings are less consistent with, but do not exclude, a role for the PDZ domain in targeting the protease to specific substrates during apoptosis.
Keywords: PDZ domain, serine protease, apoptosis, mitochondria, peptide-binding module
HtrA2/Omi is a mitochondrial serine protease with extensive homology with bacterial high-temperature requirement A protease (HtrA) (Faccio et al. 2000). Bacterial HtrA has a dual role: at normal temperatures it acts as a chaperone, while at elevated temperatures it acts as a protease that disposes of denatured proteins and allows for cell survival following heat shock or other stress (Spiess et al. 1999). The proteolytic activity of HtrA2/Omi is also markedly up-regulated in response to heat shock or stress (Gray et al. 2000).
HtrA2/Omi is synthesized as a precursor protein with an N-terminal signal peptide that directs localization to the mitochondria, where proteolytic removal of the N-terminal domain exposes an inhibitor of apoptosis protein (IAP) binding motif (Suzuki et al. 2001; Martins 2002; Martins et al. 2002; van Loo et al. 2002). Indeed, HtrA2/Omi has been identified as an IAP-binding protein (Suzuki et al. 2001) and is believed to act as a promoter of apoptosis by virtue of its ability to disrupt interactions between IAPs and caspases (Suzuki et al. 2001; Hegde et al. 2002; van Loo et al. 2002; Verhagen et al. 2002). Additional studies indicated that HtrA2/Omi could also induce cell death, either apoptosis (Verhagen et al. 2002; Egger et al. 2003; Martins et al. 2003) or necrosis (Okada et al. 2003), in a caspase-independent manner through its protease activity, and X-linked IAP has been shown to be a proteolytic substrate in vitro.
Despite these seemingly compelling lines of evidence implicating HtrA2/Omi as a central player in apoptosis, recent genetic studies have cast doubt on this hypothesis. Cells from mice in which HtrA2/Omi has been inactivated, either by a point mutation (Jones et al. 2003) or by gene knockout (Martins et al. 2004), are not resistant to apoptosis, but rather, are more sensitive to cell death. Indeed, mice with an HtrA2/Omi inactivating point mutation are known as mnd2 (motor neuron degeneration 2) mice, because they suffer from a neurodegenerative disease caused by progressive mitochondrial damage (Jones et al. 2003). These findings suggest that the major function of HtrA2/Omi is the maintenance of proper mitochondrial function by handling misfolded proteins in the intermembrane space, and this role is analogous to that of bacterial HtrA in the periplasm. Thus, it appears that HtrA2/Omi functions mainly to promote cell survival and plays only a peripheral role in apoptosis, along with other mitochondrial proteins that are released into the cytoplasm after the cell has already committed to the apoptotic death program (Ekert and Vaux 2005).
Mature HtrA2/Omi contains a protease domain and a PDZ (PSD-95/Discs-large/ZO-1) domain (HtrA2-PDZ). The crystal structure of HtrA2/Omi reveals that the PDZ domain packs against the protease domain with its ligand-binding cleft buried against the protease active site (Li et al. 2002). Access to the protease catalytic site is therefore blocked by the PDZ domain. Proteolytic activity can be greatly enhanced by disruption of the PDZ/protease interaction, either by mutation of the PDZ/protease interface (Li et al. 2002) or by engagement of HtrA2-PDZ with a peptide ligand (Martins et al. 2003). Thus, it is reasonable to postulate that the proteolytic activity of HtrA2/Omi in vivo may be regulated by the binding of peptides to HtrA2-PDZ. Characterization of the ligand specificity and structure/function relationships for HtrA2-PDZ may be useful in identifying potential endogenous substrates and also for helping to understand the physiological roles of HtrA2/Omi.
As PDZ domains typically bind peptide ligands with free C termini, a synthetic peptide library of this type has been used to study the binding specificity of HtrA2-PDZ (Martins et al. 2003), and it was found that the PDZ domain recognizes predominantly hydrophobic sequences that require a free C terminus for binding. However, the library did not contain Trp residues, and several studies have shown that many PDZ domains prefer ligands that contain Trp side chains, particularly at position −1. Also, some PDZ domains can recognize ligands through interactions that do not involve a free C terminus (Hillier et al. 1999; Wittchen et al. 1999; Wong et al. 2003), but this possibility was not investigated for HtrA2-PDZ. Furthermore, the energetics of the interaction between HtrA2-PDZ and peptide ligands were not analyzed in detail, and the structural basis for ligand recognition was not elucidated.
Herein we present a detailed structural and functional analysis of the interactions between human HtrA2-PDZ and optimal peptide ligands. While ligands with a free C terminus are preferred, we find that HtrA2-PDZ can also recognize internal motifs. In both types of ligands, HtrA2-PDZ recognizes extended hydrophobic motifs, consistent with a role for HtrA2/Omi in the recognition and handling of misfolded proteins. Affinity measurements with series of synthetic peptides were used to map the binding contributions of individual ligand residues, and an efficient combinatorial alanine-scanning approach was used to identify the side chains in HtrA2-PDZ that contribute energetically to ligand binding. Finally, we solved the X-ray crystal structure for HtrA2-PDZ in complex with a high-affinity ligand. Taken together, these studies provide a comprehensive view of the relationships between the structure and function of HtrA2-PDZ and provide insights into the biological function of HtrA2/Omi.
Results and Discussion
Peptides selected for binding to HtrA2-PDZ
Phage-displayed peptide libraries were used to select ligands that bind to HtrA2-PDZ. We used either a decapeptide library fused to the C terminus of the phage coat protein (Zhang et al. 2006) or an octapeptide library fused to the N terminus (Sidhu et al. 2000). In each case, the library contained NNK degenerate codons that encode for all 20 natural amino acids. In the case of the C-terminal library, the inclusion of amber stop codons in the degenerate codon also provided for the display of shorter peptides. Each library was cycled through three rounds of binding selection with immobilized GST-HtrA2-PDZ fusion protein as the capture target.
The sequencing of 80 clones from the C-terminal library revealed 24 unique sequences (Fig. 1). Overall, the sequences are highly hydrophobic in character, although at most positions, no single hydrophobic residue type was highly preferred over others. Valine was most prevalent at the C-terminal position (position 0)2, but other small hydrophobes (Leu, Ile, Ala) also occurred, indicating that site 0 accommodates the small aliphatic side chains typical of most PDZ domain ligands (Appleton et al. 2006; Zhang et al. 2006). Positions −1, −2, and −3 were dominated by the large hydrophobes Trp and Phe; Trp was dominant at position −1, while Phe was dominant at position −3. There was no consensus or hydrophobic bias at position −4, suggesting that this site does not contribute to binding. Finally, there was a bias for large hydrophobes at position −5, and no consensus or bias was evident beyond this position. Overall, these results indicate that HtrA2-PDZ recognizes hydrophobic C termini, and binding is likely mediated by contacts extending across the last six residues of the peptide ligand.
Figure 1.

Peptide ligands for HtrA2-PDZ selected from phage-displayed libraries. Sequences selected from libraries fused to the C or N terminus of a phage coat protein are shown above or below the dashed line, respectively. Hydrophobic residues are shown in bold text, and the conserved, hydrophobic tripeptide common to the C- and N-terminal ligands is shaded gray. The number of times each sequence occurred (n) is shown to the right. Positions in the peptide ligand are numbered according to the convention for C-terminal PDZ domain ligands; residues from the C terminus to the N terminus are designated 0, −1, −2, ....
Binding selections with the N-terminal library were also successful, and sequencing of 89 clones revealed 13 unique sequences (Fig. 1). The sequences were hydrophobic in character, and Trp residues were particularly prevalent. In this case, homology alignment was less clear-cut than for the sequences from the C-terminal library, which was facilitated by the C-terminal residue functioning as an anchor position. However, in many of the sequences, we noticed a conserved, hydrophobic tripeptide motif that was also present in many of the C-terminal sequences at the −3, −2, and −1 positions. Thus, we aligned the N-terminal sequences on the basis of this conserved motif, and in so doing, revealed additional homology at position 0, which was occupied by either Gly or Asp residues. Taken together, the results with the two libraries suggest that HtrA2-PDZ can recognize both C-terminal and internal peptide sequences that contain extended hydrophobic stretches.
Affinity assays with synthetic peptides
Peptides corresponding to the most prevalent selected sequences were synthesized and assayed for binding to HtrA2-PDZ (Table 1). A peptide derived from the sequence selected from the C-terminal library (H2-C1) bound with high affinity (IC50 = 3.3μM). Amidation of the C terminus (H2-C1a) abolished binding, demonstrating the importance of an interaction between HtrA2-PDZ and the terminal carboxylate of the ligand. To investigate the energetic contributions of the side chains at each position, we conducted an alanine scan of H2-C1. The replacement of Thr−4 with Ala (H2-C1f) did not effect binding, and this is consistent with the lack of a consensus at this position among the selected sequences. Ala replacements for Val0 (H2-C1b) or Met−3 (H2-C1e) had moderate impact on binding, with IC50 values increased threefold or eightfold, respectively. Alanine replacement for Phe−2 (H2-C1d) had a somewhat greater effect, as the IC50 value was increased ∼15-fold. The largest effects on binding were due to the substitution of Ala for either of the two Trp residues. The Trp−1 side chain was particularly crucial, as binding of the Ala-substituted peptide (H2-C1c) was barely detectable (>300-fold reduction). The replacement of Trp−5 reduced binding by >30-fold.
Table 1.
IC50 values for synthetic peptides binding to HtrA2-PDZ

The relative importance of the peptide side chains was also confirmed by a series of peptides that tested the effects of N-terminal deletions. Deletion of Trp−5 (H2-C1h) resulted in a 15-fold reduction in binding, while further deletion of Thr−4 (H2-C1i) actually regained some activity, as the binding was only sevenfold reduced in comparison with H2-C1. The further deletion of Met−3 had a fivefold deleterious effect on binding, and the C-terminal tripeptide (H2-C1j) bound with an IC50 value in the hundred micromolar range.
Taken together, these results demonstrate that HtrA2-PDZ recognizes peptide H2-C1 through interactions with the C-terminal carboxylate and all five hydrophobic residues. Among these hydrophobic residues, it is notable that the least important side chain is that of the C-terminal Val0 residue. This contrasts with results for other PDZ domains, such as the Erbin PDZ domain (Erbin-PDZ) (Skelton et al. 2003) and the first PDZ domain of ZO-1 (ZO1-PDZ1) (Zhang et al. 2006), in which interactions with the C-terminal aliphatic side chain are a critical source of binding energy. It appears that, while site 0 of HtrA2-PDZ excludes nonhydrophobic side chains, the hydrophobic binding pocket does not contribute significant energy to ligand recognition, and we speculate that this may partly explain the ability of HtrA2-PDZ to recognize internal ligands (see below). The most crucial sources of binding energy are the C-terminal carboxylate group and the Trp side chain at position −1, and an additional satellite of binding energy is provided by the Trp side chain at position −5. At the −3 and −2 positions, the two intervening hydrophobic residues between the dominant Trp residues further augment affinity. It should be noted that the potential contributions of position −3 may be underestimated by our analysis, since the Met−3 found in peptide H2-C1 is not the dominant residue among the selected sequences (Fig. 1), and, for example, it is possible that substitution of Met−3 by a Phe residue may improve binding further.
We also measured the affinities of synthetic peptides corresponding to the most prevalent sequences selected from the N-terminal library. In this case, an additional C-terminal Gly residue was added to the sequence to mimic the linker region of the peptide fusion displayed on phage and, for all peptides in this series, the C terminus was blocked by amidation. We synthesized two different peptides (H2-N1 and H2-N2), which bound to HtrA2-PDZ with IC50 values in the 20–30 μM range. We next measured the effects of deleting residues from either end of peptide H2-N1. Starting from the C terminus, two residues could be deleted without affecting binding (H2-N1a and H2-N1b), but deletion of Trp2 completely abolished binding (H2-N1c). Surprisingly, further deletion of Gly1 and Gly0 produced a tetrapeptide (H2-N1e) that exhibited binding to HtrA2-PDZ. Deletions from the N terminus showed that Ser−4 does not contribute significantly to binding, but further deletion of His−3 abolished binding. Based on these data, we identified a tripeptide (H2-N1i) that bound to HtrA2-PDZ with an IC50 value in the hundred micromolar range, and notably, this tripeptide corresponded to the region that was conserved in our alignment of the ligands isolated from the C- and N-terminal peptide libraries (Fig. 1). To investigate the energetic contributions of the important side chains identified by deletion scanning, we also conducted an alanine scan of peptide H2-N1a. Ala replacement for Trp2 (H2-N1j) had a modest impact on binding affinity, as the IC50 value was increased ∼10-fold. Trp−1 and Trp−2 are crucial, as binding was completely abolished by Ala substitution at either position (H2-N1k and H2-N1l). Ala replacement for His−3 (H2-N1m) resulted in a modest fourfold reduction in binding.
These results show that HtrA2-PDZ binds to internal peptide motifs somewhat less tightly than to C-terminal motifs, but nonetheless, the affinities are well within the range of biologically relevant interactions. Alignment of the binding sequences and deletion and alanine-scanning with synthetic peptides show that both types of ligands share a conserved, hydrophobic binding core that corresponds to the −3, −2, and −1 positions of the C-terminal ligands. In the case of the C-terminal ligand, affinity is further augmented by contributions from the C-terminal carboxylate group and also from a hydrophobic side chain at position −5. In contrast, the N-terminal ligands lack a free C terminus, and instead, position0 is occupied by either a small Gly residue, which may allow the peptide to adapt a β-turn conformation, as has been observed in the structure of a PDZ domain heterodimer complex (Hillier et al. 1999); or alternatively, an Asp residue, which may mimic the carboxylate group of C-terminal ligands. Peptide H2-N1 contains an energetically important Trp residue at position2, and we speculate that this residue may contribute to binding through additional hydrophobic interactions with HtrA2-PDZ (see below). Taken together, these results show that HtrA2-PDZ is adapted to recognize extended stretches of hydrophobic sequence, and a free C terminus is not absolutely required to achieve reasonable binding affinities.
The crystal structure of HtrA2-PDZ
We determined the crystal structure of HtrA2-PDZ in complex with a peptide ligand (WTMFWVCOOH) that was derived using phage display (Table 1). Similar to other structural studies of PDZ domains (Appleton et al. 2006), we used a fusion protein in which the peptide ligand was linked to the C terminus of HtrA2-PDZ through a triglycine spacer. This fusion protein (HtrA2-PDZext) crystallized as a dimer with two molecules in the asymmetric unit and with each molecule providing its C terminus as a ligand for the other PDZ domain (Fig. 2A). The crystals diffracted to 2.75 Å resolution; the structure was solved by molecular replacement using the PDZ domain from the full-length HtrA2/Omi crystal structure (Li et al. 2002) and refined to R work and R free values of 20.8% and 25.4%, respectively (Table 2). The final model has good geometry, with 92% of the residues in the most favored region of the Ramachandran plot and no residues in the disallowed region (Laskowski et al. 1993). Both molecules including the C-terminal hexapeptide extension are well defined in the electron density; the two molecules have no obvious structural differences and superimpose with an RMSD of 0.6 Å over 99 Cα positions.
Figure 2.
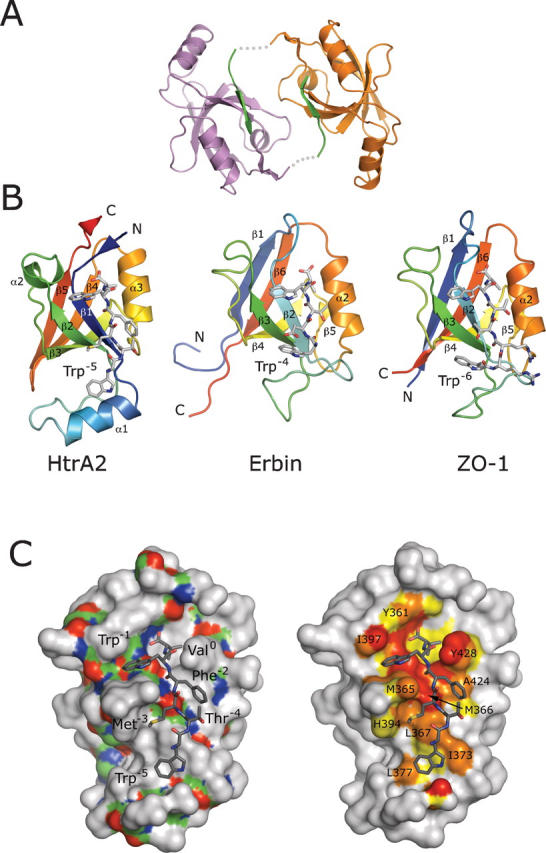
The HtrA2-PDZ crystal structure. (A) Dimer of HtrA2-PDZext; (orange or magenta) the individual PDZ domains; (green) the hexapeptides. The hexapeptides are fused to the PDZ domains via triglycine linkers (gray dotted lines), which are disordered in the structure. (B) Comparison of HtrA2-PDZ with an NMR structure of Erbin-PDZ (PDB code 1N7T) (Skelton et al. 2003) and a crystal structure of ZO1-PDZ1 (PDB code 2H2C) (Appleton et al. 2006). The main chain of each PDZ domain is colored like a rainbow from the N terminus (blue) to the C terminus (red); elements of secondary structure are labeled. Bound peptides are shown as sticks colored according to atom type: (gray) carbon; (blue) nitrogen; (red) oxygen; (yellow) sulfur. HtrA2-PDZ is circularly permuted in comparison to the other PDZ domains, but the overall folds and binding modes are similar. In each structure, the peptide ligand contains a Trp residue at position −1 and an additional Trp residue at a position upstream of position −3 (labeled). (C) HtrA2-PDZ recognizes ligands containing hydrophobic side chains at the 0, −1, −2, −3, and –5 positions. (Left panel) The molecular surface of HtrA2-PDZ is shown with the main chain colored according to atom type and the side chains gray. The peptide ligand is colored according to atom type, and the side chains are labeled. (Right panel) Atoms forming the peptide-binding surface of HtrA2-PDZ are colored according to their relative distance from the peptide ligand: (red) ≤3.5 Å; (orange) 3.6–4.0 Å; (yellow) 4.1–4.5 Å; residues of interest are labeled. Structural figures were produced with PyMOL (DeLano Scientific).
Table 2.
Data collection and refinement statistics

HtrA2-PDZ is circularly permuted relative to the canonical β-sandwich-fold found in most PDZ structures, including those of Erbin-PDZ and ZO1-PDZ1 (Fig. 2B; Skelton et al. 2003; Appleton et al. 2006). The permutation changes the order of the secondary structure elements within the β-sandwich. The ligand-binding cleft of HtrA2-PDZ is located on one edge of the sandwich between the most N-terminal β-strand (β1) and helix α3. These elements are structurally equivalent to strand β2 and helix α2 in Erbin-PDZ and ZO1-PDZ1 (Fig. 2B). HtrA2-PDZ also contains an extra helix (α1) at the “bottom” of the β-sandwich, which is not found in Erbin-PDZ and ZO1-PDZ1.
HtrA2-PDZ recognizes its binding partners in a manner similar to Erbin-PDZ (Skelton et al. 2003) and ZO1-PDZ1 (Appleton et al. 2006; Zhang et al. 2006), but there are significant differences. All three domains prefer aliphatic residues at site0 and a Trp at position−1 (Figs. 1 and 2B). Not surprisingly, binding of Trp into site−1 is common to many PDZ domains (Fuh et al. 2000; Appleton et al. 2006; Zhang et al. 2006), as all interactions are mediated by main-chain atoms of the conserved PDZ domain fold (Fig. 2C). In the HtrA2-PDZ structure, Trp−1 is bracketed by Met365 and Ile397, but alanine scanning indicates that these interactions do not contribute favorably to binding (see below). Unlike ZO1-PDZ1 and Erbin-PDZ, which are typical class 1 PDZ domains preferring Thr/Ser at position−2, HtrA2-PDZ is a class 2 PDZ domain preferring hydrophobic residues at this position (Songyang et al. 1997). In the crystal structure presented here (Fig. 2C), Phe−2 occupies a shallow groove formed by the side chains of Met366, Ala424, and Tyr428. HtrA2-PDZ also prefers hydrophobic residues at position−3 (Fig. 1), and in our crystal structure, Met−3 packs against Met365, Leu367, and His394. This contrasts with Erbin-PDZ and ZO1-PDZ1, which use hydrogen-bonding or electrostatic interactions to recognize polar or charged residues at the −2 and −3 positions. The only feature in common between HtrA2-PDZ, ZO1-PDZ1, and Erbin-PDZ upstream of site−3 is that the optimal ligand for each of the proteins contains a Trp that interacts with residues from the region connecting strands β2 and β3 (numbered according to the canonical PDZ domain fold). However, this Trp is located in the −4, −5, or −6 position of the ligand for Erbin-PDZ, HtrA2-PDZ, or ZO1-PDZ1, respectively (Fig. 2B).
Recent studies have revealed that ligand recognition by most PDZ domains involves interactions with each of the last five, six, or seven C-terminal residues of optimal ligands (Fuh et al. 2000; Kozlov et al. 2000, 2002; Karthikeyan et al. 2001, 2002; Im et al. 2003a, b; Kang et al. 2003; Skelton et al. 2003; Appleton et al. 2006; Zhang et al. 2006). Consequently, some PDZ domains like ZO1-PDZ1 and Erbin-PDZ exhibit high ligand specificity and are capable of recognizing only a limited range of sequences with high affinity. For these PDZ domains, high specificity allows them to interact with a limited number of biological partners and thus enables the formation of specific biological complexes. HtrA2-PDZ displays much lower specificity toward its ligands (Fig. 1) and has evolved to bind amino acids of general hydrophobic character at the 0, −1, −2, −3, and −5 positions (Fig. 2C). In our structure, the ligand side chains at these positions are involved in numerous van der Waals interactions with several hydrophobic residues that form the ligand-binding site of HtrA2-PDZ. Furthermore, position−4 of HtrA2-PDZ shows no preference for any amino acid type, and in our structure, Thr−4 points toward the solvent. In summary, it appears that HtrA2-PDZ is adapted to recognize a broad range of extended hydrophobic peptides, including both C-terminal and internal motifs, and is not restricted to a narrow set of peptide ligands.
We also attempted to crystallize HtrA2-PDZ in complex with an internal ligand (SHWWGGWLGG) by fusing the ligand to the PDZ domain C terminus, in a manner analogous to that used for the C-terminal ligand. We obtained crystals that diffracted to a maximum resolution of 2.5 Å, but unfortunately, the structure revealed that the proteins crystallized with the carboxylate group of the C-terminal Gly residue interacting with the carboxylate-binding loop of the PDZ domain (data not shown), even though we had added two Gly residues beyond the last residue of the ligand to mimic the linker used for phage display. This arrangement resembles the typical binding mode of PDZ domains for C-terminal ligands and thus cannot be representative of the manner in which HtrA2-PDZ recognizes internal ligands that lack a free C-terminal carboxylate. In retrospect, this was not surprising because unliganded PDZ domains often crystallize via lattice contacts from neighboring domains. As HtrA2-PDZ does not accept C-terminal hydrophilic residues (Fig. 1), we designed another fusion protein in which the same internal ligand was fused to the C terminus of HtrA2-PDZ, but the C-terminal Gly residue was replaced with a Lys residue, reasoning that this would eliminate any possibility of the C terminus occupying site0. However, despite extensive effort, this fusion protein failed to crystallize. Without an anchoring C-terminal carboxylate, the hydrophobic peptide ligand may slide into multiple registers, particularly since the internal ligand contains two pairs of hydrophobic residues (WW and WL). This would likely hinder crystallization by causing the formation of heterogeneous protein complexes. Indeed, since the extended, hydrophobic binding pocket of HtrA2-PDZ is capable of accepting a wide variety of ligand sequences (Fig. 1), it is not guaranteed that an internal ligand must necessarily bind in a single conformation, and we speculate that this may be a natural property of this PDZ domain, which unfortunately precludes facile structural analysis.
The full-length HtrA2/Omi structure is composed of an N-terminal protease domain and the C-terminal PDZ domain, which packs against and occludes the protease active site (Fig. 3A; Li et al. 2002). The PDZ domain in the full-length structure superimposes perfectly with both PDZ molecules in our structure (RMSD of 0.5–0.7 Å), indicating that the different binding modes do not require major structural rearrangements in the PDZ domain main chain (Fig. 3B). The protease contacts three discontinuous regions of the PDZ domain (Fig. 3C). These interactions are atypical in comparison to most PDZ–ligand interactions and do not involve the carboxylate-binding loop or an extension of the β-sandwich through β-type main-chain interactions. The majority of interactions between the protease and PDZ domains are mediated by four side chains of the protease (Met323, Val325, Ile329, Phe331) (Li et al. 2002) that pack against the hydrophobic region of the PDZ domain that is also responsible for binding to Met−3 and Trp−5 of the peptide ligand (Fig. 3C). In contrast, the 0, −1, and −2 sites, critical for recognition of the C-terminal peptide ligand, are not engaged in interactions with the protease domain.
Figure 3.

Comparison of HtrA2-PDZ bound to a C-terminal peptide and from the structure of the full-length HtrA2/Omi protein. (A) The crystal structure of full-length HtrA2/Omi (PDB code 1LCY) (Li et al. 2002). (Gray surface) The PDZ domain; (cyan schematic) the protease. (Arrow) The position of the β11–β12 region of the protease that contacts the PDZ domain. (B) Overlay of the HtrA2/Omi PDZ domain bound to a C-terminal peptide (blue) and from the structure of the full-length HtrA2/Omi protein (gray). (C) Atoms forming the binding surface for the peptide (left) or the protease (middle) are colored on the PDZ domain according to their relative distance from their respective binding partner: (red) ≤3.5 Å; (orange) 3.6–4.0 Å; (yellow) 4.1–4.5 Å. Although the details of binding are different (right), the β11–β12 region (cyan) of the protease domain contacts the same region as Trp−5 and Met−3 of the C-terminal peptide ligand (green).
In summary, the hydrophobic ligand-binding site of HtrA2-PDZ can accommodate several different types of ligands through predominantly hydrophobic interactions. The various ligands use some common elements of the binding groove but rely on contacts with different regions of the PDZ domain to establish favorable binding contacts. This contrasts with Erbin-PDZ and ZO1-PDZ1, which recognize highly specific ligands through interactions that include hydrophobic contacts, but also, electrostatic and hydrogen-bonding interactions.
Shotgun alanine scanning of HtrA2-PDZ
We used combinatorial shotgun alanine scanning (Weiss et al. 2000) to assess the contributions of individual HtrA2-PDZ side chains to C-terminal ligand binding (Table 3). We constructed three libraries that together mutagenized 61 residues in and around the ligand-binding site. Each position was mutagenized by a degenerate codon that ideally encoded for only the wild type and alanine, but the degeneracy of the genetic code necessitated two additional mutations at some positions. These libraries were then selected for binding to the high-affinity C-terminal peptide ligand (H2-C1), and ∼100 positive clones from each library were sequenced after two rounds of selection. The number of clones with the wild-type residue at each position was compared to the number with Ala and categorized as substitutions that reduce (ratio >1), do not affect (ratio ∼1), or improve (ratio <1) binding to peptide. To control for variation in expression or display levels for different library members, the libraries were also selected for binding to an immobilized antibody capable of recognizing an epitope tag that was displayed at the N terminus of all library members. The ratio of wild type to Ala in the peptide selection was then scaled by the ratio of wild type to Ala observed in the antibody selection to give a normalized frequency of occurrence (F).
Table 3.
HtrA2-PDZ shotgun alanine scan


The F value correlates with the contribution of each side chain to ligand binding; values greater or less than 1 indicate favorable or unfavorable contributions, respectively. The analysis identified 16 side chains for which alanine substitutions had large deleterious effects on binding (F ≥ 4), and these effects were mapped onto the structure of HtrA2-PDZ in complex with the peptide (Fig. 4). Five residues that showed significant effect on peptide binding upon alanine substitution (Tyr361, Ile362, Gly363, Val427, Val431) form the carboxylate-binding loop and hydrophobic pocket that accommodate Val0. Five other important residues (Thr368, Leu369, Ser370, Ile373, Leu374) form a patch and appear to maintain the proper conformation of helix α1 to form a hydrophobic pocket accommodating the Trp−5 side chain, which packs against several hydrophobes, including Ile373 and Leu377. Several functionally important residues are not in direct contact with the ligand but are partially (Leu392, Pro401) or completely buried (Leu437, Leu369, Leu374) in the PDZ domain fold, and it is likely that alanine substitutions at these positions cause global structural perturbations. Alanine substitutions for three additional solvent-exposed residues (Arg359, Arg380, Lys395) are also detrimental to binding. As these residues are not in contact with the ligand, these effects are not readily explained by the structure, but we speculate that the positively charged side chains may enhance binding through long-range electrostatic interactions with the negatively charged C terminus of the ligand (Selzer et al. 2000).
Figure 4.
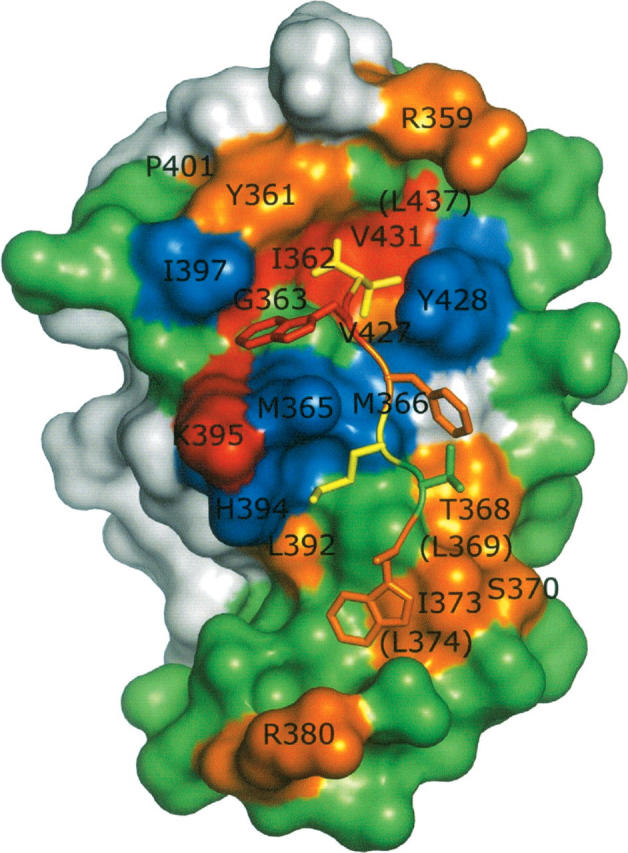
Results of shotgun alanine scanning mapped on the structure of HtrA2-PDZ. (Surface) The PDZ domain; (a tube with side-chain sticks) the peptide ligand main chain. The PDZ domain side chains are (red) F > 16; (orange) 16 > F > 4; (green) 4 > F > 0.5; (blue) F < 0.5; or (gray) not scanned. All residues with F > 4 or F < 0.5 are labeled, and in cases where the side chain is not visible, the labels are in parentheses. The peptide residues are colored according to the fold reduction in binding caused by Ala substitution: (red) >100; (orange) >10; (yellow) >3; (green) <3.
Notably, Ala is preferred to the wild type at Met365, Met366, His394, Ile397, and Tyr428 (F ≤ 0.3), suggesting the existence of steric hindrance between these side chains and the bulky side chains of Trp−1, Phe−2, and Met−3. These results underscore the importance of main chain interactions for recognition of Trp−1, and similar results were obtained previously for Erbin-PDZ, where it was found that Ala substitutions for side chains around Trp−1 were favored for binding (Skelton et al. 2003). Thus, even the high-affinity peptide ligand does not make favorable contacts with all parts of the binding site, and we speculate that this may contribute to the promiscuity of HtrA2-PDZ, as the binding of different ligands may depend on favorable contacts with different regions of the PDZ domain surface. It would be interesting to assess the contributions of these HtrA2-PDZ side chains to the binding of ligands that do not contain Trp at position−1, as it is possible that side chains that are detrimental for the binding of ligands that contain Trp−1 may nonetheless contribute favorably to the binding of other ligands.
Conclusions
Our results demonstrate that HtrA2-PDZ is capable of recognizing both C-terminal and internal hydrophobic polypeptide motifs. Recognition is achieved by multiple binding sites that stretch across the binding groove and make favorable contacts with multiple ligand side chains. No particular residue type is absolutely required at any position, and thus, HtrA2-PDZ can be considered to be a promiscuous peptide-binding module adapted to recognize unstructured stretches of hydrophobic polypeptides. This type of specificity is well suited for the major biological function of HtrA2/Omi, which is the recognition of misfolded polypeptides with exposed hydrophobic stretches that would be buried in the cores of folded proteins. However, although our results suggest that HtrA2/Omi is promiscuously activated, it is still possible that the protease acts by cleaving specific substrates in the mitochondria, as has been shown for the DegS protease in the bacterial periplasm (Wilken et al. 2004). Our findings are less in accord with the other proposed function of HtrA2-PDZ, which is the targeting of specific substrates for proteolysis during apoptosis. Thus, we conclude that the likely function of HtrA2-PDZ, in both mitochondria and in the apoptotic cascade, is to recognize a broad and diverse range of misfolded proteins rather than specific targeting to a distinct set of C-terminal ligands.
Materials and Methods
Materials
Enzymes and M13-KO7 helper phage were from New England Biolabs. Maxisorp immunoplates plates were from Nalge NUNC International. Escherichia coli XL1-Blue and BL21 were from Stratagene. Plasmids pET15b and pET22b were from Novagen. Thrombin was from Calbiochem. Bovine serum albumin (BSA) and Tween 20 were from Sigma. Horseradish peroxidase/anti-M13 antibody conjugate, horseradish peroxidase/anti-glutathione S-transferase (GST) antibody conjugate, glutathione Sapharose-4B, plasmid pGEX6P-3, and Superdex-75 were from Amersham Pharmacia Biotech. NiNTA was from QIAGEN. 3,3′,5,5′-Tetramethyl-benzidine/H2O2 peroxidase substrate was from Kirkegaard and Perry Laboratories Inc. NeutrAvidin was from Pierce Biotechnology Inc.
Synthetic peptides
Peptides were synthesized using standard 9-fluorenylmethoxycarbonyl protocols, cleaved off the resin with 2.5% triisopropylsilane and 2.5% H2O in trifluoroacetic acid, and purified by reversed-phase high-performance liquid chromatography. The purity and mass of each peptide were verified by liquid chromatography/mass spectrometry.
Protein purification
For use in peptide-phage selection experiments, a DNA fragment encoding HtrA2-PDZ (HtrA2/Omi residues 359–458) was cloned into the BamHI/XhoI sites of plasmid pGEX6P-3 to create a gene encoding a GST-HtrA2-PDZ fusion protein. A single colony of E. coli. BL21(DE3) harboring the expression plasmid was inoculated into 30 mL of LB medium supplemented with 50 μg/mL carbenicillin (LB/carb medium) and was grown overnight at 37°C. The bacteria were harvested, washed, resuspended, and inoculated into 500 mL of LB/carb medium. The culture was grown at 37°C to mid-log phase (A 600 = 0.8). Protein expression was induced with 0.4 mM isopropyl 1-thio-β-D-galactopyranoside, and the culture was grown for 16 h at 30°C. The bacteria were pelleted by centrifugation at 4000g for 15 min, washed twice with phosphate-buffered saline (PBS), and frozen for 8 h at −80°C. The pellet was resuspended in 50 mL of PBS, and the bacteria were lysed by passing through the Microfluidizer Processing Equipment. The GST-HtrA2-PDZ fusion protein was purified from the cell lysate by affinity chromatography on a 2-mL glutathione Sepharose-4B column according to the manufacturer's instructions.
For X-ray crystallography, a DNA fragment encoding residues 359–458 of HtrA2/Omi was cloned into the NdeI/BamHI sites of the pET15b expression vector, and this created an open reading frame encoding an HtrA2-PDZ fusion protein with an N-terminal His-tag followed by a thrombin cleavage site. Standard molecular biology techniques were used to fuse a nine-residue extension (GGGWTMFWV) to the C terminus of the PDZ domain to produce an open reading frame encoding a protein designated HtrA2-PDZext. E. coli BL21(DE3) harboring the expression plasmid was grown, induced, harvested, and lysed as described above. The cell lysate was loaded onto a Ni-NTA agarose column. The column was washed with 50 mM Tris (pH 8.0), 500 mM NaCl, and 20 mM imidazole, and the protein was eluted with 250 mM imidazole in the same buffer. Fractions containing HtrA2-PDZext were pooled, thrombin was added (1 unit/mg of protein), and the sample was dialyzed overnight against PBS at 4°C. The protein sample was then concentrated and further purified over a Superdex-75 column in 50 mM TrisHCl (pH 8.0), 300 mM NaCl, and 5 mM β-mercaptoethanol to remove thrombin and the cleaved His-tag.
Isolation of peptide ligands for HtrA2-PDZ
Previously described procedures were used to isolate peptides that bound to the GST-HtrA2-PDZ fusion protein. The peptides were selected from libraries fused to either the C (Zhang et al. 2006) or N terminus (Sidhu et al. 2000) of the M13 major coat protein. After three rounds of selection, individual clones were grown in a 96-well format in 500 μL of 2YT broth supplemented with carbenicillin (50 μg/mL), kanamycin (25 μg/mL), and M13-KO7 helper phage. The culture supernatants were used directly in phage enzyme-linked immunosorbent assays (ELISAs) (Sidhu et al. 2000) to detect peptides that bound specifically to HtrA2-PDZ, and the peptide sequences were determined from the sequences of the encoding DNA.
Construction of libraries for HtrA2-PDZ shotgun scanning
HtrA2-PDZ was displayed on the surface of M13 bacteriophage by modifying a previously described phagemid (pS2202b) (Skelton et al. 2003). Standard molecular biology techniques were used to replace the fragment of pS2202b encoding the Erbin PDZ domain with a DNA fragment encoding HtrA2-PDZ. The resulting phagemid (p8hOmi) contained an open reading frame that encoded for the maltose binding protein secretion signal, followed by an epitope tag (gD-tag, amino acid sequence: SMADPNRFRGKDLGS), followed by HtrA2-PDZ, and ending with the M13 major coat protein. E. coli harboring p8hOmi were coinfected with M13-KO7 helper phage, and cultures were grown at 30°C with 25 mM IPTG for protein induction, resulting in the production of phage particles that encapsulated p8hOmi DNA and displayed HtrA2-PDZ.
Libraries were constructed using previously described methods (Weiss et al. 2000) with appropriately designed “stop template” versions of p8hOmi. For each library, we used a stop template that contained TAA stop codons within the regions to be mutated. The stop template was used as the template for the Kunkel mutagenesis method (Kunkel et al. 1987) with mutagenic oligonucleotides designed to simultaneously repair the stop codons and introduce mutations with degenerate “alanine-scanning shotgun” codons, as described previously (Weiss et al. 2000). Three libraries were constructed, and each library mutated a discrete region of HtrA2-PDZ as follows: library 1, positions 359–379; library 2, positions 380–401; and library 3, positions 419–439. Libraries 1, 2, and 3 contained 7.8 × 1010, 5.5 × 1010, and 4.0 × 1010 unique members, respectively.
Sorting and analysis of HtrA2-PDZ shotgun scanning libraries
Phage from the libraries described above were propagated in E. coli XL1-blue with the addition of M13-KO7 helper phage. After overnight growth at 30°C, phage were concentrated by precipitation with PEG/NaCl and resuspended in PBS, 0.5% BSA, and 0.1% Tween 20, as described previously (Sidhu et al. 2000). Phage solutions (1012 phage/mL) were added to 96-well maxisorp immunoplates that had been coated with capture target and blocked with BSA. Two different targets were used; for the display selection, the target was an immobilized antibody that recognized the gD-tag epitope fused to the N terminus of HtrA2-PDZ; whereas for the function selection, a biotinylated peptide (biotin-SWTMFWV) that binds to HtrA2-PDZ with high affinity was immobilized on NeutrAvidin-coated plates. Following a 2-h incubation to allow for phage binding, the plates were washed 10 times with PBS and 0.05% Tween 20. Bound phage were eluted with 0.1 M HCl for 10 min, and the eluent was neutralized with 1.0 M Tris base. Eluted phage were amplified in E. coli XL1-blue and used for further rounds of selection.
Individual clones from each round of selection were grown in a 96-well format in 500 μL of 2YT broth supplemented with carbenicillin, kanamycin, and M13-KO7 helper phage, and the culture supernatants were used directly in phage ELISAs (Sidhu et al. 2000) to detect phage-displayed HtrA2-PDZ variants that bound to either the biotinylated peptide or the anti-gD-tag antibody. Clones that exhibited phage ELISA signals at least twofold greater than signals on control plates coated with BSA were considered as positive clones, and they were subjected to DNA sequence analysis.
The sequences were analyzed with the program SGCOUNT (Weiss et al. 2000). SGCOUNT aligned each DNA sequence against the wild-type DNA sequence by using a Needleman-Wunch pairwise alignment algorithm, translated each aligned sequence of acceptable quality, and tabulated the occurrence of each natural amino acid at each position. Approximately 100 sequences were analyzed for each library sorted against each target.
Affinity assays
The binding affinities of peptides for HtrA2-PDZ were determined as IC50 values using a previously described competition ELISA (Zhang et al. 2006). The IC50 value was defined as the concentration of peptide that blocked 50% of PDZ domain binding to immobilized peptide. Assay plates were prepared by immobilizing an N-terminally biotinylated peptide (biotin-GWTMFWV) on maxisorp plates coated with NeutrAvidin and blocked with BSA. A fixed concentration of GST-HtrA2-PDZ fusion protein (20 nM) in PBS, 0.5% BSA, and 0.1% Tween 20 (PBT buffer) was preincubated for 1 h with serial dilutions of peptide and then transferred to the assay plates. After 1 h of incubation, the plates were washed with PBS and 0.05% Tween 20, incubated for 30 min with HRP/anti-GST antibody (1:10,000) in PBT buffer, washed again, and detected with 3,3′,5,5′-tetramethyl-benzidine/H2O2 peroxidase substrate.
Crystallization, data collection, and refinement
HtrA2-PDZext was crystallized in sitting drops at 19°C by mixing equal volumes of protein solution (5.8 mg/mL) and crystallization buffer (0.1 M sodium citrate and 1.0 M mono-ammonium dihydrogen phosphate at pH 5.6). Crystals appeared overnight and grew to a maximum size of 175 μm × 175 μm × 125 μm. Before data collection, crystals were transferred to a cryosolution containing crystallization buffer plus 20% ethylene glycol and flash-frozen in liquid nitrogen. Data were collected to a maximum resolution of 2.75 Å at beamline 9-1 (Stanford Synchrotron Radiation Laboratory, California) and processed with HKL2000 (Otwinowski and Minor 1997). The crystals belong to spacegroup P41212 (a = b = 90.1 Å, c = 83.5 Å) and contain two molecules in the asymmetric unit. The structure was solved by molecular replacement with the program AMoRe (Navaza 1994) and the coordinates of the PDZ domain from an HtrA2/Omi crystal structure (PDB code 1LCY) (Li et al. 2002). The atomic model was built with the program O (Jones et al. 1991) and refined with the program REFMAC (Murshudov et al. 1997) while applying noncrystallographic symmetry (NCS) restraints.
Protein Data Bank deposition
The atomic coordinates and structure factor (code 2PZD) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, New Jersey (http://www.rcsb.org/).
Acknowledgments
We thank Yan Wu for help with protein purification, Bridget Currell for DNA sequencing, and Clifford Quan for peptide synthesis. Portions of this research were carried out at the Stanford Synchrotron Radiation Laboratory, a national user facility operated by Stanford University on behalf of the U.S. Department of Energy, Office of Basic Energy Sciences.
Footnotes
The C terminus of a PDZ domain ligand is designated as position 0, and the remaining positions are numbered with negative integers whose absolute value decreases toward the C terminus. The corresponding binding sites on the PDZ domain are numbered in a corresponding manner as site 0, site −1, etc.
Reprint requests to: Sachdev S. Sidhu, Department of Protein Engineering, Genentech, Inc., 1 DNA Way, South San Francisco, CA 94080, USA; e-mail: sidhu@gene.com; fax: (650) 225-3734; or Christian Wiesmann, Department of Protein Engineering, Genentech, Inc., 1 DNA Way, South San Francisco, CA 94080, USA; e-mail: chw@gene.com; fax: (650) 225-3734.
Abbreviations: BSA, bovine serum albumin; ELISA, enzyme-linked immunosorbent assay; GST, glutathione S-transferase; HRP, horseradish peroxidase; HtrA, high-temperature requirement A; HtrA2-PDZ, PDZ domain of HtrA2/Omi; IAP, inhibitor of apoptosis protein; PBS, phosphate-buffered saline; PDZ, PSD-95/Discs-large/ZO-1; RMSD, root mean square deviation.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.072833207.
References
- Appleton B.A., Zhang, Y., Wu, P., Yin, J.P., Hunziker, W., Skelton, N.J., Sidhu, S.S., and Wiesmann, C. 2006. A comparative structural analysis of the erbin PDZ domain and the first PDZ domain of ZO-1: Insights into determinants of PDZ domain specificity. J. Biol. Chem. 281: 22312–22320. [DOI] [PubMed] [Google Scholar]
- Egger L., Schneider, J., Rheme, C., Tapernoux, M., Hacki, J., and Borner, C. 2003. Serine proteases mediate apoptosis-like cell death and phagocytosis under caspase-inhibiting conditions. Cell Death Differ. 10: 1188–1203. [DOI] [PubMed] [Google Scholar]
- Ekert P.G. and Vaux, D.L. 2005. The mitochondrial death squad: Hardened killers or innocent bystanders? Curr. Opin. Cell Biol. 17: 626–630. [DOI] [PubMed] [Google Scholar]
- Faccio L., Fusco, C., Chen, A., Martinotti, S., Bonventre, J.V., and Zervos, A.S. 2000. Characterization of a novel human serine protease that has extensive homology to bacterial heat shock endoprotease HtrA and is regulated by kidney ischemia. J. Biol. Chem. 275: 2581–2588. [DOI] [PubMed] [Google Scholar]
- Fuh G., Pisabarro, M.T., Li, Y., Quan, C., Lasky, L.A., and Sidhu, S.S. 2000. Analysis of PDZ domain-ligand interactions using carboxyl-terminal phage display. J. Biol. Chem. 275: 21486–21491. [DOI] [PubMed] [Google Scholar]
- Gray C.W., Ward, R.V., Karran, E., Turconi, S., Rowles, A., Viglienghi, D., Southan, C., Barton, A., Fantom, K.G., West, A., et al. 2000. Characterization of human HtrA2, a novel serine protease involved in the mammalian cellular stress response. Eur. J. Biochem. 267: 5699–5710. [DOI] [PubMed] [Google Scholar]
- Hegde R., Srinivasula, S.M., Zhang, Z., Wassell, R., Mukattash, R., Cilenti, L., DuBois, G., Lazebnik, Y., Zervos, A.S., Fernandes-Alnemri, T., et al. 2002. Identification of Omi/HtrA2 as a mitochondrial apoptotic serine protease that disrupts inhibitor of apoptosis protein-caspase interaction. J. Biol. Chem. 277: 432–438. [DOI] [PubMed] [Google Scholar]
- Hillier B.J., Christopherson, K.S., Prehoda, K.E., Bredt, D.S., and Lim, W.A. 1999. Unexpected modes of PDZ domain scaffolding revealed by structure of nNOS-syntrophin complex. Science 284: 812–815. [PubMed] [Google Scholar]
- Im Y.J., Lee, J.H., Park, S.H., Park, S.J., Rho, S.-H., Kang, G.B., Kim, E., and Eom, S.H. 2003a. Crystal structure of the Shank PDZ–ligand complex reveals a class I PDZ interaction and a novel PDZ–PDZ dimerization. J. Biol. Chem. 278: 48099–48104. [DOI] [PubMed] [Google Scholar]
- Im Y.J., Park, S.H., Rho, S.-H., Lee, J.H., Kang, G.B., Sheng, M., Kim, E., and Eom, S.H. 2003b. Crystal structure of GRIP1 PDZ6–peptide complex reveals the structural basis for class II PDZ target recognition and PDZ domain-mediated multimerization. J. Biol. Chem. 278: 8501–8507. [DOI] [PubMed] [Google Scholar]
- Jones T.A., Zou, J.Y., Cowan, S.W., and Kjeldgaard, M. 1991. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A 47: 110–119. [DOI] [PubMed] [Google Scholar]
- Jones J.M., Datta, P., Srinivasula, S.M., Ji, W., Gupta, S., Zhang, Z., Davies, E., Hajnoczky, G., Saunders, T.L., Van Keuren, M.L., et al. 2003. Loss of Omi mitochondrial protease activity causes the neuromuscular disorder of mnd2 mutant mice. Nature 425: 721–727. [DOI] [PubMed] [Google Scholar]
- Kang B.S., Cooper, D.R., Devedjiev, Y., Derewenda, U., and Derewenda, Z.S. 2003. Molecular roots of degenerate specificity in syntenin's PDZ2 domain: Reassessment of the PDZ recognition paradigm. Structure 11: 845–853. [DOI] [PubMed] [Google Scholar]
- Karthikeyan S., Leung, T., and Ladias, J.A.A. 2001. Structural basis of the Na+/H+ exchanger factor PDZ1 interaction with the carboxyl-terminal region of the cystic fibrosis transmembrane conductance regulator. J. Biol. Chem. 276: 19683–19686. [DOI] [PubMed] [Google Scholar]
- Karthikeyan S., Leung, T., and Ladias, J.A.A. 2002. Structural determinants of the Na+/H+ exchanger regulatory factor interaction with the β2 adrenergic and platelet-derived growth factor receptors. J. Biol. Chem. 277: 18973–18978. [DOI] [PubMed] [Google Scholar]
- Kozlov G., Gehring, K., and Ekiel, I. 2000. Solution structure of the PDZ2 domain from human phosphatase hPTP1E and its interactions with C-terminal peptides from the Fas receptor. Biochemistry 39: 2572–2580. [DOI] [PubMed] [Google Scholar]
- Kozlov G., Banville, D., Gehring, K., and Ekiel, I. 2002. Solution structure of the PDZ2 domain from cytosolic human phosphatase hPTP1E complexed with a peptide reveals contribution of the β2–β3 loop to PDZ domain–ligand interactions. J. Mol. Biol. 320: 813–820. [DOI] [PubMed] [Google Scholar]
- Kunkel T.A., Roberts, J.D., and Zakour, R.A. 1987. Rapid and efficient site-specific mutagenesis without phenotypic selection. Methods Enzymol. 154: 333–363. [DOI] [PubMed] [Google Scholar]
- Laskowski R.A., MacAurthur, M.W., Moss, D.S., and Thornton, J.M. 1993. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26: 283–291. [Google Scholar]
- Li W., Srinivasula, S.M., Chai, J., Li, P., Wu, J.W., Zhang, Z., Alnemri, E.S., and Shi, Y. 2002. Structural insights into the pro-apoptotic function of mitochondrial serine protease HtrA2/Omi. Nat. Struct. Biol. 9: 436–441. [DOI] [PubMed] [Google Scholar]
- Martins L.M. 2002. The serine protease Omi/HtrA2: A second mammalian protein with a Reaper-like function. Cell Death Differ. 9: 699–701. [DOI] [PubMed] [Google Scholar]
- Martins L.M., Iaccarino, I., Tenev, T., Gschmeissner, S., Totty, N.F., Lemoine, N.R., Savopoulos, J., Gray, C.W., Creasy, C.L., Dingwall, C., et al. 2002. The serine protease Omi/HtrA2 regulates apoptosis by binding XIAP through a reaper-like motif. J. Biol. Chem. 277: 439–444. [DOI] [PubMed] [Google Scholar]
- Martins L.M., Turk, B.E., Cowling, V., Borg, A., Jarrell, E.T., Cantley, L.C., and Downward, J. 2003. Binding specificity and regulation of the serine protease and PDZ domains of HtrA2/Omi. J. Biol. Chem. 278: 49417–49427. [DOI] [PubMed] [Google Scholar]
- Martins L.M., Morrison, A., Klupsch, K., Fedele, V., Moisoi, N., Teismann, P., Abuin, A., Grau, E., Geppert, M., Live, G.P., et al. 2004. Neuroprotective role of the reaper-related serine protease HtrA2/Omi revealed by targeted deletion in mice. Mol. Cell. Biol. 24: 9848–9862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov G.N., Vagin, A.A., and Dodson, E.J. 1997. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53: 240–255. [DOI] [PubMed] [Google Scholar]
- Navaza J. 1994. AMoRe: An automated package for molecular replacement. Acta Crystallogr. A 50: 157–163. [Google Scholar]
- Okada M., Adachi, S., Imai, T., Watanabe, K.I., Toyokuni, S.Y., Ueno, M., Zervos, A.S., Kroemer, G., and Nakahata, T. 2003. A novel mechanism for imatinib mesylate-induced cell death of BCR-ABL-positive human leukemic cells: Caspase-independent, necrosis-like programmed cell death mediated by serine protease activity. Blood 103: 2299–2307. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z. and Minor, W. 1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276: 307–326. [DOI] [PubMed] [Google Scholar]
- Selzer T., Albeck, S., and Schreiber, G. 2000. Rational design of faster associating and tighter binding protein complexes. Nat. Struct. Biol. 7: 537–541. [DOI] [PubMed] [Google Scholar]
- Sidhu S.S., Lowman, H.B., Cunningham, B.C., and Wells, J.A. 2000. Phage display for selection of novel binding peptides. Methods Enzymol. 328: 333–363. [DOI] [PubMed] [Google Scholar]
- Skelton N.J., Koehler, M.F.T., Zobel, K., Wong, W.L., Yeh, S., Pisabarro, M.T., Yin, J.P., Lasky, L.A., and Sidhu, S.S. 2003. Origins of PDZ domain ligand specificity: Structure determination and mutagenesis of the Erbin PDZ domain. J. Biol. Chem. 278: 7645–7654. [DOI] [PubMed] [Google Scholar]
- Songyang Z., Fanning, A.S., Fu, C., Xu, J., Marfatia, S.M., Chishti, A.H., Crompton, A., Chan, A.C., Anderson, J.M., and Cantley, L.C. 1997. Recognition of unique carboxyl-terminal motifs by distinct PDZ domains. Science 275: 73–77. [DOI] [PubMed] [Google Scholar]
- Spiess C., Beil, A., and Ehrmann, M. 1999. A temperature-dependent switch from chaperone to protease in a widely conserved heat shock protein. Cell 97: 339–347. [DOI] [PubMed] [Google Scholar]
- Suzuki Y., Imai, Y., Nakayama, H., Takahashi, K., Takio, K., and Takahashi, R. 2001. A serine protease, HtrA2, is released from the mitochondria and interacts with XIAP, inducing cell death. Mol. Cell 8: 613–621. [DOI] [PubMed] [Google Scholar]
- van Loo G., van Gurp, M., Depuydt, B., Srinivasula, S.M., Rodriguez, I., Alnemri, E.S., Gevaert, K., Vandekerckhove, J., Declercq, W., and Vandenabeele, P. 2002. The serine protease Omi/HtrA2 is released from mitochondria during apoptosis. Omi interacts with caspase-inhibitor XIAP and induces enhanced caspase activity. Cell Death Differ. 9: 20–26. [DOI] [PubMed] [Google Scholar]
- Verhagen A.M., Silke, J., Ekert, P.G., Pakusch, M., Kaufmann, H., Connolly, L.M., Day, C.L., Tikoo, A., Burke, R., Wrobel, C., et al. 2002. HtrA2 promotes cell death through its serine protease activity and its ability to antagonize inhibitor of apoptosis proteins. J. Biol. Chem. 277: 445–454. [DOI] [PubMed] [Google Scholar]
- Weiss G.A., Watanabe, C.K., Zhong, A., Goddard, A., and Sidhu, S.S. 2000. Rapid mapping of protein functional epitopes by combinatorial alanine scanning. Proc. Natl. Acad. Sci. 97: 8950–8954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilken C., Kitzing, K., Kurzbauer, R., Ehrmann, M., and Clausen, T. 2004. Crystal structure of the DegS stress sensor: How a PDZ domain recognizes misfolded protein and activates a protease. Cell 117: 483–494. [DOI] [PubMed] [Google Scholar]
- Wittchen E.S., Haskins, J., and Stevenson, B.R. 1999. Protein interactions at the tight junction. Actin has multiple binding partners, and ZO-1 forms independent complexes with ZO-2 and ZO-3. J. Biol. Chem. 274: 35179–35185. [DOI] [PubMed] [Google Scholar]
- Wong H.-C., Bourdelas, A., Krauss, Y., Lee, H.-J., Shao, Y., Wu, D., Mlodzik, M., Shi, D.-L., and Zheng, J. 2003. Direct binding of the PDZ domain of dishevelled to a conserved internal sequence in the C-terminal region of frizzled. Mol. Cell 12: 1251–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Yeh, S., Appleton, B.A., Held, H.A., Kausalya, P.J., Phua, D.C.Y., Wong, W.L., Lasky, L.A., Wiesmann, C., Hunziker, W., et al. 2006. Convergent and divergent ligand specificity amongst the PDZ domains of the LAP and ZO families. J. Biol. Chem. 281: 22299–22311. [DOI] [PubMed] [Google Scholar]