

Abstract
Despite its recognized utility, the extent to which evolutionary sequence conservation-based approaches may systematically overlook functional noncoding sequences remains unclear. We have tiled across sequence encompassing the zebrafish phox2b gene, ultimately evaluating 48 amplicons corresponding to all noncoding sequences therein for enhancer activity in zebrafish. Post hoc analyses of this interval utilizing five commonly used measures of evolutionary constraint (AVID, MLAGAN, SLAGAN, phastCons, WebMCS) demonstrate that each systematically overlooks regulatory sequences. These established algorithms detected only 29%–61% of our identified regulatory elements, consistent with the suggestion that many regulatory sequences may not be readily detected by metrics of sequence constraint. However, we were able to discriminate functional from nonfunctional sequences based upon GC composition and identified position weight matrices (PWM), demonstrating that, in at least one case, deleting sequences containing a subset of these PWMs from one identified regulatory element abrogated its regulatory function. Collectively, these data demonstrate that the noncoding functional component of vertebrate genomes may far exceed estimates predicated on evolutionary constraint.
Regulatory sequences play significant roles in development (Stathopoulos and Levine 2005; Davidson and Erwin 2006), disease (Lettice et al. 2003; Emison et al. 2005; Kleinjan and van Heyningen 2005), and interspecific phenotypic variation (Levine and Tjian 2003; Stathopoulos and Levine 2005; Davidson and Erwin 2006). Efforts to identify regulatory sequences have been heavily weighted on the use of evolutionary sequence conservation through comparative sequence analysis (Marshall et al. 1994; Aparicio et al. 1995; de la Calle-Mustienes et al. 2005; Grice et al. 2005; Woolfe et al. 2005; Fisher et al. 2006a; Pennacchio et al. 2006; Prabhakar et al. 2006; Venkatesh et al. 2006; Pennacchio et al. 2007) because, in contrast to coding sequences, we are unable to reliably predict the identity of regulatory noncoding regions based on sequence alone. However, no single evolutionary distance or metric of constraint has been shown to reliably capture all regulatory sequence intervals. Although some studies rely heavily upon stringent conservation (e.g., 100% identity over 200 base pairs [bp]) across great evolutionary distances (human versus fugu) to identify putative regulatory sequences (Bejerano et al. 2004; Pennacchio et al. 2006), many functional sequences have been identified under less rigorous parameters or closer evolutionary distances (Frazer et al. 1995; Fisher et al. 2006a). Additionally, a small number of examples exist of regulatory sequences that are not conserved, even among mammals (Bejerano et al. 2004; King et al. 2005; Siepel et al. 2005; Taylor et al. 2006; The ENCODE Project Consortium 2007).
Some straightforward questions remain unanswered in studies of this type. First, how efficiently does a metric of constraint actually detect functional information? Second, with what frequency are functional sequences overlooked when analyses are restricted to a metric of constraint? Insight into these issues requires the comprehensive evaluation of the regulatory activity of all noncoding sequences surrounding a gene, irrespective of their sequence conservation.
To directly address this issue we focused our efforts on the zebrafish phox2b locus, employing a transgenic enhancer assay in zebrafish (Fisher et al. 2006a) to determine the regulatory activity of 48 amplicons tiled across a 40.7-kb interval encompassing this gene. The phox2b gene has three exons spanning 3.1 kb; it encodes a paired homeobox transcription factor whose expression is both critical for autonomic neuron specification and tightly controlled (Pattyn et al. 1997, 1999; Amiel et al. 2003; Benailly et al. 2003; Trochet et al. 2005).
Results
phox2b is expressed in autonomic neurons during development
We anticipated that sequences acting as enhancers of phox2b expression would drive green fluorescent protein (GFP) expression in vivo consistent with the endogenous gene. Thus we first determined the developmental expression pattern of phox2b in wild-type zebrafish embryos between the 12 hours post-fertilization (hpf) and 4 days post-fertilization (dpf) (Fig. 1). phox2b is expressed throughout the noradrenergic neuronal populations of vertebrate embryos prior to 12 hpf (data not shown). By 24 hpf expression can be clearly detected throughout the developing hindbrain, in the anterior spinal cord/medulla oblongata, ventral diencephalon, and cranial sensory neurons, and persists in these populations at 48 hpf (Fig. 1). It is also less robustly detected in the locus coeruleus, the epibranchial arches, and throughout the spinal column at the same time points (Fig. 1). Consistent with its role in the genesis and pathogenesis of the enteric nervous system, phox2b is robustly expressed in migrating enteric neuroblasts, beginning at 3 dpf (Elworthy et al. 2005) and is maintained at 4 dpf (Fig. 1).
Figure 1.

In situ hybridization (ISH) of endogenous phox2b expression. ISH was performed on wild-type zebrafish embryos from 24 to 96 hpf using a dig-labeled phox2b RNA probe. (a) Dorsal view of 24-hpf embryo, illustrating phox2b expression in the hindbrain and anterior spinal column. (b) Dorsal view of 48-hpf embryo. hb, hindbrain; mo, medulla oblongata; ventral diencephalon (filled arrowhead), locus coeruleus (open arrowhead). (c) Lateral view of 48-hpf embryo. Rhombomeres of the hindbrain are numbered; mo, medulla oblongata; locus coeruleus (open arrowhead); cranial ganglia (black arrow). (d) Lateral view of the trunk of a 96-hpf embryo. Spinal cord (open dotted arrow) and ENS (open arrows).
Construction of a comprehensive tiling path across the phox2b locus
In order to assess the degree to which conservation identifies regulatory sequences, we first set out to capture and evaluate the regulatory activity of all noncoding sequences in the phox2b interval. We constructed a nonoverlapping tiling path of 33 sequence intervals, comprising 37.6/40.7 kb (92.4%) of the nucleotides in the phox2b locus. To facilitate this we established two broad sequence classes: zebrafish conserved sequences (ZCS) (Fig. 2a, green boxes) or zebrafish nonconserved sequences (ZNCS) (Fig. 2a, red boxes). ZCS correspond to zebrafish sequence intervals for which the MULTIZ alignment tool detects alignment with fugu, tetraodon, human, and/or mouse sequence; ZNCS display no such alignment. The selected sequence intervals (ZCS, n = 20; ZNCS, n = 13; Fig. 2a; Supplemental Table S1) were amplified and subcloned into GFP reporter transgene constructs (Fisher et al. 2006a, b). Importantly, MULTIZ detects intervals of sequence alignment that may be said to display conservation (Blanchette et al. 2004). It is not, however, a metric of evolutionary sequence constraint, as defined by sequences displaying evidence of purifying selection (Cooper et al. 2005; King et al. 2007; Margulies et al. 2007). We have used the terms conserved and constrained throughout the text to reflect this distinction.
Figure 2.

Generation of a nonoverlapping tiling path across the phox2b locus and their expression domains. (a) The zebrafish phox2b interval (chr9: 25101456–25141976; Zv5) was divided into 33 amplicons (total size of 39.3 kb) according to whether intervals could be aligned by MULTIZ to any of four comparator orthologous intervals (fugu, tetraodon, human, or mouse; see Methods). The phox2b exons were excluded. Amplicons are identified based on their distance from the phox2b transcriptional start site and are displayed as custom tracks on the UCSC Genome Browser (genome.ucsc.edu). (b) G0 embryos were raised to sexual maturity, mated with AB fish, and screened for germline transmission of enhancer activity. Expression is noted in the black diamonds below each construct. Black refers to strong expression, gray to weaker expression, and white to no expression. For each positive amplicon at least two G1 founders have been identified that display the reported expression. Those having only one identified founder, thus far, are designated with the *. VDi, ventral diencephalon; LC, locus coeruleus; r, rhombomeres of the hindbrain; CG, cranial ganglia; PA, pharyngeal arches; SC, spinal cord; ENS, enteric nervous system.
ZCS and ZNCS modulate phox2b-appropriate expression
To examine the regulatory activity of all noncoding sequences within the phox2b interval, all constructs were injected into zebrafish embryos (Methods) and examined in at least 200 G0 mosaic embryos (data not shown). G0 embryos that displayed reporter signal (24 hpf to 4 dpf) were raised to sexual maturity and their offspring screened for GFP expression. Reporter patterns in G1 embryos (Fig. 2b) were consistent with the corresponding G0 embryos for all constructs; multiple independent G1 founders were identified for each construct unless otherwise noted (Fig. 2b).
We identified 17 sequence intervals, 13/20 ZCS (61%) and 4/13 ZNCS (31%), directing tissue-specific reporter expression, including the ventral diencephalon, locus coeruleus, hindbrain, cranial sensory neurons, epibranchial arches, spinal cord, and enteric nervous system. Indeed, all directed expression in phox2b positive tissues (Fig. 2b), with most directing expression in the hindbrain (15/17) (Figs. 2b, 3b,c) among other structures. Some amplicons drove expression in discrete subsets of rhombomeres. ZCS −5.5 directed expression in rhombomeres 1 and 2; ZCS −11.2 directed reporter expression predominated in rhombomeres 6–7 and extending into the anterior spinal cord (Fig. 3b). Three elements, ZCS −1.4, ZCS −16.6, and ZNCS −27.9, drove expression in the locus coeruleus; these constructs shared additional expression domains in the hindbrain and spinal cord. ZNCS −27.9 also directed expression in cranial sensory neuron populations (Fig. 2b). Expression in the ventral diencephalon was controlled by ZCS −1.4, ZCS +9.7, ZCS −8.3, ZCS −13.9, and ZNCS +5.6.
Figure 3.

Amplicons across the phox2b display significant functional overlap. The total interval assayed is depicted by color-coded rectangles: red (ZCS), green (ZNCS), purple (phox2b exons), and blue (phox2b introns). (a,b) Images corresponding to functional ZCS constructs. (a) 72-hpf zebrafish embryos with arrow marking ENS expression (ZCS −8.3 and ZCS −23.7). (b) Images indicating the range of hindbrain expression patterns driven by ZCS constructs. (c) Images indicating the range of hindbrain expression patterns driven by ZNCS constructs.
Although the above regulatory control in adrenergic populations may all be discerned by evaluation of embryos from 12 to 48 hpf, phox2b is also required for the formation of the cholinergic neurons of the enteric nervous system (Elworthy et al. 2005), which is not detected until 3 dpf (Elworthy et al. 2005). We identified two sequence intervals with regulatory control that included the enteric nervous system (ENS) (ZCS −8.3 and ZCS −23.7; Figs. 2, 3). Additionally, eight sequence intervals directed expression in domains beyond the endogenous pattern of phox2b expression. This observation is consistent with similar transgenic enhancer assays conducted in vertebrate systems (Nobrega and Pennacchio 2004; Woolfe et al. 2005; Fisher et al. 2006a; Pennacchio et al. 2007) and may simply be an artifact of regulatory DNA taken out of genomic context and thus removed from potential cooperative interactions that may refine their expression. Alternatively, these sequences may regulate neighboring genes whether exclusive from or in addition to their regulation of phox2b. However, expression of the most proximal 5′ neighboring gene (ZGC113342; 40 kb) is restricted to the notocord (data not shown) and is therefore not consistent with this hypothesis.
As defined by MULTIZ, 13/20 ZCS and 4/13 ZNCS sequences display tissue-specific regulatory control. The parameters implemented by algorithms commonly used to identify sequences under purifying selection are, however, more stringent than those imposed by MULTIZ. To determine what fraction of tested functional sequences may therefore be defined as constrained or nonconstrained, we applied several commonly used metrics (phastCons, PipMaker, AVID, MLAGAN, Shuffle-LAGAN) to this interval (Fig. 4), comparing the zebrafish reference sequence to the orthologous interval in all available vertebrates (chimpanzee, baboon, rhesus macaque, cow, pig, cat, dog, rat, mouse, opossum, chicken, frog, fugu, and tetraodon).
Figure 4.

Implementation and comparison of multiple sequence alignment strategies. Predictions made by AVID, MLAGAN, SLAGAN, phastCons, PipMaker, and WebMCS were compared with functional data generated in this study and with each other. Some analyses were predicated on comparisons using † zebrafish versus fugu comparisons, ‡ zebrafish versus human comparisons as described in the text. Results of these analyses are displayed as custom tracks on the UCSC Genome Browser (genome.ucsc.edu).
Metrics of evolutionary constraint capture some but not all regulatory sequences
To determine the success of calls made at the phox2b locus by the AVID, MLAGAN, SLAGAN, PipMaker, and phastCons algorithms, we set out to establish multiple characteristics of their efficiency (sensitivity, specificity, and positive and negative predictive values). First, sensitivity, defined as the fraction of experimentally validated functional elements detected. Second, specificity, defined as how effective the algorithms correctly identify the nonfunctional elements. Third, positive predictive value, defined as the rate at which a positive prediction of function is true. Fourth, negative predictive value, defined as the rate at which a prediction of nonfunction is true.
All algorithms were tested on alignments generated using the zebrafish phox2b interval as the reference sequence under default parameters (75% identity; ≥100-bp windows; Methods). AVID, MLAGAN, and SLAGAN construct iterative pairwise alignments with a common reference sequence and slide a window of fixed width along the aligned sequences seeking sequence stretches that satisfy certain criteria of length and identity. When these algorithms were applied in comparisons between zebrafish and fugu, they displayed a sensitivity of 41% (Table 1), contrasting with the sensitivity of MULTIZ (76%). As expected, when comparing the above metrics of constraint with conserved sequences identified using MULTIZ, specificity increases from 56% to 81%. Furthermore, these metrics display positive predictive values of 70%, marginally higher than MULTIZ (65%), in comparisons of zebrafish to fugu, with negative predictive values of 57%. In general, greater confidence can be placed in positive calls from all algorithms measuring constraint than the negative calls. The observation that MULTIZ provides more reliable negative prediction likely reflects the fact that the other algorithms predict a greater number of amplicons nonfunctional (13 versus 19–23). Sequences identified by the above algorithms are schematically displayed in Figure 4.
Table 1.
Quantitative analysis of multiple sequence alignment strategies

The predictions made by AVID, MLAGAN, SLAGAN, phastCons, PipMaker, and WebMCS were scored as True Positive (TP), False Positive (FP), True Negative (TN), and False Negative (FN) according to how they corresponded to our functional assay for the initial 33 amplicon tiling path. Sensitivity is calculated as the ratio of the number of TP divided by the number of functional amplicons. Specificity is calculated as the ratio of the number of TN divided by the number of assayed nonfunctional amplicons. Positive predictive value is calculated as the ratio of TP divided by predicted functional amplicons (TP + TN). Negative predictive value is calculated as the ratio of TN divided by predicted nonfunctional amplicons (TN + FN).
aZebrafish versus Fugu comparisons.
bZebrafish versus Human comparisons.
To evaluate whether any of these characteristics improved when comparisons over greater evolutionary distances were considered, we repeated these analyses comparing the zebrafish and human PHOX2B loci. In short, specificity increased for AVID and MLAGAN from 81% to 88% and remained unchanged at 81% for SLAGAN (Table 1). In contrast, sensitivity decreased from 41% to 29%–35%, although the drop in sensitivity is perhaps not surprising given the increased evolutionary gap between these organisms. The corresponding positive and negative predictive values did not change substantially (Table 1). To summarize, the above measures of constraint failed to detect 47%–71% of the functional amplicons identified in our assay (Sensitivity, Table 1); none detected any functional ZNCS amplicons.
To exclude the possibility that our observations were biased by the use of global alignment approaches, we then applied the PipMaker, WebMCS, and phastCons algorithms to this interval. PipMaker is a local alignment strategy predicated on the use of BLASTZ (Schwartz et al. 2003). WebMCS uses BLASTZ sequence alignments and phylogeny to calculate the probability of observing a given number of sequence identities at each nucleotide position (Margulies et al. 2003). In contrast, phastCons utilizes the MULTIZ threaded blockset multispecies alignment strategy (Blanchette et al. 2004), implementing a hidden Markov model to maximize the identification of known functional sequences. Importantly, both WebMCS and phastCons can impute significance to aligned sequence stretches under 100 bp (Margulies et al. 2003; Siepel et al. 2005). As anticipated, when compared to the above global alignment metrics, phastCons displayed by far the highest sensitivity (53% versus 29%–41%; Table 1) and lowest specificity (69% versus 80%–88%; Table 1). However, despite identifying more constrained amplicons than the other algorithms, phastCons displayed largely similar positive and negative predictive values (Table 1). The PipMaker algorithm performed strongly, displaying sensitivity close to that of phastCons (47%) and counter intuitively, relatively high specificity (81%). This algorithm performed equally when being applied to sequence comparisons between zebrafish and fugu, and those between zebrafish and human. PipMaker also had very strong relative positive and negative predictive values (73% and 59%). However, although PipMaker is an alignment algorithm and not a measure of constraint, it does provide the alignment substrate on which WebMCS operates. In contrast, the MCS predictions display the lowest sensitivity (33%) of all metrics of constraint assayed, and also the lowest specificity (80%). However, as with phastCons, the MCS predictions display positive and negative predictive values within the range of all other tested measures of constraint (Table 1).
Additionally we also noted that sequence analyses using these metrics identified multiple independent constrained noncoding sequence intervals within several of our initial 33 amplicons. To determine whether regulatory control (Fig. 3a,b) could be attributed to one or multiple constrained sequences therein, we selected three amplicons for further analysis (ZCS −1.4, ZCS −8.3, and ZCS −23.7; Fig. 5). We also wanted to establish how splitting functional elements into multiple smaller elements based on alignment therein would affect our initial observations. In two instances (ZCS −1.4 and ZCS −8.3) the primary amplicon could be separated into fragments containing sequence conserved across the vertebrate radiation and sequence conserved solely among teleosts. Thus we set out to ask whether regulatory control exerted by the primary amplicon could be explained by a single derived fragment conserved over the greater evolutionary distance. ZCS −1.4 was reduced to two smaller sequences (ZCS −1.4a and b): ZCS −1.4a aligned with all four vertebrate species (human, mouse, tetraodon, and fugu; Fig. 5f) and ZCS −1.4b aligned only among teleosts (tetraodon and fugu; Fig. 5f). Both displayed tissue-dependent regulatory control. ZCS −8.3 was separated into three nonoverlapping sequence fragments (ZCS −8.3a, b, and c; Fig. 5g). ZCS −8.3b aligned with all four vertebrate orthologs and was detected by all algorithms used and in all comparisons; sequence intervals ZCS −8.3a and ZCS −8.3c aligned only to the teleosts (Fig. 5g) and were detected by all algorithms used but only in comparisons among teleosts. Interestingly, only ZCS −8.3b and ZCS −8.3c displayed regulatory control similar to the original ZCS −8.3 (Fig. 5g and data not shown); ZCS −8.3a did not display reporter expression. Consistent with our above observations, sequence constraint detected in comparisons among teleosts displayed no reduced predictive value.
Figure 5.

Functional dissection of biological activity in identified regulatory intervals. (a) Sequences with high predicted affinity to the identified motifs which discriminate functional from nonfunctional regions within the assayed phox2b interval and their rank order as identified in Supplemental Table S4. (b–d) Logos representing position weight matrices M6, M36, and M39, respectively. Regulatory amplicons ZNCS +6.7, ZCS −1.4, ZCS −8.3, ZCS −23.7, and ZNCS −27.9 were reduced in size and evaluated as component amplicons to determine the sequence origins of their observed activity (e–h). Boxes shaded red denote functional ZNCS and those shaded green denote functional ZCS; open boxes denote no observed activity.
Similarly, we separated ZCS −23.7 into five pieces (ZCS −23.7a–e; Fig. 5h), detected by all algorithms in all comparisons. All but one of the five sequence fragments demonstrated hindbrain expression in G0 mosaic embryos, consistent with the original ZCS −23.7 fragment (Supplemental Fig. S1; data not shown). This reflects our observations with ZCS −1.4 and ZCS −8.3, suggesting that identified regulatory sequences may occasionally be comprised of modules with discrete but overlapping and potentially cooperative function. Consistent with their independent function in this assay, these ZCS −23.7a–e sequences are scattered over an interval ≥20 kb in the human genome in stark contrast to their tight distribution in the zebrafish genome (Fig. 4). Furthermore, although ZCS −23.7d did not display reporter expression at any of the times examined, we cannot rule out other roles for this sequence, perhaps later in development or in refining the regulatory control of other identified phox2b enhancers.
We then repeated our analyses using the expanded amplicon set; the result was a marginal improvement in sensitivity and positive predictive value for all algorithms and a corresponding marginal decrease in specificity and negative predictive values (Supplemental Table S2). Overall, these data indicate that analyses restricted by commonly used metrics of constraint risk a potentially significant loss of functional information, ranging between 39% and 71% at this critical locus.
Dissection of functional regulatory nonconserved sequences
The fact that nonaligned sequence intervals, not simply those with insufficient identity to be designated as constrained, were observed to display tissue-specific regulatory control is quite unexpected. However, similar examples are known; four nonconserved functional sequences are known in the human HBB complex (King et al. 2005), and recently the ENCODE project reported ∼50% of functional elements identified in vitro were nonconserved across mammals (The ENCODE Project Consortium 2007). Several parsimonious explanations may exist. First, the unaligned interval might be duplicated in the reference sequence and therefore preclude alignment. Second, the sequence interval may be rearranged in the reference sequence and therefore fail to align. Third, the boundaries of functional aligned regions may be imprecisely defined, excluding functional sequence components from overtly aligned intervals because they fall below a required threshold or diffuse enhancer modules comprised of transcription factor binding sites that are used in concert but separated by sequence under reduced selective pressure and might not be detected as conserved blocks of sequence. Either example may potentially be considered an example of a diffuse enhancer module. Fourth, the sequence may correspond to a species-specific regulatory element.
To address the first two possibilities, we aligned shorter 500, 1000, and 2000 bp overlapping tiling windows across the phox2b locus against the genomes of mouse, human, fugu, and tetraodon (fr1, tetNig1, mm8, and hg18). Surprisingly, we did not detect any additional significant alignments that were missed by aligning the whole locus (data not shown). Additionally, we determined that paralogous regions within the zebrafish genome were not aligned with the reference genome and therefore were not obscuring alignments within the phox2b locus.
To address the third possibility we selected two intervals, ZNCS +6.7 and ZNCS −27.9, making systematically smaller constructs by progressively excluding 5′ and 3′ flanking sequence (Fig. 5e,h). ZNCS +6.7 regulatory control was extinguished when it was reduced by 594 bp (ZNCS +6.7 [−594]). We then made two further constructs, one restoring the deleted sequence 3′ to ZNCS +6.7[left], i.e., adjacent to ZCS +9.7, and the other restoring the deleted sequence 5′ to the ZNCS +6.7[right], i.e., adjacent to ZCS +6.4. Only the latter construct restored regulatory control expression in G0 zebrafish (Fig. 5e). These data are consistent with imprecise definition of regulatory element boundaries. However, the regulatory control displayed by ZNCS +6.7 and ZCS +6.4 do not overlap. Furthermore, we have independently tested whether smaller 500-bp sequence intervals can align with any portion of the locus and do not observe any alignment within ZNCS +6.7 with any fragment of the four comparison genomes. Therefore, the boundary of ZNCS +6.7 appears conservatively defined. In concert these data suggest the above sequences may cooperate in exerting their regulatory output or that the regulatory control of ZNCS +6.7 is truly independent and may be better explained as a diffuse regulatory module.
The proposed connection between sequence constraint and biological function is straightforward; functional modules harbor sequences that are intolerant of substitution and are therefore conserved. The constrained sites in cis-regulatory modules are presumed transcription factor binding sites (TFBS). Because these are frequently short (6–20 bp), the more densely they are distributed, the easier they are to detect by alignment. Conversely, regulatory modules whose TFBS are not densely distributed (diffuse enhancers) (Pennacchio et al. 2006) are more difficult to detect by alignment. We sought to determine whether nonaligned sequences also contained conserved TFBS that would discriminate them from nonfunctional sequence intervals, albeit distributed at insufficient density to permit their detection based on alignment alone. These TFBS can be represented by position weight matrices (PWMs) or oligomers (strings of nucleotides of arbitrary length). To this end we used AlignACE, an algorithm to find PWMs overrepresented in functional regions (Hughes et al. 2000). Although this approach detected significant differences between motifs found in functional versus nonfunctional regions, most of these differences could be attributed to the higher GC content of the functional regions (data not shown). The mechanistic basis of this correlation is unclear, but it is consistent with previous reports that functional mammalian enhancers frequently display higher GC content than nonfunctional sequences such as ancient repeats (Taylor et al. 2006). While transcription factors frequently bind degenerate sites that are more realistically modeled by PWMs, using oligomers significantly reduces the dimensionality of the search space and is not subject to GC bias. We therefore sought to determine which oligomers (6, 7, or 8 mers) were able to most clearly distinguish functional from nonfunctional sequence intervals. Several oligomers were present exclusively in functional intervals, but the frequency of occurrence of these oligomers was not above that found in the control experiment in which the sequence across the locus is randomized (data not shown). Finally, we used these oligomers to seed PWMs, which were then optimized using simulated annealing (Beer and Tavazoie 2004; see Supplemental Materials). This approach yielded 80 PWMs that had sites suggestive of strong binding affinity in all functional regions and weak affinity sites in nonfunctional regions. The best match in any nonfunctional region is weaker than the worst match in any functional region. The magnitude of the difference between the scores in functional and nonfunctional regions is reflected in the log-likelihood (see Supplemental Materials). The density and distribution of these sites are shown in Figure 4. In contrast, searching for PWMs in 20 independent randomizations of the total assayed sequence space yielded significantly fewer such PWMs (52.7 ± 13), establishing that the PWMs found in the functional regions were statistically significant at P < 0.02, assuming a normal distribution (for examples, see Supplemental Fig. S2; Supplemental Table S3). Of the 80 predictive PWMs many are similar; for the purposes of presentation we distilled this list to 66 PWMs that are distinct by a similarity threshold of 0.8 (Supplemental Table S4; Hughes et al. 2000). While it is unlikely that these are all functional TFBS, the possibility that they are functionally significant is strengthened by the observation that significantly fewer PWMs are found on randomized sequence. Consistent with this interpretation, deletion of sequences with predicted strong binding affinity to the novel motifs (M36, M7, M6, M39, M46, M75, M28, M77; Fig. 5a–d) from ZNCS +6.7, to create ZNCS +6.7[left], abrogates regulatory control (Fig. 5e; Supplemental Table S4).
However, we also identify motifs in aligned portions of ZNCS −27.9, although they have low power to discriminate functional from nonfunctional sequences and lie outside the core functional interval; reduction of ZNCS −27.9 by 165 bp or by 305 bp failed to overtly compromise its regulatory control (Fig. 5h). Interestingly, ZNCS −27.9 (−305) displays no sequence similarity with other evaluated vertebrates, consistent with our fifth postulate of a lineage-specific regulatory element. We were surprised to find many PWMs that were present in all functional regions and absent from all nonfunctional regions; it is likely that the relatively small set of positive and negative functional regions in this study prevents the identification of binding sites which are present in subsets of the functional regions. This may also explain the absence of identified motifs within ZNCS −27.9 (−305).
Discussion
There is broad consensus that constrained, noncoding sequences frequently display regulatory control of tissue-specific expression. The development of in silico tools for the identification of noncoding functional regulatory sequences frequently makes use of training sets of empirically identified functional sequences, the vast majority of which correspond to constrained sequences. In order to establish whether such assumptions are valid, it is thus crucial to better understand what information may be missed by restricting analyses to constrained sequences.
We can now provide robust data to address our two initial questions. First, we demonstrate that, at phox2b, the evaluated parameters for constraint detect functional elements at rates ranging from 29% to 61%. Second, we also demonstrate in vivo that relying on standard methods to detect evolutionary constraint overlooks significant functional information with frequencies ranging from 42% to 51%; these data are consistent with emerging in vitro data and in silico predictions from the ENCODE project (The ENCODE Project Consortium 2007; King et al. 2007). Critically, we also show that functional elements within this interval share sequence characteristics, even when they are not identifiably constrained. Although our data clearly establish a precedent, there is a need to interpret these observations with caution. The teleost radiation occurred 3–400 million years ago; thus, zebrafish and fugu are separated by a much larger evolutionary distance than mammals and this may also, in part, explain the lack of observable alignment or constraint. Similar analyses of other loci, including those in mammals, will be needed before the degree to which this study may be extrapolated becomes clear. However, these data state clearly the importance of exhaustive functional analyses and also demonstrate a need for new computational techniques to uncover a broader spectrum of functional noncoding sequences. In summary, these data have the potential to significantly impact the manner in which mutation detection is conducted in the context of human disease and establish a data set from which further refinement of existing algorithms may be leveraged.
Methods
Selection and amplification of zebrafish noncoding sequences
Genomic sequence corresponding to chr9: 25101456–25141976 of the zebrafish genome (Zv5) was utilized for this study. Comparison of the Zv6 and the Zv5 builds of the phox2b interval revealed inconsistency between them at sequences within the interval between ZNCS −12.8 and ZCS −13.9. At this point Zv5 and Zv6 differ in the contig placed 5′ to ZNCS −12.8. To determine which build was correct we assayed the breakpoints predicted by both Zv5 and Zv6 using PCR amplicons bridging the interval and confirmed that sequence within Zv5 at this point is an accurate reflection of the endogenous genome of the AB zebrafish strain (data not shown). The interval selected displayed uninterupted synteny among vertebrates. The four vertebrate genomes presently aligned by MULTIZ (teleosts tetraodon and fugu, mouse, and human) were used to identify regions that aligned with this genome build for zebrafish. Amplicons were designed to exclusively amplify zebrafish sequence intervals aligned with other vertebrate sequences (ZCS) and those determined not to align with other vertebrate sequences (ZNCS). Sequences were amplified from AB zebrafish genomic DNA under standard conditions (sequences specified in Supplemental Table S1). Each amplicon was subcloned into the Tol2-based transgene reporter construct pGW-cfosEGFP (Fisher et al. 2006a, b).
Fish care
Zebrafish were raised and bred in accordance with standard conditions (Kimmel et al. 1995; Westerfield 2000). Embryos were maintained at 28°C and staged in accordance with standard methods (Kimmel et al. 1995; Westerfield 2000). Embryos for in situ hybridization were raised in embryo medium containing 0.003% phenylthiocarbamide to prevent pigmentation.
Embryo injections and analysis
Constructs were generated and injected into wild-type G0 embryos (n ≥ 200) as previously described (Fisher et al. 2006b). Injected embryos were evaluated for reporter expression at 24, 48, 72, and 96 hpf. Embryos displaying consistent expression were selected and allowed to mature to facilitate germline transmission and evaluation of reporter expression. Embryos were analyzed and imaged using a Carl Zeiss Lumar V12 Stereo microscope with AxioVision version 4.5 software.
In situ hybridization
Embryos were collected from matings from wild-type zebrafish, fixed at desired time points 24, 48, 72, and 96 hpf and fixed for in situ hybridization using standard protocols. Plasmid from which the phox2b riboprobe was generated was a kind gift from Robert Kelsh (University of Bath); digoxin-labeled antisense probe was prepared using T7 RNA polymerase after NotI cleavage of the plasmid.
Informatics
Sequence corresponding to zebrafish chr9: 25101456–25141976 (Zv5) was submitted to the VISTA browser (www-gsd.lbl.gov/vista/) in concert with ∼200 kb of sequence encompassing the orthologous interval from each of 14 vertebrates (chimp, baboon, rhesus, cow, pig, cat, dog, rat, mouse, opossum, chicken, frog, fugu, tetraodon; see Supplemental Table S2 for sequence intervals). Sequences were subjected to AVID, SLAGAN, MLAGAN analyses under default parameters (75% identity, ≥100 bp; Supplemental Figs. S3–S5) using the zebrafish sequence as reference. Sequence intervals identified within Zebrafish–Human and Zebrafish–Fugu comparisons by these analyses were submitted to the UCSC browser as custom tracks for visual comparison.
Alignments were obtained by running BLASTZ on overlapping tiling windows across the phox2b locus (0–500 bp, 250–750 bp, 500–1000 bp, 750–1250 bp, etc.) using the HoxD55q similarity matrix ([91–90–25–100], [−90 100–100–25], [−25–100 100–90], [−100–25–90 91]) and BLASTZ parameters which match those used in the UCSC genome browser alignments: dr versus mm or hg (K = 2200 L = 6000 H = 2000), dr versus fugu (K = 3000 L = 2200 H = 2000) dr versus tetNig (K = 3000 L = 2200 H = 2500) dr versus dr (K = 3000 L = 2200 H = 2000).
We used the most significant 100 6, 7, and 8 mers to seed PWMs which were then optimized using simulated annealing in an approach modified from Beer and Tavazoie (2004) with 10 different random seeds (Beer and Tavazoie 2004). Matrices were randomized a column at a time and scored according to the likelihood, L, that strong sites are found in functional regions and weak sites are found in nonfunctional regions. Changes were accepted if L increased, and rejected if L decreased with probability eΔlogL/T where T0 = 1, and T decreased exponentially with a half-time of 2000 iterations. The likelihood is given by the product over the 33 tested regions, L = 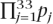 , where pj is the probability of finding a site in a region. The strength of the sites were modeled with the sigmoidal function, qi = 1/(1 + e−20(xi−0.75)), where xi is the best normalized matrix score in the sequence, and pj = qi if the region j with site i is functional, and pj = 1 − qi if the region is nonfunctional. In this way PWMs which score highly on functional regions and weakly on nonfunctional regions receive higher probability, P. This analysis further underscores the flexibility of PWMs as opposed to k-mers, which while generally advantageous can be more prone to overfitting.
, where pj is the probability of finding a site in a region. The strength of the sites were modeled with the sigmoidal function, qi = 1/(1 + e−20(xi−0.75)), where xi is the best normalized matrix score in the sequence, and pj = qi if the region j with site i is functional, and pj = 1 − qi if the region is nonfunctional. In this way PWMs which score highly on functional regions and weakly on nonfunctional regions receive higher probability, P. This analysis further underscores the flexibility of PWMs as opposed to k-mers, which while generally advantageous can be more prone to overfitting.
Acknowledgments
This study was supported by funds from the NIH (R01GM071648) and March of Dimes to A.S.M. D.M.M. was also supported by NIH pre-doctoral training grant 5T32GM07814. We also thank Drs. D.J. Cutler and K.D. Smith for critical reading of this manuscript, and Anthony Antonellis and Elliott Margulies for their computational insights.
Footnotes
[Supplemental material is available online at www.genome.org.]
Article published online before print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.6929408
References
- Amiel J., Laudier B., Attie-Bitach T., Trang H., de Pontual L., Gener B., Trochet D., Etchevers H., Ray P., Simonneau M., Laudier B., Attie-Bitach T., Trang H., de Pontual L., Gener B., Trochet D., Etchevers H., Ray P., Simonneau M., Attie-Bitach T., Trang H., de Pontual L., Gener B., Trochet D., Etchevers H., Ray P., Simonneau M., Trang H., de Pontual L., Gener B., Trochet D., Etchevers H., Ray P., Simonneau M., de Pontual L., Gener B., Trochet D., Etchevers H., Ray P., Simonneau M., Gener B., Trochet D., Etchevers H., Ray P., Simonneau M., Trochet D., Etchevers H., Ray P., Simonneau M., Etchevers H., Ray P., Simonneau M., Ray P., Simonneau M., Simonneau M., et al. Polyalanine expansion and frameshift mutations of the paired-like homeobox gene PHOX2B in congenital central hypoventilation syndrome. Nat. Genet. 2003;33:459–461. doi: 10.1038/ng1130. [DOI] [PubMed] [Google Scholar]
- Aparicio S., Morrison A., Gould A., Gilthorpe J., Chaudhuri C., Rigby P., Krumlauf R., Brenner S., Morrison A., Gould A., Gilthorpe J., Chaudhuri C., Rigby P., Krumlauf R., Brenner S., Gould A., Gilthorpe J., Chaudhuri C., Rigby P., Krumlauf R., Brenner S., Gilthorpe J., Chaudhuri C., Rigby P., Krumlauf R., Brenner S., Chaudhuri C., Rigby P., Krumlauf R., Brenner S., Rigby P., Krumlauf R., Brenner S., Krumlauf R., Brenner S., Brenner S. Detecting conserved regulatory elements with the model genome of the Japanese puffer fish, Fugu rubripes. Proc. Natl. Acad. Sci. 1995;92:1684–1688. doi: 10.1073/pnas.92.5.1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beer M.A., Tavazoie S., Tavazoie S. Predicting gene expression from sequence. Cell. 2004;117:185–198. doi: 10.1016/s0092-8674(04)00304-6. [DOI] [PubMed] [Google Scholar]
- Bejerano G., Haussler D., Blanchette M., Haussler D., Blanchette M., Blanchette M. Into the heart of darkness: Large-scale clustering of human non-coding DNA. Bioinformatics. 2004;20:I40–I48. doi: 10.1093/bioinformatics/bth946. (Suppl. 1) [DOI] [PubMed] [Google Scholar]
- Benailly H.K., Lapierre J.M., Laudier B., Amiel J., Attie T., De Blois M.C., Vekemans M., Romana S.P., Lapierre J.M., Laudier B., Amiel J., Attie T., De Blois M.C., Vekemans M., Romana S.P., Laudier B., Amiel J., Attie T., De Blois M.C., Vekemans M., Romana S.P., Amiel J., Attie T., De Blois M.C., Vekemans M., Romana S.P., Attie T., De Blois M.C., Vekemans M., Romana S.P., De Blois M.C., Vekemans M., Romana S.P., Vekemans M., Romana S.P., Romana S.P. PMX2B, a new candidate gene for Hirschsprung’s disease. Clin. Genet. 2003;64:204–209. doi: 10.1034/j.1399-0004.2003.00105.x. [DOI] [PubMed] [Google Scholar]
- Blanchette M., Kent W.J., Riemer C., Elnitski L., Smit A.F., Roskin K.M., Baertsch R., Rosenbloom K., Clawson H., Green E.D., Kent W.J., Riemer C., Elnitski L., Smit A.F., Roskin K.M., Baertsch R., Rosenbloom K., Clawson H., Green E.D., Riemer C., Elnitski L., Smit A.F., Roskin K.M., Baertsch R., Rosenbloom K., Clawson H., Green E.D., Elnitski L., Smit A.F., Roskin K.M., Baertsch R., Rosenbloom K., Clawson H., Green E.D., Smit A.F., Roskin K.M., Baertsch R., Rosenbloom K., Clawson H., Green E.D., Roskin K.M., Baertsch R., Rosenbloom K., Clawson H., Green E.D., Baertsch R., Rosenbloom K., Clawson H., Green E.D., Rosenbloom K., Clawson H., Green E.D., Clawson H., Green E.D., Green E.D., et al. Aligning multiple genomic sequences with the threaded blockset aligner. Genome Res. 2004;14:708–715. doi: 10.1101/gr.1933104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper G.M., Stone E.A., Asimenos G., Green E.D., Batzoglou S., Sidow A., Stone E.A., Asimenos G., Green E.D., Batzoglou S., Sidow A., Asimenos G., Green E.D., Batzoglou S., Sidow A., Green E.D., Batzoglou S., Sidow A., Batzoglou S., Sidow A., Sidow A. Distribution and intensity of constraint in mammalian genomic sequence. Genome Res. 2005;15:901–913. doi: 10.1101/gr.3577405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson E.H., Erwin D.H., Erwin D.H. Gene regulatory networks and the evolution of animal body plans. Science. 2006;311:796–800. doi: 10.1126/science.1113832. [DOI] [PubMed] [Google Scholar]
- de la Calle-Mustienes E., Feijoo C.G., Manzanares M., Tena J.J., Rodriguez-Seguel E., Letizia A., Allende M.L., Gomez-Skarmeta J.L., Feijoo C.G., Manzanares M., Tena J.J., Rodriguez-Seguel E., Letizia A., Allende M.L., Gomez-Skarmeta J.L., Manzanares M., Tena J.J., Rodriguez-Seguel E., Letizia A., Allende M.L., Gomez-Skarmeta J.L., Tena J.J., Rodriguez-Seguel E., Letizia A., Allende M.L., Gomez-Skarmeta J.L., Rodriguez-Seguel E., Letizia A., Allende M.L., Gomez-Skarmeta J.L., Letizia A., Allende M.L., Gomez-Skarmeta J.L., Allende M.L., Gomez-Skarmeta J.L., Gomez-Skarmeta J.L. A functional survey of the enhancer activity of conserved non-coding sequences from vertebrate Iroquois cluster gene deserts. Genome Res. 2005;15:1061–1072. doi: 10.1101/gr.4004805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elworthy S., Pinto J.P., Pettifer A., Cancela M.L., Kelsh R.N., Pinto J.P., Pettifer A., Cancela M.L., Kelsh R.N., Pettifer A., Cancela M.L., Kelsh R.N., Cancela M.L., Kelsh R.N., Kelsh R.N. Phox2b function in the enteric nervous system is conserved in zebrafish and is sox10-dependent. Mech. Dev. 2005;122:659–669. doi: 10.1016/j.mod.2004.12.008. [DOI] [PubMed] [Google Scholar]
- Emison E.S., McCallion A.S., Kashuk C.S., Bush R.T., Grice E., Lin S., Portnoy M.E., Cutler D.J., Green E.D., Chakravarti A., McCallion A.S., Kashuk C.S., Bush R.T., Grice E., Lin S., Portnoy M.E., Cutler D.J., Green E.D., Chakravarti A., Kashuk C.S., Bush R.T., Grice E., Lin S., Portnoy M.E., Cutler D.J., Green E.D., Chakravarti A., Bush R.T., Grice E., Lin S., Portnoy M.E., Cutler D.J., Green E.D., Chakravarti A., Grice E., Lin S., Portnoy M.E., Cutler D.J., Green E.D., Chakravarti A., Lin S., Portnoy M.E., Cutler D.J., Green E.D., Chakravarti A., Portnoy M.E., Cutler D.J., Green E.D., Chakravarti A., Cutler D.J., Green E.D., Chakravarti A., Green E.D., Chakravarti A., Chakravarti A. A common sex-dependent mutation in a RET enhancer underlies Hirschsprung disease risk. Nature. 2005;434:857–863. doi: 10.1038/nature03467. [DOI] [PubMed] [Google Scholar]
- The ENCODE Project Consortium Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;447:799–816. doi: 10.1038/nature05874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher S., Grice E.A., Vinton R.M., Bessling S.L., McCallion A.S., Grice E.A., Vinton R.M., Bessling S.L., McCallion A.S., Vinton R.M., Bessling S.L., McCallion A.S., Bessling S.L., McCallion A.S., McCallion A.S. Conservation of RET regulatory function from human to zebrafish without sequence similarity. Science. 2006a;312:276–279. doi: 10.1126/science.1124070. [DOI] [PubMed] [Google Scholar]
- Fisher S., Grice E.A., Vinton R.M., Bessling S.L., Urasaki A., Kawakami K., McCallion A.S., Grice E.A., Vinton R.M., Bessling S.L., Urasaki A., Kawakami K., McCallion A.S., Vinton R.M., Bessling S.L., Urasaki A., Kawakami K., McCallion A.S., Bessling S.L., Urasaki A., Kawakami K., McCallion A.S., Urasaki A., Kawakami K., McCallion A.S., Kawakami K., McCallion A.S., McCallion A.S. Evaluating the biological relevance of putative enhancers using Tol2 transposon-mediated transgenesis in zebrafish. Nat. Protoc. 2006b;1:1297–1305. doi: 10.1038/nprot.2006.230. [DOI] [PubMed] [Google Scholar]
- Frazer K.A., Narla G., Zhang J.L., Rubin E.M., Narla G., Zhang J.L., Rubin E.M., Zhang J.L., Rubin E.M., Rubin E.M. The apolipoprotein(a) gene is regulated by sex hormones and acute-phase inducers in YAC transgenic mice. Nat. Genet. 1995;9:424–431. doi: 10.1038/ng0495-424. [DOI] [PubMed] [Google Scholar]
- Grice E.A., Rochelle E.S., Green E.D., Chakravarti A., McCallion A.S., Rochelle E.S., Green E.D., Chakravarti A., McCallion A.S., Green E.D., Chakravarti A., McCallion A.S., Chakravarti A., McCallion A.S., McCallion A.S. Evaluation of the RET regulatory landscape reveals the biological relevance of a HSCR-implicated enhancer. Hum. Mol. Genet. 2005;14:3837–3845. doi: 10.1093/hmg/ddi408. [DOI] [PubMed] [Google Scholar]
- Hughes J.D., Estep P.W., Tavazoie S., Church G.M., Estep P.W., Tavazoie S., Church G.M., Tavazoie S., Church G.M., Church G.M. Computational identification of cis-regulatory elements associated with groups of functionally related genes in Saccharomyces cerevisiae. J. Mol. Biol. 2000;296:1205–1214. doi: 10.1006/jmbi.2000.3519. [DOI] [PubMed] [Google Scholar]
- Kimmel C.B., Ballard W.W., Kimmel S.R., Ullmann B., Schilling T.F., Ballard W.W., Kimmel S.R., Ullmann B., Schilling T.F., Kimmel S.R., Ullmann B., Schilling T.F., Ullmann B., Schilling T.F., Schilling T.F. Stages of embryonic development of the zebrafish. Dev. Dyn. 1995;203:253–310. doi: 10.1002/aja.1002030302. [DOI] [PubMed] [Google Scholar]
- King D.C., Taylor J., Elnitski L., Chiaromonte F., Miller W., Hardison R.C., Taylor J., Elnitski L., Chiaromonte F., Miller W., Hardison R.C., Elnitski L., Chiaromonte F., Miller W., Hardison R.C., Chiaromonte F., Miller W., Hardison R.C., Miller W., Hardison R.C., Hardison R.C. Evaluation of regulatory potential and conservation scores for detecting cis-regulatory modules in aligned mammalian genome sequences. Genome Res. 2005;15:1051–1060. doi: 10.1101/gr.3642605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King D.C., Taylor J., Zhang Y., Cheng Y., Lawson H.A., Martin J., Chiaromonte F., Miller W., Hardison R.C., Taylor J., Zhang Y., Cheng Y., Lawson H.A., Martin J., Chiaromonte F., Miller W., Hardison R.C., Zhang Y., Cheng Y., Lawson H.A., Martin J., Chiaromonte F., Miller W., Hardison R.C., Cheng Y., Lawson H.A., Martin J., Chiaromonte F., Miller W., Hardison R.C., Lawson H.A., Martin J., Chiaromonte F., Miller W., Hardison R.C., Martin J., Chiaromonte F., Miller W., Hardison R.C., Chiaromonte F., Miller W., Hardison R.C., Miller W., Hardison R.C., Hardison R.C. Finding cis-regulatory elements using comparative genomics: Some lessons from ENCODE data. Genome Res. 2007;17:775–786. doi: 10.1101/gr.5592107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinjan D.A., van Heyningen V., van Heyningen V. Long-range control of gene expression: Emerging mechanisms and disruption in disease. Am. J. Hum. Genet. 2005;76:8–32. doi: 10.1086/426833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lettice L.A., Heaney S.J., Purdie L.A., Li L., de Beer P., Oostra B.A., Goode D., Elgar G., Hill R.E., de Graaff E., Heaney S.J., Purdie L.A., Li L., de Beer P., Oostra B.A., Goode D., Elgar G., Hill R.E., de Graaff E., Purdie L.A., Li L., de Beer P., Oostra B.A., Goode D., Elgar G., Hill R.E., de Graaff E., Li L., de Beer P., Oostra B.A., Goode D., Elgar G., Hill R.E., de Graaff E., de Beer P., Oostra B.A., Goode D., Elgar G., Hill R.E., de Graaff E., Oostra B.A., Goode D., Elgar G., Hill R.E., de Graaff E., Goode D., Elgar G., Hill R.E., de Graaff E., Elgar G., Hill R.E., de Graaff E., Hill R.E., de Graaff E., de Graaff E. A long-range Shh enhancer regulates expression in the developing limb and fin and is associated with preaxial polydactyly. Hum. Mol. Genet. 2003;12:1725–1735. doi: 10.1093/hmg/ddg180. [DOI] [PubMed] [Google Scholar]
- Levine M., Tjian R., Tjian R. Transcription regulation and animal diversity. Nature. 2003;424:147–151. doi: 10.1038/nature01763. [DOI] [PubMed] [Google Scholar]
- Margulies E.H., Blanchette M., Haussler D., Green E.D., Blanchette M., Haussler D., Green E.D., Haussler D., Green E.D., Green E.D. Identification and characterization of multi-species conserved sequences. Genome Res. 2003;13:2507–2518. doi: 10.1101/gr.1602203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margulies E.H., Cooper G.M., Asimenos G., Thomas D.J., Dewey C.N., Siepel A., Birney E., Keefe D., Schwartz A.S., Hou M., Cooper G.M., Asimenos G., Thomas D.J., Dewey C.N., Siepel A., Birney E., Keefe D., Schwartz A.S., Hou M., Asimenos G., Thomas D.J., Dewey C.N., Siepel A., Birney E., Keefe D., Schwartz A.S., Hou M., Thomas D.J., Dewey C.N., Siepel A., Birney E., Keefe D., Schwartz A.S., Hou M., Dewey C.N., Siepel A., Birney E., Keefe D., Schwartz A.S., Hou M., Siepel A., Birney E., Keefe D., Schwartz A.S., Hou M., Birney E., Keefe D., Schwartz A.S., Hou M., Keefe D., Schwartz A.S., Hou M., Schwartz A.S., Hou M., Hou M., et al. Analyses of deep mammalian sequence alignments and constraint predictions for 1% of the human genome. Genome Res. 2007;17:760–774. doi: 10.1101/gr.6034307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall H., Studer M., Popperl H., Aparicio S., Kuroiwa A., Brenner S., Krumlauf R., Studer M., Popperl H., Aparicio S., Kuroiwa A., Brenner S., Krumlauf R., Popperl H., Aparicio S., Kuroiwa A., Brenner S., Krumlauf R., Aparicio S., Kuroiwa A., Brenner S., Krumlauf R., Kuroiwa A., Brenner S., Krumlauf R., Brenner S., Krumlauf R., Krumlauf R. A conserved retinoic acid response element required for early expression of the homeobox gene Hoxb-1. Nature. 1994;370:567–571. doi: 10.1038/370567a0. [DOI] [PubMed] [Google Scholar]
- Nobrega M.A., Pennacchio L.A., Pennacchio L.A. Comparative genomic analysis as a tool for biological discovery. J. Physiol. 2004;554:31–39. doi: 10.1113/jphysiol.2003.050948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattyn A., Morin X., Cremer H., Goridis C., Brunet J.F., Morin X., Cremer H., Goridis C., Brunet J.F., Cremer H., Goridis C., Brunet J.F., Goridis C., Brunet J.F., Brunet J.F. Expression and interactions of the two closely related homeobox genes Phox2a and Phox2b during neurogenesis. Development. 1997;124:4065–4075. doi: 10.1242/dev.124.20.4065. [DOI] [PubMed] [Google Scholar]
- Pattyn A., Morin X., Cremer H., Goridis C., Brunet J.F., Morin X., Cremer H., Goridis C., Brunet J.F., Cremer H., Goridis C., Brunet J.F., Goridis C., Brunet J.F., Brunet J.F. The homeobox gene Phox2b is essential for the development of autonomic neural crest derivatives. Nature. 1999;399:366–370. doi: 10.1038/20700. [DOI] [PubMed] [Google Scholar]
- Pennacchio L.A., Ahituv N., Moses A.M., Prabhakar S., Nobrega M.A., Shoukry M., Minovitsky S., Dubchak I., Holt A., Lewis K.D., Ahituv N., Moses A.M., Prabhakar S., Nobrega M.A., Shoukry M., Minovitsky S., Dubchak I., Holt A., Lewis K.D., Moses A.M., Prabhakar S., Nobrega M.A., Shoukry M., Minovitsky S., Dubchak I., Holt A., Lewis K.D., Prabhakar S., Nobrega M.A., Shoukry M., Minovitsky S., Dubchak I., Holt A., Lewis K.D., Nobrega M.A., Shoukry M., Minovitsky S., Dubchak I., Holt A., Lewis K.D., Shoukry M., Minovitsky S., Dubchak I., Holt A., Lewis K.D., Minovitsky S., Dubchak I., Holt A., Lewis K.D., Dubchak I., Holt A., Lewis K.D., Holt A., Lewis K.D., Lewis K.D., et al. In vivo enhancer analysis of human conserved noncoding sequences. Nature. 2006;444:499–502. doi: 10.1038/nature05295. [DOI] [PubMed] [Google Scholar]
- Pennacchio L.A., Loots G.G., Nobrega M.A., Ovcharenko I., Loots G.G., Nobrega M.A., Ovcharenko I., Nobrega M.A., Ovcharenko I., Ovcharenko I. Predicting tissue-specific enhancers in the human genome. Genome Res. 2007;17:201–211. doi: 10.1101/gr.5972507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prabhakar S., Poulin F., Shoukry M., Afzal V., Rubin E.M., Couronne O., Pennacchio L.A., Poulin F., Shoukry M., Afzal V., Rubin E.M., Couronne O., Pennacchio L.A., Shoukry M., Afzal V., Rubin E.M., Couronne O., Pennacchio L.A., Afzal V., Rubin E.M., Couronne O., Pennacchio L.A., Rubin E.M., Couronne O., Pennacchio L.A., Couronne O., Pennacchio L.A., Pennacchio L.A. Close sequence comparisons are sufficient to identify human cis-regulatory elements. Genome Res. 2006;16:855–863. doi: 10.1101/gr.4717506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz S., Elnitski L., Li M., Weirauch M., Riemer C., Smit A., Green E.D., Hardison R.C., Miller W., Elnitski L., Li M., Weirauch M., Riemer C., Smit A., Green E.D., Hardison R.C., Miller W., Li M., Weirauch M., Riemer C., Smit A., Green E.D., Hardison R.C., Miller W., Weirauch M., Riemer C., Smit A., Green E.D., Hardison R.C., Miller W., Riemer C., Smit A., Green E.D., Hardison R.C., Miller W., Smit A., Green E.D., Hardison R.C., Miller W., NISC Comparative Sequencing Program. Green E.D., Hardison R.C., Miller W., Hardison R.C., Miller W., Miller W. MultiPipMaker and supporting tools: Alignments and analysis of multiple genomic DNA sequences. Nucleic Acids Res. 2003;31:3518–3524. doi: 10.1093/nar/gkg579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siepel A., Bejerano G., Pedersen J.S., Hinrichs A.S., Hou M., Rosenbloom K., Clawson H., Spieth J., Hillier L.W., Richards S., Bejerano G., Pedersen J.S., Hinrichs A.S., Hou M., Rosenbloom K., Clawson H., Spieth J., Hillier L.W., Richards S., Pedersen J.S., Hinrichs A.S., Hou M., Rosenbloom K., Clawson H., Spieth J., Hillier L.W., Richards S., Hinrichs A.S., Hou M., Rosenbloom K., Clawson H., Spieth J., Hillier L.W., Richards S., Hou M., Rosenbloom K., Clawson H., Spieth J., Hillier L.W., Richards S., Rosenbloom K., Clawson H., Spieth J., Hillier L.W., Richards S., Clawson H., Spieth J., Hillier L.W., Richards S., Spieth J., Hillier L.W., Richards S., Hillier L.W., Richards S., Richards S., et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005;15:1034–1050. doi: 10.1101/gr.3715005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stathopoulos A., Levine M., Levine M. Genomic regulatory networks and animal development. Dev. Cell. 2005;9:449–462. doi: 10.1016/j.devcel.2005.09.005. [DOI] [PubMed] [Google Scholar]
- Taylor J., Tyekucheva S., King D.C., Hardison R.C., Miller W., Chiaromonte F., Tyekucheva S., King D.C., Hardison R.C., Miller W., Chiaromonte F., King D.C., Hardison R.C., Miller W., Chiaromonte F., Hardison R.C., Miller W., Chiaromonte F., Miller W., Chiaromonte F., Chiaromonte F. ESPERR: Learning strong and weak signals in genomic sequence alignments to identify functional elements. Genome Res. 2006;16:1596–1604. doi: 10.1101/gr.4537706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trochet D., O’Brien L.M., Gozal D., Trang H., Nordenskjold A., Laudier B., Svensson P.J., Uhrig S., Cole T., Niemann S., O’Brien L.M., Gozal D., Trang H., Nordenskjold A., Laudier B., Svensson P.J., Uhrig S., Cole T., Niemann S., Gozal D., Trang H., Nordenskjold A., Laudier B., Svensson P.J., Uhrig S., Cole T., Niemann S., Trang H., Nordenskjold A., Laudier B., Svensson P.J., Uhrig S., Cole T., Niemann S., Nordenskjold A., Laudier B., Svensson P.J., Uhrig S., Cole T., Niemann S., Laudier B., Svensson P.J., Uhrig S., Cole T., Niemann S., Svensson P.J., Uhrig S., Cole T., Niemann S., Uhrig S., Cole T., Niemann S., Cole T., Niemann S., Niemann S., et al. PHOX2B genotype allows for prediction of tumor risk in congenital central hypoventilation syndrome. Am. J. Hum. Genet. 2005;76:421–426. doi: 10.1086/428366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatesh B., Kirkness E.F., Loh Y.H., Halpern A.L., Lee A.P., Johnson J., Dandona N., Viswanathan L.D., Tay A., Venter J.C., Kirkness E.F., Loh Y.H., Halpern A.L., Lee A.P., Johnson J., Dandona N., Viswanathan L.D., Tay A., Venter J.C., Loh Y.H., Halpern A.L., Lee A.P., Johnson J., Dandona N., Viswanathan L.D., Tay A., Venter J.C., Halpern A.L., Lee A.P., Johnson J., Dandona N., Viswanathan L.D., Tay A., Venter J.C., Lee A.P., Johnson J., Dandona N., Viswanathan L.D., Tay A., Venter J.C., Johnson J., Dandona N., Viswanathan L.D., Tay A., Venter J.C., Dandona N., Viswanathan L.D., Tay A., Venter J.C., Viswanathan L.D., Tay A., Venter J.C., Tay A., Venter J.C., Venter J.C., et al. Ancient noncoding elements conserved in the human genome. Science. 2006;314:1892. doi: 10.1126/science.1130708. [DOI] [PubMed] [Google Scholar]
- Westerfield M. The zebrafish book. University of Oregon Press; Eugene, OR: 2000. [Google Scholar]
- Woolfe A., Goodson M., Goode D.K., Snell P., McEwen G.K., Vavouri T., Smith S.F., North P., Callaway H., Kelly K., Goodson M., Goode D.K., Snell P., McEwen G.K., Vavouri T., Smith S.F., North P., Callaway H., Kelly K., Goode D.K., Snell P., McEwen G.K., Vavouri T., Smith S.F., North P., Callaway H., Kelly K., Snell P., McEwen G.K., Vavouri T., Smith S.F., North P., Callaway H., Kelly K., McEwen G.K., Vavouri T., Smith S.F., North P., Callaway H., Kelly K., Vavouri T., Smith S.F., North P., Callaway H., Kelly K., Smith S.F., North P., Callaway H., Kelly K., North P., Callaway H., Kelly K., Callaway H., Kelly K., Kelly K., et al. Highly conserved non-coding sequences are associated with vertebrate development. PLoS Biol. 2005;3:e7. doi: 10.1371/journal.pbio.0030007. [DOI] [PMC free article] [PubMed] [Google Scholar]