

Abstract
Recent computational scans for non-coding RNAs (ncRNAs) in multiple organisms have relied on existing multiple sequence alignments. However, as sequence similarity drops, a key signal of RNA structure—frequent compensating base changes—is increasingly likely to cause sequence-based alignment methods to misalign, or even refuse to align, homologous ncRNAs, consequently obscuring that structural signal. We have used CMfinder, a structure-oriented local alignment tool, to search the ENCODE regions of vertebrate multiple alignments. In agreement with other studies, we find a large number of potential RNA structures in the ENCODE regions. We report 6587 candidate regions with an estimated false-positive rate of 50%. More intriguingly, many of these candidates may be better represented by alignments taking the RNA secondary structure into account than those based on primary sequence alone, often quite dramatically. For example, approximately one-quarter of our predicted motifs show revisions in >50% of their aligned positions. Furthermore, our results are strongly complementary to those discovered by sequence-alignment-based approaches—84% of our candidates are not covered by Washietl et al., increasing the number of ncRNA candidates in the ENCODE region by 32%. In a group of 11 ncRNA candidates that were tested by RT-PCR, 10 were confirmed to be present as RNA transcripts in human tissue, and most show evidence of significant differential expression across tissues. Our results broadly suggest caution in any analysis relying on multiple sequence alignments in less well-conserved regions, clearly support growing appreciation for the biological significance of ncRNAs, and strongly support the argument for considering RNA structure directly in any searches for these elements.
The main objective of the ENCyclopedia Of DNA Elements (ENCODE) project is to identify all functional elements in the human genome sequence. For this purpose, 30 Mb, or roughly 1% of the total genome, have been selected as ENCODE regions for this pilot project. The Pilot Project involves close interactions between computational and experimental scientists to evaluate various methods for annotating the human genome (The ENCODE Project Consortium 2007). A major challenge in the project is to annotate the large number of non-coding RNAs (ncRNAs), which are difficult to find by computational or experimental means. The discovery of a steadily increasing number of untranslated RNAs since the late 1990s has dramatically changed views on the roles and importance of ncRNAs.
The task of computationally finding ncRNAs is difficult because one has to consider secondary structure as well as nucleotide sequence. With only one sequence available, one can fold the sequence using single sequence folding methods (Hofacker et al. 1994; Zuker 2003; Ding et al. 2004), but structure can be detected more reliably from a set of related sequences, if available (Westhof and Michel 1994; Westhof et al. 1996). Predicting the RNA secondary structure is a necessity when searching for structured ncRNAs, and this makes RNA search algorithms computationally expensive. The seminal approach of Sankoff (1985) performs simultaneous alignment and structure inference, but it remains too computationally expensive for broad use. Various approximations to it have been developed, including FOLDALIGN (Havgaard et al. 2007), Dynalign (Harmanci et al. 2007), Stemloc (Holmes 2005), and Consan (Dowell and Eddy 2006), all attempting to increase performance without sacrificing accuracy, but even these procedures remain relatively computationally expensive. A natural alternative approach is to align the sequences first and then do RNA structure inference based on the alignment. This strategy is particularly attractive now that high-quality whole-genome multiple sequence alignments are available for 17 or more vertebrates (e.g., see Blanchette et al. 2004). Two recently developed programs, RNAz (Washietl et al. 2005a, b) and EvoFold (Pedersen et al. 2006), exploited these alignments to search for ncRNAs. These timely scans resulted in thousands of putative novel structured ncRNAs. The initial RNAz and EvoFold scan restricted attention to those portions of the multiple alignments that were defined to be highly conserved (Siepel et al. 2005), thus minimizing the number of alignment errors. This year the RNAz and EvoFold developers joined forces to scan all multiple alignments in the ENCODE regions for putative ncRNAs, not only the most conserved ones (Washietl et al. 2007), resulting in many additional candidates in these regions (albeit with estimated false-positive rates on the order of 50%).
Although these programs have significant strengths, their false-negative rates and other limitations of these studies are essentially unexplored. A particular concern is exactly the reliance on existing multiple sequence alignments, which are based on DNA sequence similarity alone. Unfortunately, as sequence similarity drops, a key feature of RNA structure—frequent compensating base changes—is increasingly likely to cause sequence-based alignment methods that are ignorant of RNA structure to misalign, or even refuse to align, homologous ncRNAs, consequently obscuring that structural signal. As illustrated by an example below, even modest misalignments in moderately well-conserved sequences can have an adverse effect. Torarinsson et al. (2006) provide even deeper evidence, by using FOLDALIGN to show the apparent presence of thousands of RNA structures conserved between human and mouse in regions not aligned in the UCSC MULTIZ alignments. An additional concern is that RNAz and EvoFold generally assume that an RNA structure, if present, is present in all sequences in the alignment, ignoring the possibility of gain or loss on some branches of the phylogeny. Finally, both programs initially evaluate only global alignments within fixed-width sliding windows, which further reduces sensitivity since a given placement of the window may include extraneous sequence flanking a given RNA structure, may include only part of the structure, or both.
In short, reliance on sequence-based alignments (and current tools) both biases away from regions that are conserved in structure but not sequence, while not fully protecting from alignment errors that also mask structure conservation. These observations lead us to apply CMfinder (Yao et al. 2006) to the ENCODE regions as a complement to the RNAz/EvoFold scans. CMfinder searches a set of (presumably) orthologous, unaligned sequences for local patterns indicative of conserved RNA sequence and structure. We do not rely on externally supplied alignments (except to indicate orthology), do not use a sliding-window approach, and can ignore diverged sequences that do not appear to share the discovered RNA motif.
CMfinder has been very successfully used in discovering ncRNAs in bacteria. In a genome-wide study in the Firmicutes (Yao et al. 2007), CMfinder’s top-ranking motifs included most known Firmicute RNA elements, and it achieved high accuracy in both membership prediction and secondary structure prediction in comparison to the hand-curated motif models from the Rfam database (Griffith-Jones et al. 2003). In addition, CMfinder predictions have led to discovery of many novel regulatory elements in this and other bacterial groups, including several new families of riboswitches (Weinberg et al. 2007).
In agreement with the previous studies, we find a large number of potential RNA structures in the ENCODE regions. We report 6587 candidate regions with an estimated false-positive rate of 50%. More intriguingly, many of our predicted motifs may be better represented by alignments taking the RNA secondary structure into account than those based on primary sequence alone, often quite dramatically. For example, approximately one-quarter of our motifs show revisions in >50% of their positions, in comparison to the sequence-based MULTIZ alignments. Furthermore, our candidate regions are largely complementary to the results of the RNAz/EvoFold scans—while overlap with the candidates generated by those scans is much greater than would be expected by chance, 84% of our candidate regions do not overlap results of previous scans (Washietl et al. 2007). These results broadly suggest caution in any analysis relying on multiple sequence alignments in less well-conserved regions, clearly support growing appreciation for the biological significance of ncRNAs, and strongly support the argument for taking RNA structure directly into account in any searches for these elements.
Results
The candidates
We scanned 2 × 56,017 (forward/reverse) multiple alignment blocks from the UCSC MULTIZ multiple alignment (.maf) files, one block at a time (155 nt long on average). Since previous studies were presumed to be effective in well-conserved regions, we restricted analysis to alignment blocks that overlap neither exons nor the most conserved elements (as defined by the PhastCons Conserved Elements; Siepel et al. 2005). These alignments covered 8.68 Mb of human sequence (out of the total of 30 Mb in the ENCODE regions), and included 3.87 Mb of repetitive sequence as defined by the RepeatMasker (http://www.repeatmasker.org) track of the UCSC alignments. We included alignments in repeat regions in human because many of the known ncRNAs are found there. This resulted in 10,106 predicted motifs that met our cutoff criteria: a composite score >5 and Gibbs energy >−5 kcal/mol (see Methods). We estimated a false-positive rate of 50% by repeating the analysis on shuffled alignments (see Methods). Composite score and energy distributions for randomized versus original alignments are depicted in Figure 1, showing a slight shift in the distribution toward lower energy and higher score for our native predictions. Some of these predicted motifs overlap or are sense/antisense to each other. Considering these as a single candidate region, we have 6587 candidate regions. Our candidate regions average 80 nt in length, collectively covering a total of 0.53 Mb, or 6.1% of our human input sequence. Candidate regions are approximately twice as dense (per nucleotide) in nonrepetitive regions (0.38 Mb of 4.81 Mb, or 7.9%) than in repeat regions (0.15 Mb of 3.87 Mb, or 3.9% of the repetitive input data set).
Figure 1.

Score distribution of the full CMfinder input set (A) composite score and (B) consensus minimum free energies for the native and random (shuffled) sequences. There is a slight shift toward lower energy and higher score for our native data.
Known ncRNAs
As noted by Washietl et al. (2007), the ENCODE regions are surprisingly poor in annotated ncRNAs. In fact, when studying Rfam (Griffiths-Jones et al. 2003), the Functional RNA project (http://www.ncRNA.org), and the snoRNA and miRNA tracks that have been mapped to the human genome by the UCSC Genome Browser (Kent et al. 2002), we could only find one ncRNA that fully overlapped our input alignments. This was the miRNA hsa-miR-483 on chromosome 11 identified by Fu et al. (2005) in fetal liver in human. In addition, miR-483 has been annotated in mouse and rat “by similarity” in mirBase (Griffiths-Jones 2004; Griffiths-Jones et al. 2006). This miRNA was detected in our scan (composite score 8.6, energy −31.4) and was scored highly as an miRNA by RNAmicro (Hertel and Stadler 2006), which we ran on all our predictions. Our prediction, in addition to human, rat, and mouse, also includes dog, cow, and rabbit. Hsa-miR-483 was also detected by RNAz but was not in the input set for EvoFold (Washietl et al. 2007).
Transcription data and purifying selection
Using oligonucleotide tiling-array techniques, transcription maps of TARs (transcriptionally active regions) (Bertone et al. 2004) and transfrags (transcribed fragments) (Cheng et al. 2005) have been generated. We compared our predictions to TARs and transfrags generated as a part of the ENCODE project, which used 11 human tissues (The ENCODE Project Consortium 2007). Note that these maps were derived from RNA fragments >200 nt. TARs and transfrags were only generated for the RepeatMasked regions of the genome, whereas we included the repeat regions, thus candidates in repeat regions (25% of our total candidate regions) were ignored in calculating the following numbers. Of these candidate regions, 16.9% overlap TARs/transfrags. At the nucleotide level, 11.8% of the bases in the predictions overlap a TAR or a transfrag, compared to 7.0% of the input bases (i.e., our whole RepeatMasked input data). In a recent study by Kapranov et al. (2007), the genomic origins and the relations of human nuclear and cytosolic polyadenylated RNAs >200 nt (lRNA) in eight cell lines and whole-cell RNAs <200 nt (sRNA) in two cell lines were investigated. Comparing our candidate regions to these new transfrags, on the nucleotide level, 3.0% and 27.4% of our candidates were overlapped by short and long RNAs, respectively, compared to 1.5% and 16.0% of the input bases. The increased overlap with TARs/transfrags, sRNA, and lRNA is highly significant with P-values of 10−40, 10−24, and 10−86, respectively. Still, one has to be cautious since, as noted by Washietl et al. (2007), the tiling-array studies may be more sensitive on G+C-rich regions and the TARs/transfrags are very G+C-rich. With this in mind, we divided our input data into five bins based on G+C content (0%–35%, 35%–40%, 40%–45%, 45%–50%, 50%–100% G+C ranges, chosen to contain similar numbers of alignment blocks) and repeated our analysis on each bin separately. Surprisingly, none of the five G+C bins show statistically significant overlap with the tiling-array data. Basically, the explanation is that our predictions, the tiling-array predictions, and the observed overlap between them are all concentrated in the high G+C range, and controlling for this bias erases the apparent significance of the overall overlap. We did the same analysis for the RNAz and EvoFold candidates that are in our input data, and came to the same conclusion for their candidates. Although our analysis included only a portion of their candidates, it does suggest that there is not a significant overlap with TARs/transfrags when considering G+C content—the apparent overall significance of overlap with the tiling-array data is seemingly explained by the G+C biases. However, Washietl et al. (2007) further point out that it is unclear whether the G+C bias for tiling-array data has a biological explanation or is a technical artifact. Additionally, they note that secondary structure may affect detection performance on tiling arrays, considering the observation of several examples where highly stable ncRNAs result in negative signal “holes” in tiling-array data (Cheng et al. 2005). Together, these observations leave open whether to expect tiling-array technology to sensitively identify structured ncRNAs.
Lunter et al. (2006) have identified non-coding regions apparently under purifying selection on the basis of lack of indels. We compared our candidate regions to their set of Indel Purified Segments (IPSs) on human assembly hg18. For our two most G+C-rich bins (where the majority of our candidate regions lie), there is a significant overlap to the IPSs (P <10−8 and P <10−31), indicating that many of our candidate regions are under purifying selection.
GENCODE
We also compared our candidate regions to the GENCODE annotations (Harrow et al. 2006), which aim to identify all human protein-coding genes in the ENCODE regions. We find that 40% of our candidates are intergenic, whereas 60% overlap some non-exonic part of a protein-coding gene (see Table 1). We also analyzed whether introns, 3′ UTRs, or 5′ UTRs were enriched for our candidate regions, again stratified by G+C. Significant enrichment of predicted candidate regions is seen only in the highest G+C bin of 5′ UTRs (P < 10−6).
Table 1.
GENCODE overlaps: Total number and percentage of candidates overlapping non-exonic GENCODE annotations

There are also 23 candidates that overlap with an exon, because we use the GENCODE annotation here, whereas our initial filtering was done with UCSC known genes annotation.
RNAz and EvoFold
As mentioned above, a similar scan to ours was performed with the global, alignment-dependent programs RNAz and EvoFold (Washietl et al. 2007). Note that they use the TBA (Threaded Blockset Aligner) RepeatMasked multiple sequence alignments with up to 28 species as prepared by the ENCODE alignment group (Margulies et al. 2007), whereas we used the MULTIZ alignments (with autoMZ driver) with up to 17 species available at the UCSC Genome Browser. In both cases, the alignments are prepared using the TBA/MULTIZ software (Blanchette et al. 2004). We used the latest assemblies (human hg18), whereas Washietl et al. (2007) use earlier assemblies (human hg17) because the TBA ENCODE alignments are only available for hg17. We used hg18 because it was the latest assembly with genome-wide multiple alignments available. Furthermore, the input alignments for RNAz and EvoFold were pre-processed according to different preferences of these programs (Washietl et al. 2007).
To compare our predictions with those of RNAz and EvoFold, we used all their candidates (low and high confidence) that overlapped neither exons nor the PhastCons conserved elements (38% of their total predictions) (Siepel et al. 2005) and compared them to our 4933 (75% of our total candidate regions) candidates in non-repetitive regions. Only 6.7% of these candidate regions overlap with EvoFold predictions, whereas 17.2% overlap with RNAz candidates (see Fig. 2). To estimate the significance of this overlap, we calculated P-values for our five G+C bins. For the two most G+C-rich G+C bins (45%–50% and 50%–100%, which contain the majority of our candidates), the overlap with EvoFold was significant (P < 10−5 in both bins). The overlap with RNAz was significant in all five G+C bins (P < 10−22, P < 10−17, P < 10−28, P < 10−27, and P < 10−39, ordered by increasing G%+C%.) In the regions that do not overlap exons, PhastCons conserved elements, or repeat regions, we add 3861 new candidates to the 6071 RNAz or EvoFold candidates. Furthermore, we predict 1654 candidates in regions that are in repeat regions in human (excluded by Washietl et al. 2007) and thereby add 5515 candidates to the 17,046 RNAz or EvoFold candidates in the ENCODE regions, corresponding to 32% of the total number of candidates.
Figure 2.

Overlap of predictions made by CMfinder, RNAz, and EvoFold. Only predictions that are not highly conserved (phastCons), outside exons, and repeat regions are considered, since these regions are the common subset of the input regions to these three programs. The total number for each program is indicated in parentheses below the label.
EvoFold has a strong preference for TA-rich regions, whereas RNAz prefers G+C-rich regions since the minimum free energy is important to RNAz. The CMfinder predictions are approximately normally distributed, centered on 53% GC content. Still, when considering that the background G+C content is 43%, it is clear that CMfinder also prefers G+C-rich regions that tend to be more structurally stable.
Candidate database
All of our candidate regions are available in an online database (http://genome.ku.dk/resources/cmf_encode). The database includes a variety of additional annotations such as the overlaps described above, occurrences such as conserved tetraloop motifs, and predicted microRNA using RNAmicro (Hertel and Stadler 2006). The database also supports easy access to subsets of the candidates with different features. For example, one can easily retrieve all candidates overlapping TARs/transfrags or all miRNA predictions. Furthermore, each candidate region is linked directly to the UCSC Genome Browser. Despite the relatively high false-positive rate, it is possible and simple to use the information in our database to select higher confidence predictions through the “Database Search” link. For example, one can choose predictions that overlap with EvoFold/RNAz predictions and/or overlap TARs/transfrags.
Realigning parts of the genomes
A benchmark study by Gardner et al. (2005) compared the relative performances of structure- versus sequence-based methods when aligning pairs of known tRNAs. The study revealed a dramatic divergence in performance for sequences with identity below ∼60%; that is, sequence-based methods were dramatically worse below this threshold. Note that Gardner et al. define pairwise sequence identity as IDENTITIES/MIN(length A, length B) for sequences A and B (Paul P. Gardner, pers. comm.), whereas we, dealing with multiple alignments, define this as IDENTITIES/MAX(length A, length B). IDENTITIES is the number of identical positions in the alignment, and the length is the gap-free length of the sequence. For example, the sequences ATGC and AG are 100% identical by the former definition, but only 50% identical by the latter. Applying our definition to Gardner et al.’s data lowers the pairwise sequence identities by 3% on average. Although Gardner et al.’s observation is based on pairwise alignments on tRNAs, it is reasonable to assume that there exists a sequence identity threshold, for sequence-based multiple alignment tools, below which the generated alignments will be suboptimal when considering structured ncRNAs. This means that one should be careful when searching for structured ncRNAs in sequence-based alignments when the sequence similarity is below this threshold, because these alignments will contain many more errors that will propagate through alignment-dependent methods. CMfinder considers both sequence and structure information and is therefore expected to perform better on regions with low sequence similarity. Considering that our input alignments have 50% average pairwise sequence similarity, it is clear that when RNA secondary structure is of importance, these alignments will often benefit from being realigned, taking structure into account. We calculated how much of the sequence is being realigned by CMfinder, compared to the original sequence-based alignment; as expected, the degree of realignment correlates with sequence similarity (Pearson correlation of −0.77) (see Fig. 3). Approximately one-quarter of the alignments show realignment in >50% of positions (see Methods).
Figure 3.

Average pairwise sequence similarity of the predicted motifs versus the fraction that has been realigned compared to the original alignments.
Most of the known ncRNA families probably exhibit artificially high sequence similarities because of ascertainment bias—members are often discovered based on sequence similarity. To demonstrate possible benefits of structure-aware alignment, we examined MULTIZ multiple alignment blocks identified by Wang et al. (2007) to contain matches to Rfam ncRNAs (Griffiths-Jones et al. 2003), with good matches to the Rfam model in all species in the same region of the alignment. In one example containing 10 mammals, with fairly high sequence identity (∼72%), neither EvoFold nor RNAz reports a candidate there. However, CMfinder identifies a candidate (composite score >5 and energy <−5) in all 10 species in good general agreement with the H/ACA snoRNA known there (Rfam accession RF00402). CMfinder’s alignment of the region differs from the MULTIZ alignment in only 13% of positions, yet this change is sufficient to flip the RNAz prediction from negative (“RNA probability” 0.11, based on using their script to select six organisms) to strongly positive (probability 0.98) (see Supplemental material). EvoFold did not predict anything for either alignment. While this is just one example, it does highlight the fact that even reasonably solid sequence-based alignments may not suffice for RNA discovery. Considering the high number of ENCODE region alignments with relatively low sequence similarities, it is reasonable to expect CMfinder, in many cases, to perform better on these alignments than sequence-alignment-dependent tools.
Furthermore, it should be noted that RNAz and EvoFold remove individual sequences with >25% and 20% gaps, respectively, as compared to human. This is not necessary when using CMfinder since it is alignment-independent. CMfinder found motifs in 1408 and 673 individual sequences that would have been removed because of too many gaps by EvoFold and RNAz, respectively. Also RNAz is limited to four to six sequences, thus they sample six sequences (repeated three times if there are more than 10 sequences in the alignment), optimizing the selected sequences to have sequence similarity as close to 80% as possible. EvoFold considers every sequence in the alignment, resulting in a lower score if any sequence is missing the motif. In contrast, although the number of species is a factor in its composite score, CMfinder can ignore a sequence if it does not contain the motif and still report a high-scoring motif for the rest of the sequences.
Experimental verification
An increasing number of ncRNAs are reported to be implicated in tissue-specific developmental and disease processes (for review, see Costa 2005), yet the precise biological function of most ncRNAs remains elusive. To further explore the biological relevance of our prediction method, we selected 11 high-scoring ncRNA candidates for experimental verification. We selected high confidence predictions by setting stricter score cutoffs (composite score >9 and energy <−15) and by requiring a minimum length of 60 and required more than five compensating base changes, indicating a possible evolutionary pressure to maintain the structure. We tested the expression of these 11 candidates in human RNA pools using strand-specific primers (see Methods). We found that eight out of 11 ncRNA candidates, indeed, could be detected in human RNA samples by reverse-transcription PCR (RT-PCR; ncRNA candidates #1, #2, #4, #7, #8, #9, #10, and #11) (Fig. 4A). Such expression may simply reflect transcriptional noise, yet current literature suggests that mammalian ncRNAs exhibit highly tissue-specific expression profiles, which is likely to be indicative of specialized functions in the organism (Ravasi et al. 2006; Sasaki et al. 2007). Hence, in order to expand our analysis and identify potential spatial and functional roles of our predicted set of ncRNAs, we performed an extensive expression analysis in 22 human tissues by RT-PCR totaling more than 250 separate duplicated reactions (see Methods). Our analysis demonstrated that 10 out of the 11 candidates are, indeed, expressed in one or more human tissues (Fig. 4B). Interestingly, this analysis showed that seven of 10 confirmed candidates exhibited a highly tissue-specific expression profile, whereas only two ncRNAs were more ubiquitously expressed (#10 and #11) (Fig. 4B). Hence, in agreement with the current consensus, we believe that the predicted ncRNAs may have highly defined biological roles (Ravasi et al. 2006; Sasaki et al. 2007). In addition, the highly differential expression patterns of the ncRNA candidates strongly suggest that the expression is real and not merely transcriptional noise, thus supporting the validity of our prediction method.
Figure 4.

Expression of predicted ncRNA candidates by RT-PCR and Northern blot analysis. (A) Strand-specific RT-PCR analysis of ncRNA candidates on human RNA pools (see Methods). β-Actin was used as control, yielding PCR products in the presence of reverse transcriptase (RT+), but not in its absence (RT−). (B) Tissue-specific expression of ncRNA candidates as evaluated by RT-PCR analysis of human RNA samples. The same β-actin controls as for A were used. (C) Expression of ncRNA candidates within the human CNS as evaluated by RT-PCR analysis. The same β-actin controls as for A and B were used. (D) Expression of ncRNA candidate #6 as evaluated by Northern blotting of human RNA samples from 11 tissues.
An interesting observation is that nine out of 11 ncRNA candidates were detected in brain (Fig. 4B). In fact, a similar enrichment of ncRNA expression in brain versus other tissues has previously been demonstrated in mouse (Ravasi et al. 2006), and several reports on the involvement and relative abundance of ncRNA in human CNS function and development have recently emerged (Cavaille et al. 2001; French et al. 2001; Pollard et al. 2006; Sone et al. 2007). Furthermore, an RNAz screen of porcine EST sequences revealed that developmental brain tissue seems to contain more ncRNAs than other tissues (Seemann et al. 2007). In order to examine the expression profile of our CNS-expressed candidates in more detail, we performed RT-PCR analysis on human RNA purified from total brain, fetal brain, cerebellum, hippocampus, and spinal cord (Fig. 4C). Again, distinct expression profiles were identified. For example, as observed in the other tissues, candidate #11 was expressed in all the investigated nervous tissues (Fig. 4C). Candidate #8, on the other hand, showed a more restricted expression profile, detected in fetal brain and, although less pronounced, hippocampus of adult brain (Fig. 4C). Hence, even within a single organ, the predicted ncRNA candidates appear to have highly specialized expression profiles, which is suggestive of a distinct biological function.
To expand our analysis, Northern blot analysis was performed for the 10 ncRNA candidates, confirmed by RT-PCR, on human RNA from 11 different tissues (Fig. 4D). In general, detection of ncRNAs by Northern blotting has proven very difficult as the majority of ncRNAs are low-abundance transcripts (Sasaki et al. 2007). However, we were able to detect bands for ncRNA candidate #6 (Fig. 4D), and the expression of candidate #6 was confirmed to be strictly brain-specific by the Northern blot analysis. The 2.8-kb-long transcript is located within a 4-kb-long intron of synapsin III (SYN3) along with five more non-overlapping CMfinder-predicted motifs on the same strand. In Figure 4D we have removed four tissues because of a high level of background noise, interfering with the results.
Next, we investigated the precise genomic locations of the ncRNAs; five of the ncRNA candidates (#1, #2, #6, #9, and #10) are located within intronic sequences of known genes, all but candidate #1 on the same strand. Overall, we find a good correlation between our ncRNA expression analysis and database searches for the predicted host mRNA; for instance, candidate #6 is located within an intron of synapsin III (SYN3), which is neuron-specific and predominantly expressed in the brain (Kao et al. 1998). This expression profile is well confirmed by both our RT-PCR and Northern blot analysis showing a clear brain-specific expression of ncRNA #6. Furthermore, candidate #9 is located within an intron of the GRM8 (glutamate receptor metabotropic 8) precursor encoding a G-protein-coupled metabotropic glutamate receptor expressed in the central nervous systems (Duvoisin et al. 1995). Again, our RT-PCR analysis confirms candidate #9 expression both in spinal cord and in most compartments of the brain and (Fig. 4B,C). Finally, candidate #10 is located within the primary TIMP3 RNA transcript (which lies antisense to intron 5 of SYN3) that encodes an inhibitor of matrix metalloproteinases (GenBank accession NM_000362). TIMP3 mRNA is rather broadly expressed predominantly in brain, kidney, and lung (Leco et al. 1994), which correlates well with the expression patterns of candidate #10 as evaluated by our RT-PCR analysis (Fig. 4B). In conclusion, we find by both RT-PCR and Northern blot analysis that predicted ncRNA candidates are expressed in a highly tissue-specific manner, which is likely indicative of specialized biological functions and thus supports the validity of our prediction method.
Discussion
Non-coding RNAs are receiving increasing attention in genome science. This study describes the first large-scale search for structured ncRNAs in several vertebrate genomes using a local structural motif finding algorithm, which has identified several thousand novel candidate ncRNAs. Our work complements a previous pairwise scan for local structured RNA elements in corresponding unaligned regions of the human and mouse genomes (Torarinsson et al. 2006) by extending it to multiple genomes and including a wider range of sequence similarities. Furthermore, except to indicate orthology, the scan was not dependent on sequence-based pre-aligned genomic regions, as is the case with RNAz and EvoFold scans (Washietl et al. 2007), allowing us to increase the number of ncRNAs candidates in the ENCODE regions by 32%. With a growing number of sequenced genomes, and with improving genome alignment methods that are capable of capturing orthology among phylogenetically diverse species, analysis of syntenic yet diverse regions becomes more feasible (Margulies et al. 2006). Alignments of increasingly diverse regions often mean decreasing average pairwise sequence similarity. This is problematic for sequence-based alignment methods. When searching for structured ncRNAs, one can therefore benefit from disregarding these alignments and realign the regions considering sequence and structure, often resulting in better alignments. Indeed, it has been shown, for pairwise alignments of tRNAs, that it is preferable also to consider structure when aligning these if sequence similarity is below ∼60% (Gardner et al. 2005).
There are several remaining challenges in this field. Extending the analysis to (presumably) syntenic unaligned regions adjacent to aligned regions is one important direction. The main obstacles in doing this is data collection complexity and increased computation time. Candidate scoring is another challenge. Although useful, we don’t believe that any of the methods used to date constitute the last word on this topic. Even seemingly simple issues like the dinucleotide composition of shuffled alignments used as null examples are problematic. Additionally, we expect many functionally important ncRNA motifs to be repeated in the genome, for example, cis-regulatory elements controlling several genes in a common pathway or multiple members of as-yet-unknown RNA families. There has been limited work to date attempting to identify or cluster repeated motifs predicted by genome-scale RNA discovery approaches (Torarinsson et al. 2007; Will et al. 2007). The CMfinder-based approach we have described in this paper potentially provides an efficient alternative to these clustering approaches. Since each of our RNA motifs is described by a covariance model, in principle, we could use each to scan the genome for additional instances. Pragmatically, using each to scan the set of sequences representing each other motif should be effective and fast enough to be feasible (Weinberg and Ruzzo 2006), since we would expect reasonable cross-species conservation of each motif instance. However, completion of a full-genome CMfinder scan is a prerequisite. Finally, there is big need for high-throughput methods, computational and experimental, to identify a potential function for the tens of thousands of candidates that have resulted from scans like this.
Methods
Data
The multiple alignments from the ENCODE regions were obtained from the UCSC Genome Browser, more specifically, the multiple alignments of 16 vertebrate genomes with the human genome (assembly hg18, March 2006). We post-processed these alignments to remove all alignments blocks that overlapped with exons of known genes (http://hgdownload.cse.ucsc.edu/goldenPath/hg18/database/knownGene.txt.gz) or the highly conserved PhastCons elements (http://hgdownload.cse.ucsc.edu/goldenPath/hg18/database/phastConsElements17way.txt.gz) in human. Furthermore, we made an additional set with the reverse complementary sequences of each sequence in the alignment. GENCODE, TARs, transfrags, EST, and IPS data were obtained from UCSC’s Table Browser (http://genome.ucsc.edu/cgi-bin/hgTables) and converted, when needed, from assembly hg17 to hg18 using their liftOver software (http://genome.ucsc.edu/cgi-bin/hgLiftOver). sRNA and lRNA data were obtained at http://transcriptome.affymetrix.com/publication/hs_whole_genome. EvoFold and RNAz candidates were obtained at http://www.tbi.univie.ac.at/papers/SUPPLEMENTS/ENCODE. Repetitive regions were defined by the UCSC RepeatMasker track for human (hg18).
False-positive rate
In order to estimate the false-positive rate, we shuffled all of our input alignments and ran CMfinder on them. The alignments were shuffled as described by Washietl and Hofacker (2004), resulting in random alignments of the same base composition, sequence conservation, and gap patterns. The shuffling method we used retains a coarse-grained pattern of conservation (only columns with mean pairwise identity >0.5 and <0.5 were shuffled with each other, respectively) (Washietl et al. 2007). Note that this shuffling does not conserve the dinucleotide frequencies, which is an unsolved problem for shuffling multiple alignments. Dinucleotide frequencies have an effect on the Gibbs free energies due to stacking interactions. Since the Gibbs free energy plays a role in our scoring of the candidates, this has an unknown effect on our estimated false-positive rate.
Running CMfinder
We ran CMfinder (version 0.2) separately on each alignment block in the MULTIZ alignment as well as the reverse complement of each such block. When running CMfinder, we output up to five single stem predictions (size range 30–100 bp) and five predictions containing two stems (size range 40–100 bp). This corresponds to running CMfinder with the options “-n 5 -m 30 -M 100” and then with the options “-n 5 -s 2 -m 40 -M 100.” Then we tried to combine the motifs using the greedy heuristics implemented in CMfinder’s CombMotif.pl procedure, which estimates alignment scores for concatenation of all pairs of motifs and combines them progressively by merging the two motifs with the highest concatenation score. See Yao et al. (2006) for more details about these options.
We ranked all CMfinder motifs using a heuristic scoring function that favors motifs with instances in diverged species and stable consensus secondary structure. CMfinder sometimes identifies purely structural motifs (e.g., alignments of single hairpins) that could easily arise by chance. Such motifs are usually scored well by both EvoFold and RNAz. To discriminate against such, likely spurious, structural motifs with no sequence conservation, we consider local sequence conservation in the scoring function. This is based on the observation that most known ncRNA motifs, even the ones with low sequence conservation, contain mosaic patterns of local sequence conservation, which are plausibly interaction sites for other molecules under strong selection. On the other hand, we penalize global sequence conservation, as highly similar sequences are more likely to be conserved by selection pressure on primary sequence than on structure. The final score is defined as
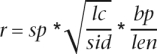 |
where sp is the number of species in which the motif occurs, lc the local sequence conservation score (see Supplemental Material for details), sid the global average pairwise sequence identity, bp the number of base pairs in the consensus structure, and len the alignment length. This score is referred to as the “composite score” (see Supplemental Material for details). A variant of this somewhat ad hoc scoring scheme performed well on ncRNA discovery in bacteria (Weinberg et al. 2007; Yao et al. 2007). The score used here is length-normalized to favor motifs with compact RNA structure. We have tried a few alternatives, including RNAz and Evofold, both of which strongly favor short, stable stem–loop motifs with low sequence similarity that are very likely to be aligned by chance. We have also tried to integrate our motif features for scoring by machine-learning algorithms including support vector machine (SVM) and logistic regression, but these methods did not perform well, probably because of the heterogeneity of the features and limitations of available training data.
After systematically studying various cutoffs, we chose to focus on candidates with a composite score >5 and Gibbs energy <−5, which resulted in a large number of candidates with a reasonable false-positive rate (see Supplemental Material for details). The energy is computed as the average energy of each sequence in the alignment as calculated by RNAfold (Hofacker et al. 1994) when constrained to the secondary structure annotated by CMfinder.
P-value calculation
To calculate the P-values, we counted the number of candidate regions whose center nucleotide overlaps the data we are testing against, that is, TARs. To get a P-value, we compare it to the null model that each candidate is a dart thrown randomly onto the genome. If the TARs cover a fraction P of the ENCODE nucleotides in MAF blocks (our input data), then it is a simple binomial model: each of the N darts has probability P of hitting a TAR. For N candidates, the expected number of hits is μ = N * P, with a standard deviation
We then calculate the P-value using the normal approximation to the binomial distribution, pnorm function in R [pnorm (observed, μ, σ, lower.tail = F)]. Out of a concern that various edge effects might distort the statistics, we also calculated the P-values using the leftmost and rightmost nucleotide, instead of the center nucleotide. This gives very similar results, although, when comparing to RNAz and EvoFold, the P-values were a bit worse, probably because they are global and use window lengths, whereas CMfinder is local, therefore an overlap with our candidates’ central nucleotide to RNAz and EvoFold candidates seems more likely. See Supplemental material for all the P-values.
Realignment calculation
To quantify how much has been realigned by CMfinder in a given motif compared to the original multiple alignment (see Fig. 3), we calculate the following quantities. Let sp be the number of sequences in the CMfinder alignment, and define m to be the number of matched positions in that alignment, that is, the number of quadruples (s, t, i, j) with 1 ≤ s < t ≤ sp and such that position i of sequence s is aligned with position j of sequence t. Let v be the number of those matches that are realigned relative to the MULTIZ alignment, that is, the number of quadruples as above for which position i of s is matched to position j of t in the CMfinder alignment, but not in the MULTIZ alignment (i and j are aligned either to nucleotides in different positions or to gaps). The overall realignment fraction we report is v/m. For example, if we have two multiple alignments, A and B, of four sequences that are all 10 bp long, we will compare all six possible sequence pairs (all pair combinations of the four sequences). If we have, say, six columns that are aligned differently in alignments A and B between sequences 1 and 3 and that the rest is aligned alike, then we would say that 10% [6/(6*10)] of alignment B is realigned compared to alignment A.
Experiments
The tissue-specific expression profiles of 11 candidate ncRNAs were determined by RT-PCR using purified total RNA from 22 different human tissues (adrenal gland, bone marrow, brain [whole, fetal, cerebellum, and hippocampus], kidney, liver [fetal], lung, prostate, salivary gland, skeletal muscle, spleen, testis, thymus, thyroid gland, trachea, uterus, colon, and small intestine). cDNA was generated by reverse transcription (RT) using M-MLV SuperScript III Reverse Transcriptase (Invitrogen). The RT was carried out according to the supplied standard protocol using either random hexamer primers (Fig. 4B) or gene-specific primers to test for strand specificity (Fig. 4A) (see the Supplemental Material for the primer list). A total of 5 pmol of primer and ∼1 μg of RNA was used per 20-μL RT reaction. Directly upon completion of the RT, the cDNA was amplified by PCR using HotStarTaq DNA polymerase (Qiagen) according to the supplied protocol. The PCR was carried out on ∼10% of the total cDNA (by mass per 20-μL RT reaction) using the following program: 6 min at 95°C denaturing (denaturing for 30 sec at 95°C; annealing for 30 sec at 54°–56°C; elongation for 30 sec at 72°C) (40 cycles); and elongation for 10 min at 72°C. A primer set for β-actin was used as a positive control. Blank and negative “no RT” RNA controls (equal mass of RNA to cDNA) were also included to test for DNA contamination of the RNA samples. The PCR products were visualized by ethidium bromide staining on a 2% agarose gel. The complete procedure of RT-PCR and gel visualization was performed at least twice for each candidate in each individual tissue. The identity of the detected DNA fragments was confirmed by sequencing using the BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems) according to the supplied protocol.
For Northern blot analysis of ncRNA expression, Nylon membranes with pre-blotted human RNA samples (15 μg/tissue; Zyagen) were hybridized at 37°C in Ultrahyb hybridization buffer (Ambion) with 80-nt end-labeled probes antisense to the predicted ncRNAs. Upon overnight hybridization, membranes were washed in 2× SSC, 0.1% SDS, and bands were visualized by PhosphorImaging.
Acknowledgments
We thank Phil Green, Graham McVicker, Jakob H. Havgaard, and Luc Jaeger for useful discussions. We acknowledge funding from the Danish Research Council for production and technology and the Danish Center for Scientific Computation. Wilhelm Johannsen Centre for Functional Genome Research is established by the Danish National Research Foundation.
Footnotes
[Supplemental material is available online at www.genome.org.]
Article published online before print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.6887408
References
- Bertone P., Stoc V., Royce T.E., Rozowsky J.S., Urban A.E., Zhu X., Rinn J.L., Tongprasit W., Samanta M., Weissman S., Stoc V., Royce T.E., Rozowsky J.S., Urban A.E., Zhu X., Rinn J.L., Tongprasit W., Samanta M., Weissman S., Royce T.E., Rozowsky J.S., Urban A.E., Zhu X., Rinn J.L., Tongprasit W., Samanta M., Weissman S., Rozowsky J.S., Urban A.E., Zhu X., Rinn J.L., Tongprasit W., Samanta M., Weissman S., Urban A.E., Zhu X., Rinn J.L., Tongprasit W., Samanta M., Weissman S., Zhu X., Rinn J.L., Tongprasit W., Samanta M., Weissman S., Rinn J.L., Tongprasit W., Samanta M., Weissman S., Tongprasit W., Samanta M., Weissman S., Samanta M., Weissman S., Weissman S., et al. Global identification of human transcribed sequences with genome tiling arrays. Science. 2004;306:2242–2246. doi: 10.1126/science.1103388. [DOI] [PubMed] [Google Scholar]
- Blanchette M., Kent W.J., Riemer C., Elnitski L., Smit A.F., Roskin K.M., Baertsch R., Rosenbloom K., Clawson H., Green E.D., Kent W.J., Riemer C., Elnitski L., Smit A.F., Roskin K.M., Baertsch R., Rosenbloom K., Clawson H., Green E.D., Riemer C., Elnitski L., Smit A.F., Roskin K.M., Baertsch R., Rosenbloom K., Clawson H., Green E.D., Elnitski L., Smit A.F., Roskin K.M., Baertsch R., Rosenbloom K., Clawson H., Green E.D., Smit A.F., Roskin K.M., Baertsch R., Rosenbloom K., Clawson H., Green E.D., Roskin K.M., Baertsch R., Rosenbloom K., Clawson H., Green E.D., Baertsch R., Rosenbloom K., Clawson H., Green E.D., Rosenbloom K., Clawson H., Green E.D., Clawson H., Green E.D., Green E.D., et al. Aligning mulitple genomic sequences with the threaded blockset aligner. Genome Res. 2004;14:708–715. doi: 10.1101/gr.1933104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavaille J., Vitali P., Basyuk E., Huttenhofer A., Bachellerie J.P., Vitali P., Basyuk E., Huttenhofer A., Bachellerie J.P., Basyuk E., Huttenhofer A., Bachellerie J.P., Huttenhofer A., Bachellerie J.P., Bachellerie J.P. A novel brain-specific box C/D small nucleolar RNA processed from tandemly repeated introns of a noncoding RNA gene in rats. J. Biol. Chem. 2001;276:26374–26383. doi: 10.1074/jbc.M103544200. [DOI] [PubMed] [Google Scholar]
- Cheng J., Kapranov P., Drenkow J., Dike S., Brubaker S., Patel S., Long J., Stern D., Tammana H., Helt G., Kapranov P., Drenkow J., Dike S., Brubaker S., Patel S., Long J., Stern D., Tammana H., Helt G., Drenkow J., Dike S., Brubaker S., Patel S., Long J., Stern D., Tammana H., Helt G., Dike S., Brubaker S., Patel S., Long J., Stern D., Tammana H., Helt G., Brubaker S., Patel S., Long J., Stern D., Tammana H., Helt G., Patel S., Long J., Stern D., Tammana H., Helt G., Long J., Stern D., Tammana H., Helt G., Stern D., Tammana H., Helt G., Tammana H., Helt G., Helt G., et al. Transcriptional maps of 10 human chromosomes at 5-nucleotide resolution. Science. 2005;308:1149–1154. doi: 10.1126/science.1108625. [DOI] [PubMed] [Google Scholar]
- Costa F.F. Non-coding RNAs: New players in eukaryotic biology. Gene. 2005;357:83–94. doi: 10.1016/j.gene.2005.06.019. [DOI] [PubMed] [Google Scholar]
- Ding Y., Chan C.Y., Lawrence C.E., Chan C.Y., Lawrence C.E., Lawrence C.E. Sfold web server for statistical folding and rational design of nucleic acids. Nucleic Acids Res. 2004;32:W135–W141. doi: 10.1093/nar/gkh449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowell R.D., Eddy S.R., Eddy S.R. Efficient pairwise RNA structure prediction and alignment using sequence alignment constraints. BMC Bioinformatics. 2006;7:400. doi: 10.1186/1471-2105-7-400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duvoisin R.M., Zhang C., Ramonell K., Zhang C., Ramonell K., Ramonell K. A novel metabotropic glutamate receptor expressed in the retina and olfactory bulb. J. Neurosci. 1995;15:3075–3083. doi: 10.1523/JNEUROSCI.15-04-03075.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The ENCODE Project Consortium Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;447:799–816. doi: 10.1038/nature05874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- French P.J., Bliss T.V., O’Connor V., Bliss T.V., O’Connor V., O’Connor V. Ntab, a novel non-coding RNA abundantly expressed in rat brain. Neuroscience. 2001;108:207–215. doi: 10.1016/s0306-4522(01)00408-0. [DOI] [PubMed] [Google Scholar]
- Fu H., Tie Y., Xu C., Zhang Z., Zhu J., Shi Y., Jiang H., Sun Z., Zheng X., Tie Y., Xu C., Zhang Z., Zhu J., Shi Y., Jiang H., Sun Z., Zheng X., Xu C., Zhang Z., Zhu J., Shi Y., Jiang H., Sun Z., Zheng X., Zhang Z., Zhu J., Shi Y., Jiang H., Sun Z., Zheng X., Zhu J., Shi Y., Jiang H., Sun Z., Zheng X., Shi Y., Jiang H., Sun Z., Zheng X., Jiang H., Sun Z., Zheng X., Sun Z., Zheng X., Zheng X. Identification of human fetal liver miRNAs by a novel method. FEBS Lett. 2005;579:3849–3854. doi: 10.1016/j.febslet.2005.05.064. [DOI] [PubMed] [Google Scholar]
- Gardner P.P., Wilm A., Washietl S., Wilm A., Washietl S., Washietl S. A benchmark of multiple sequence alignment programs upon structural RNAs. Nucleic Acids Res. 2005;33:2433–2439. doi: 10.1093/nar/gki541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths-Jones S. The microRNA Registry. Nucleic Acids Res. 2004;32:D109–D111. doi: 10.1093/nar/gkh023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths-Jones S., Bateman A., Marshall M., Khanna A., Eddy S.R., Bateman A., Marshall M., Khanna A., Eddy S.R., Marshall M., Khanna A., Eddy S.R., Khanna A., Eddy S.R., Eddy S.R. Rfam: An RNA family database. Nucleic Acids Res. 2003;31:439–441. doi: 10.1093/nar/gkg006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths-Jones S., Grocock R.J., van Dongen S., Bateman A., Enright A.J., Grocock R.J., van Dongen S., Bateman A., Enright A.J., van Dongen S., Bateman A., Enright A.J., Bateman A., Enright A.J., Enright A.J. miRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Res. 2006;34:D140–D144. doi: 10.1093/nar/gkj112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harmanci A.O., Sharma G., Mathews D.H., Sharma G., Mathews D.H., Mathews D.H. Efficient pairwise RNA structure prediction using probabilistic alignment constraints in Dynalign. Bioinformatics. 2007;8:130. doi: 10.1186/1471-2105-8-130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrow J., Denoeud F., Frankish A., Reymond A., Chen C.K., Chrast J., Lagarde J., Gilbert J.G., Storey R., Swarbreck D., Denoeud F., Frankish A., Reymond A., Chen C.K., Chrast J., Lagarde J., Gilbert J.G., Storey R., Swarbreck D., Frankish A., Reymond A., Chen C.K., Chrast J., Lagarde J., Gilbert J.G., Storey R., Swarbreck D., Reymond A., Chen C.K., Chrast J., Lagarde J., Gilbert J.G., Storey R., Swarbreck D., Chen C.K., Chrast J., Lagarde J., Gilbert J.G., Storey R., Swarbreck D., Chrast J., Lagarde J., Gilbert J.G., Storey R., Swarbreck D., Lagarde J., Gilbert J.G., Storey R., Swarbreck D., Gilbert J.G., Storey R., Swarbreck D., Storey R., Swarbreck D., Swarbreck D., et al. GENCODE: Producing a reference annotation for ENCODE. Genome Biol. 2006;7:1–9. doi: 10.1186/gb-2006-7-s1-s4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Havgaard J.H., Torarinsson E., Gorodkin J., Torarinsson E., Gorodkin J., Gorodkin J. Fast pairwise structural RNA alignments by pruning of the dynamical programming matrix. PLoS Comput. Biol. 2007;3:e193. doi: 10.1371/journal.pcbi.0030193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hertel J., Stadler P.F., Stadler P.F. Hairpins in a haystack: Recognizing miRNA precursors in comparative genomics data. Bioinformatics. 2006;22:e197–e202. doi: 10.1093/bioinformatics/btl257. [DOI] [PubMed] [Google Scholar]
- Hofacker I.L., Fontana W., Stadler P.F., Bonhoeffer L.S., Tacker M., Schuster P., Fontana W., Stadler P.F., Bonhoeffer L.S., Tacker M., Schuster P., Stadler P.F., Bonhoeffer L.S., Tacker M., Schuster P., Bonhoeffer L.S., Tacker M., Schuster P., Tacker M., Schuster P., Schuster P. Fast folding and comparison of RNA secondary structures. Monatsh. Chem. 1994;125:167–188. [Google Scholar]
- Holmes I. Accelerated probabilistic inference of RNA structure evolution. BMC Bioinformatics. 2005;6:73. doi: 10.1186/1471-2105-6-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao H.T., Porton B., Czernik A.J., Feng J., Yiu G., Haring M., Benfenati F., Greengard P., Porton B., Czernik A.J., Feng J., Yiu G., Haring M., Benfenati F., Greengard P., Czernik A.J., Feng J., Yiu G., Haring M., Benfenati F., Greengard P., Feng J., Yiu G., Haring M., Benfenati F., Greengard P., Yiu G., Haring M., Benfenati F., Greengard P., Haring M., Benfenati F., Greengard P., Benfenati F., Greengard P., Greengard P. A third member of the synapsin gene family. Proc. Natl. Acad. Sci. 1998;95:4667–4672. doi: 10.1073/pnas.95.8.4667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapranov P., Cheng J., Dike S., Nix D.A., Duttagupta R., Willingham A.T., Stadler P.F., Hertel J., Hackermüller J., Hofacker I.L., Cheng J., Dike S., Nix D.A., Duttagupta R., Willingham A.T., Stadler P.F., Hertel J., Hackermüller J., Hofacker I.L., Dike S., Nix D.A., Duttagupta R., Willingham A.T., Stadler P.F., Hertel J., Hackermüller J., Hofacker I.L., Nix D.A., Duttagupta R., Willingham A.T., Stadler P.F., Hertel J., Hackermüller J., Hofacker I.L., Duttagupta R., Willingham A.T., Stadler P.F., Hertel J., Hackermüller J., Hofacker I.L., Willingham A.T., Stadler P.F., Hertel J., Hackermüller J., Hofacker I.L., Stadler P.F., Hertel J., Hackermüller J., Hofacker I.L., Hertel J., Hackermüller J., Hofacker I.L., Hackermüller J., Hofacker I.L., Hofacker I.L., et al. RNA maps reveal new RNA classes and a possible function for pervasive transcription. Science. 2007;316:1484–1488. doi: 10.1126/science.1138341. [DOI] [PubMed] [Google Scholar]
- Kent W.J., Sugnet C.W., Furey T.S., Roskin K.M., Pringle T.H., Zahler A.M., Haussler D., Sugnet C.W., Furey T.S., Roskin K.M., Pringle T.H., Zahler A.M., Haussler D., Furey T.S., Roskin K.M., Pringle T.H., Zahler A.M., Haussler D., Roskin K.M., Pringle T.H., Zahler A.M., Haussler D., Pringle T.H., Zahler A.M., Haussler D., Zahler A.M., Haussler D., Haussler D. The Human Genome Browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leco K.J., Khokha R., Pavloff N., Hawkes S.P., Edwards D.R., Khokha R., Pavloff N., Hawkes S.P., Edwards D.R., Pavloff N., Hawkes S.P., Edwards D.R., Hawkes S.P., Edwards D.R., Edwards D.R. Tissue inhibitor of metalloproteinases-3 (TIMP-3) is an extracellular matrix-associated protein with a distinctive pattern of expression in mouse cells and tissues. J. Biol. Chem. 1994;269:9352–9360. [PubMed] [Google Scholar]
- Lunter G., Ponting C.P., Hein J., Ponting C.P., Hein J., Hein J. Genome-wide identification of human functional DNA using a neutral indel model. PLoS Comput. Biol. 2006;2:e5. doi: 10.1371/journal.pcbi.0020005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margulies E.H., Chen C.W., Green E.D., Chen C.W., Green E.D., Green E.D. Differences between pair-wise and multi-sequence alignment methods affect vertebrate genome comparisons. Trends Genet. 2006;22:187–193. doi: 10.1016/j.tig.2006.02.005. [DOI] [PubMed] [Google Scholar]
- Margulies E.H., Cooper G.M., Asimenos G., Thomas D.J., Dewey C.N., Siepel A., Birney E., Keefe D., Schwartz A.S., Hou M., Cooper G.M., Asimenos G., Thomas D.J., Dewey C.N., Siepel A., Birney E., Keefe D., Schwartz A.S., Hou M., Asimenos G., Thomas D.J., Dewey C.N., Siepel A., Birney E., Keefe D., Schwartz A.S., Hou M., Thomas D.J., Dewey C.N., Siepel A., Birney E., Keefe D., Schwartz A.S., Hou M., Dewey C.N., Siepel A., Birney E., Keefe D., Schwartz A.S., Hou M., Siepel A., Birney E., Keefe D., Schwartz A.S., Hou M., Birney E., Keefe D., Schwartz A.S., Hou M., Keefe D., Schwartz A.S., Hou M., Schwartz A.S., Hou M., Hou M., et al. Analyses of deep mammalian sequence alignments and constraint predictions for 1% of the human genome. Genome Res. 2007;17:746–759. doi: 10.1101/gr.6034307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen J.S., Bejerano G., Siepel A., Rosenbloom K., Lindblad-Toh K., Lander E., Rogers J., Kent J., Miller W., Haussler D., Bejerano G., Siepel A., Rosenbloom K., Lindblad-Toh K., Lander E., Rogers J., Kent J., Miller W., Haussler D., Siepel A., Rosenbloom K., Lindblad-Toh K., Lander E., Rogers J., Kent J., Miller W., Haussler D., Rosenbloom K., Lindblad-Toh K., Lander E., Rogers J., Kent J., Miller W., Haussler D., Lindblad-Toh K., Lander E., Rogers J., Kent J., Miller W., Haussler D., Lander E., Rogers J., Kent J., Miller W., Haussler D., Rogers J., Kent J., Miller W., Haussler D., Kent J., Miller W., Haussler D., Miller W., Haussler D., Haussler D. Identification and classification of conserved RNA secondary structures in the human genome. PLoS Comput. Biol. 2006;2:e33. doi: 10.1371/journal.pcbi.0020033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard K.S., Salama S.R., Lambert N., Lambot M.A., Coppens S., Pedersen J.S., Katzman S., King B., Onodera C., Siepel A., Salama S.R., Lambert N., Lambot M.A., Coppens S., Pedersen J.S., Katzman S., King B., Onodera C., Siepel A., Lambert N., Lambot M.A., Coppens S., Pedersen J.S., Katzman S., King B., Onodera C., Siepel A., Lambot M.A., Coppens S., Pedersen J.S., Katzman S., King B., Onodera C., Siepel A., Coppens S., Pedersen J.S., Katzman S., King B., Onodera C., Siepel A., Pedersen J.S., Katzman S., King B., Onodera C., Siepel A., Katzman S., King B., Onodera C., Siepel A., King B., Onodera C., Siepel A., Onodera C., Siepel A., Siepel A., et al. An RNA gene expressed during cortical development evolved rapidly in humans. Nature. 2006;443:167–172. doi: 10.1038/nature05113. [DOI] [PubMed] [Google Scholar]
- Ravasi T., Suzuki H., Pang K.C., Katayama S., Furuno M., Okunishi R., Fukuda S., Ru K., Frith M.C., Gongora M.M., Suzuki H., Pang K.C., Katayama S., Furuno M., Okunishi R., Fukuda S., Ru K., Frith M.C., Gongora M.M., Pang K.C., Katayama S., Furuno M., Okunishi R., Fukuda S., Ru K., Frith M.C., Gongora M.M., Katayama S., Furuno M., Okunishi R., Fukuda S., Ru K., Frith M.C., Gongora M.M., Furuno M., Okunishi R., Fukuda S., Ru K., Frith M.C., Gongora M.M., Okunishi R., Fukuda S., Ru K., Frith M.C., Gongora M.M., Fukuda S., Ru K., Frith M.C., Gongora M.M., Ru K., Frith M.C., Gongora M.M., Frith M.C., Gongora M.M., Gongora M.M., et al. Experimental validation of the regulated expression of large numbers of non-coding RNAs from the mouse genome. Genome Res. 2006;16:11–19. doi: 10.1101/gr.4200206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sankoff D. Simultaneous solution of the RNA folding, alignment and protosequence problems. SIAM J. Appl. Math. 1985;45:810–825. [Google Scholar]
- Sasaki Y.T., Sano M., Ideue T., Kin T., Asai K., Hirose T., Sano M., Ideue T., Kin T., Asai K., Hirose T., Ideue T., Kin T., Asai K., Hirose T., Kin T., Asai K., Hirose T., Asai K., Hirose T., Hirose T. Identification and characterization of human non-coding RNAs with tissue-specific expression. Biochem. Biophys. Res. Commun. 2007;357:991–996. doi: 10.1016/j.bbrc.2007.04.034. [DOI] [PubMed] [Google Scholar]
- Seemann S.E., Gilchrist M.J., Hofacker I.L., Stadler P.F., Gorodkin J., Gilchrist M.J., Hofacker I.L., Stadler P.F., Gorodkin J., Hofacker I.L., Stadler P.F., Gorodkin J., Stadler P.F., Gorodkin J., Gorodkin J. Detection of RNA structures in porcine EST data and related mammals. BMC Genomics. 2007;8:316. doi: 10.1186/1471-2164-8-316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siepel A., Bejerano G., Pedersen J.S., Hinrichs A.S., Hou M., Rosenbloom K., Clawson H., Spieth J., Hillier L.W., Richards S., Bejerano G., Pedersen J.S., Hinrichs A.S., Hou M., Rosenbloom K., Clawson H., Spieth J., Hillier L.W., Richards S., Pedersen J.S., Hinrichs A.S., Hou M., Rosenbloom K., Clawson H., Spieth J., Hillier L.W., Richards S., Hinrichs A.S., Hou M., Rosenbloom K., Clawson H., Spieth J., Hillier L.W., Richards S., Hou M., Rosenbloom K., Clawson H., Spieth J., Hillier L.W., Richards S., Rosenbloom K., Clawson H., Spieth J., Hillier L.W., Richards S., Clawson H., Spieth J., Hillier L.W., Richards S., Spieth J., Hillier L.W., Richards S., Hillier L.W., Richards S., Richards S., et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005;15:1034–1050. doi: 10.1101/gr.3715005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sone M., Hayashi T., Tarui H., Agata K., Takeichi M., Nakagawa S., Hayashi T., Tarui H., Agata K., Takeichi M., Nakagawa S., Tarui H., Agata K., Takeichi M., Nakagawa S., Agata K., Takeichi M., Nakagawa S., Takeichi M., Nakagawa S., Nakagawa S. The mRNA-like noncoding RNA Gomafu constitutes a novel nuclear domain in a subset of neurons. J. Cell Sci. 2007;120:2498–2506. doi: 10.1242/jcs.009357. [DOI] [PubMed] [Google Scholar]
- Torarinsson E., Sawera M., Havgaard J.H., Fredholm M., Gorodkin J., Sawera M., Havgaard J.H., Fredholm M., Gorodkin J., Havgaard J.H., Fredholm M., Gorodkin J., Fredholm M., Gorodkin J., Gorodkin J. Thousand of corresponding human and mouse genomic regions unalignable in primary sequence contain common RNA strucuture. Genome Res. 2006;16:885–889. doi: 10.1101/gr.5226606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torarinsson E., Havgaard J.H., Gorodkin J., Havgaard J.H., Gorodkin J., Gorodkin J. Multiple structural alignment and clustering of RNA sequences. Bioinformatics. 2007;23:926–932. doi: 10.1093/bioinformatics/btm049. [DOI] [PubMed] [Google Scholar]
- Wang A.X., Ruzzo W.L., Tompa M., Ruzzo W.L., Tompa M., Tompa M. How accurately is ncRNA aligned within whole-genome multiple alignments? BMC Bioinformatics. 2007;8:417. doi: 10.1186/1471-2105-8-417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washietl S., Hofacker I.L., Hofacker I.L. Consensus folding of aligned sequences as a new measure for the detection of functional RNAs by comparative genomics. J. Mol. Biol. 2004;342:19–30. doi: 10.1016/j.jmb.2004.07.018. [DOI] [PubMed] [Google Scholar]
- Washietl S., Hofacker I.L., Stadler P.F., Hofacker I.L., Stadler P.F., Stadler P.F. Fast and reliable prediction of noncoding RNAs. Proc. Natl. Acad. Sci. 2005a;102:2454–2459. doi: 10.1073/pnas.0409169102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washietl S., Hofacker I.L., Lukasser M., Hüttenhofer A., Stadler P.F., Hofacker I.L., Lukasser M., Hüttenhofer A., Stadler P.F., Lukasser M., Hüttenhofer A., Stadler P.F., Hüttenhofer A., Stadler P.F., Stadler P.F. Mapping of conserved RNA secondary structures predicts thousands of functional non-coding RNAs in the human genome. Nat. Biotechnol. 2005b;23:1383–1390. doi: 10.1038/nbt1144. [DOI] [PubMed] [Google Scholar]
- Washietl S., Pedersen J.S., Korbel J.O., Gruber A.R., Hackermuller J., Hertel J., Lindemeyer M., Reiche K., Stocsits C., Tanzer A., Pedersen J.S., Korbel J.O., Gruber A.R., Hackermuller J., Hertel J., Lindemeyer M., Reiche K., Stocsits C., Tanzer A., Korbel J.O., Gruber A.R., Hackermuller J., Hertel J., Lindemeyer M., Reiche K., Stocsits C., Tanzer A., Gruber A.R., Hackermuller J., Hertel J., Lindemeyer M., Reiche K., Stocsits C., Tanzer A., Hackermuller J., Hertel J., Lindemeyer M., Reiche K., Stocsits C., Tanzer A., Hertel J., Lindemeyer M., Reiche K., Stocsits C., Tanzer A., Lindemeyer M., Reiche K., Stocsits C., Tanzer A., Reiche K., Stocsits C., Tanzer A., Stocsits C., Tanzer A., Tanzer A., et al. Structured RNAs in the ENCODE selected regions of the human genome. Genome Res. 2007;17:852–864. doi: 10.1101/gr.5650707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg Z., Ruzzo W.L., Ruzzo W.L. Sequence-based heuristics for faster annotation of non-coding RNA families. Bioinformatics. 2006;22:35–39. doi: 10.1093/bioinformatics/bti743. [DOI] [PubMed] [Google Scholar]
- Weinberg Z., Barrick J.E., Yao Z., Roth A., Kim J.N., Gore J., Wang J.X., Lee E.R., Block K.F., Sudarsan N., Barrick J.E., Yao Z., Roth A., Kim J.N., Gore J., Wang J.X., Lee E.R., Block K.F., Sudarsan N., Yao Z., Roth A., Kim J.N., Gore J., Wang J.X., Lee E.R., Block K.F., Sudarsan N., Roth A., Kim J.N., Gore J., Wang J.X., Lee E.R., Block K.F., Sudarsan N., Kim J.N., Gore J., Wang J.X., Lee E.R., Block K.F., Sudarsan N., Gore J., Wang J.X., Lee E.R., Block K.F., Sudarsan N., Wang J.X., Lee E.R., Block K.F., Sudarsan N., Lee E.R., Block K.F., Sudarsan N., Block K.F., Sudarsan N., Sudarsan N., et al. Identification of 22 candidate structured RNAs in bacteria using the CMfinder comparative genomics pipeline. Nucleic Acids Res. 2007;35:4809–4819. doi: 10.1093/nar/gkm487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westhof E., Michel F., Michel F. Prediction and experimental investigation of RNA secondary and tertiary foldings. In: Nagai K., Mattaj I.W., Mattaj I.W., editors. RNA–protein interactions. Oxford University Press; New York: 1994. pp. 26–51. [Google Scholar]
- Westhof E., Auffinger E., Gaspin C., Auffinger E., Gaspin C., Gaspin C. DNA and RNA structure prediction. In: Bihop M.J., Rawlings C.J., Rawlings C.J., editors. DNA–protein sequence analysis. Oxford University Press; New York: 1996. pp. 255–278. [Google Scholar]
- Will S., Reiche K., Hofacker I.L., Stadler P.F., Backofen R., Reiche K., Hofacker I.L., Stadler P.F., Backofen R., Hofacker I.L., Stadler P.F., Backofen R., Stadler P.F., Backofen R., Backofen R. Inferring noncoding RNA families and classes by means of genome-scale structure-based clustering. PLoS Comput. Biol. 2007;3:e65. doi: 10.1371/journal.pcbi.0030065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Z., Weinberg Z., Ruzzo W.L., Weinberg Z., Ruzzo W.L., Ruzzo W.L. CMfinder—A covariance model based RNA motif finding algorithm. Bioinformatics. 2006;22:445–452. doi: 10.1093/bioinformatics/btk008. [DOI] [PubMed] [Google Scholar]
- Yao Z., Barrick J.E., Weinberg Z., Neph S., Breaker R.R., Tompa M., Ruzzo W.L., Barrick J.E., Weinberg Z., Neph S., Breaker R.R., Tompa M., Ruzzo W.L., Weinberg Z., Neph S., Breaker R.R., Tompa M., Ruzzo W.L., Neph S., Breaker R.R., Tompa M., Ruzzo W.L., Breaker R.R., Tompa M., Ruzzo W.L., Tompa M., Ruzzo W.L., Ruzzo W.L. A computational pipeline for high-throughput discovery of cis-regulatory noncoding RNA in prokaryotes. PLoS Comput. Biol. 2007;3:e126. doi: 10.1371/journal.pcbi.0030126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]