

Abstract
Recent accumulation of microbial genome data has demonstrated that lateral gene transfers constitute an important and universal evolutionary process in prokaryotes, while those in multicellular eukaryotes are still regarded as unusual, except for endosymbiotic gene transfers from mitochondria and plastids. Here we thoroughly investigated the bacterial genes derived from a Wolbachia endosymbiont on the nuclear genome of the beetle Callosobruchus chinensis. Exhaustive PCR detection and Southern blot analysis suggested that ∼30% of Wolbachia genes, in terms of the gene repertoire of wMel, are present on the insect nuclear genome. Fluorescent in situ hybridization located the transferred genes on the proximal region of the basal short arm of the X chromosome. Molecular evolutionary and other lines of evidence indicated that the transferred genes are probably derived from a single lateral transfer event. The transferred genes were, for the length examined, structurally disrupted, freed from functional constraints, and transcriptionally inactive. Hence, most, if not all, of the transferred genes have been pseudogenized. Notwithstanding this, the transferred genes were ubiquitously detected from Japanese and Taiwanese populations of C. chinensis, while the number of the transferred genes detected differed between the populations. The transferred genes were not detected from congenic beetle species, indicating that the transfer event occurred after speciation of C. chinensis, which was estimated to be one or several million years ago. These features of the laterally transferred endosymbiont genes are compared with the evolutionary patterns of mitochondrial and plastid genome fragments acquired by nuclear genomes through recent endosymbiotic gene transfers.
Lateral, or horizontal, gene transfer is the exchange of genetic materials between phylogenetically distant, and reproductively and genetically isolated, organisms. Conventionally, lateral gene transfer was regarded as a rather exceptional process with limited contributions to organismal evolution. However, recent accumulation of microbial genome data has revealed that, particularly in prokaryotes, lateral gene transfer is a much more universal evolutionary process than ever thought, contributing to reorganization of prokaryotic genomes and acquisition of novel biological functions (Ochman et al. 2000; Koonin et al. 2001).
Meanwhile, lateral gene transfer involved in eukaryotes is still regarded as unusual. However, as more and more genomic sequences from diverse eukaryotes become available, it has turned out that lateral gene transfers can also occur in eukaryotes. According to the comprehensive review by Andersson (2005), there are two types of gene transfers in eukaryotes: (1) transfer of genes from mitochondria and plastids, which are organelles of prokaryotic endosymbiont origin, hereafter called “endosymbiotic gene transfer,” and (2) transfer of genes from unrelated organisms, hereafter called “lateral gene transfer.”
In the nuclear genomes of diverse eukaryotes, in addition to the ancient endosymbiotic gene transfers dating back to the origin of eukaryotes, a large number of organellar genomic DNA fragments of much more recent origins are frequently found. It is now widely accepted that such endosymbiotic gene transfers are ubiquitous and currently ongoing in diverse eukaryotes (Bensasson et al. 2001; Leister 2003; Timmis et al. 2004).
By contrast, lateral gene transfers from unrelated organisms are thought to be uncommon among eukaryotes (Brown 2003; Andersson 2005). The majority of eukaryotic lateral gene transfers are found from unicellular eukaryotes (for review, see Andersson 2005). Since many unicellular eukaryotes are phagotrophic on bacteria and other micro-organisms, they have a constant intracellular exposure to prokaryotic DNA, which may predispose them to incorporate foreign genetic material into their genomes (Doolittle 1998). Meanwhile, lateral gene transfers are rarer in multicellular eukaryotes like animals (Doolittle 1998; Andersson 2005).
Here it is tempting to assume that endosymbiotic gene transfers of nonorganellar origin might have occurred in some multicellular animals that constantly harbor endocellular micro-organisms. Insects may represent such an animal group, wherein microbial endocellular associates occur universally. In particular, rickettsia-like endocellular α-proteobacteria of the genus Wolbachia are found in a wide range of arthropods and nematodes, including at least 20% of insect species. The endosymbionts are intimately associated with germ line cells of their hosts and are maternally transmitted, like organelles, through the host generations (O’Neill et al. 1997; Bourtzis and Miller 2003; Veneti et al. 2004). Considering the high levels of host–symbiont developmental integrity and ecological ubiquity, it is conceivable that such lateral gene transfers from the vertically transmitted endosymbionts might have occasionally occurred, thereby contributing to the host nuclear DNA dynamics to some extent, as does organelle-derived DNA.
Such an insect–endosymbiont lateral gene transfer was first reported from the adzuki bean beetle Callosobruchus chinensis, in which genes originating from a Wolbachia endosymbiont were genetically identified on the X chromosome of the insect (Kondo et al. 2002a). Recently, other cases of Wolbachia-derived lateral gene transfers have been emerging from genome sequencing projects of insects and nematodes (Fenn et al. 2006; Dunning-Hotopp et al. 2007). However, the evolutionary process and consequences of such host–symbiont lateral gene transfers have not yet been investigated in detail and thus are poorly understood.
In this study, we thoroughly investigated the magnitude, process, and evolution of the Wolbachia–insect lateral gene transfer in C. chinensis. A previous study suggested that the original donor bacterium of the transferred genes is a member of the Wolbachia group A (Kondo et al. 2002b), to which the Wolbachia strain wMel from Drosophila melanogaster belongs. Hence, we analyzed the laterally transferred Wolbachia genes in C. chinensis by making use of the wMel genome data (Wu et al. 2004) as reference information.
Results and Discussion
Exhaustive PCR detection of Wolbachia genes in the insect genome
We designed 222 pairs of PCR primers for specific amplification of 222 Wolbachia genes that cover the whole genome of wMel, consisting of 1195 open reading frames (ORFs) (Wu et al. 2004). When DNA samples of the Wolbachia-infected insect strain jC were subjected to PCR amplification with these primer sets, 205 pairs amplified a DNA segment of expected size while 17 pairs did not (Supplemental Table S1). When DNA samples of the tetracycline-treated, Wolbachia-free insect strain jCAus were subjected to PCR amplification with the 205 primer sets, 57 pairs amplified a DNA segment of expected size (Supplemental Table S1). DNA sequencing confirmed that all of these PCR products are certainly Wolbachia genes (Supplemental Table S1). These results suggested that a large number of Wolbachia genes are present in the insect genome.
Exhaustive Southern blot detection of Wolbachia genes in the insect genome
By using long PCR amplification with these primers, we prepared 143 partial genomic segments of wMel. These segments had an average size of 8.4 kb (ranging from 960 to 18,030 bp), and neighboring segments shared an average overlap of 0.4 kb (ranging from 180 to 3731 bp) (Supplemental Table S2). In total, these DNA segments covered 1145 kb, about 90% of the 1268-kb genome of wMel, except for two prophage regions where repetitive sequences prevented reliable PCR amplification. By using these segments as probes, DNA samples of the Wolbachia-free insect strain jCAus were subjected to Southern blot analysis. Of the 143 probes, 52 probes produced significant hybridization signals (Supplemental Table S2), suggesting that large regions of the Wolbachia genome are present in the insect genome.
Mapping of transferred Wolbachia genes on the wMel genome
Figure 1 shows a mapping of the PCR-positive sites and the Southern-positive regions on the wMel genome. In general, the Southern-positive regions showed good agreement with the PCR-positive sites. Namely, in each of the regions of the 43 positive probes, one or more PCR-positive sites were located. In the regions of the other nine positive probes, no PCR-positive sites were identified. Probably these regions contain only a short stretch of Wolbachia gene, or Wolbachia genes in these regions have accumulated too many mutations to be amplified by PCR. Both the PCR-positive and Southern-positive regions were remarkably concentrated on the left half of the wMel genome (see Fig. 1). These patterns suggested that a large Wolbachia genome segment was integrated into the genome of the jCAus insect.
Figure 1.

Wolbachia genes detected from the bean beetle Callosobruchus chinensis strain jCAus mapped on the whole genome of the Wolbachia strain wMel from Drosophila melanogaster. (Inner circle) PCR detection of Wolbachia genes. Red and black lines indicate PCR-positive and PCR-negative genes, respectively. (Outer circle) Southern blot detection of Wolbachia genome regions. Green and white blocks indicate Southern-positive and Southern-negative regions, respectively. Pink blocks show prophage regions.
Estimating the magnitude of lateral transfer of Wolbachia genes
Of 205 Wolbachia genes inspected by PCR, 57 genes were successfully amplified (Supplemental Table S1). DNA sequencing confirmed that all these amplifications were true positives. There is no doubt that some of the other genes were not amplified due to primer mismatches, comprising pseudonegatives. Hence, the value of 28% (57/205) gene transfer in terms of the wMel genome must be an underestimate. Of 143 probes that covered nearly the whole genome of wMel, 52 probes gave positive signals in the Southern blot analysis of jCAus DNA (Supplemental Table S2). There is no doubt that, in many cases, only a partial region of these probes hybridized to the transferred Wolbachia genes located on the insect genome. Hence, the value of 36% (52/143) gene transfer in terms of the wMel genome must be an overestimate. On the basis of these data, we suggest that around 30% (28%–36%) of the wMel genes are present in the genome of the insect strain jCAus. Given that the wMel genome is 1268 kb in size, encoding 1195 ORFs (Wu et al. 2004), about 380 kb of the Wolbachia genomic region, encoding 360 genes, is expected to be located on the insect genome.
Physical mapping of transferred Wolbachia genes on the insect chromosomes
By using either the PCR-positive products or the Southern-positive regions as probes, we performed fluorescent in situ hybridization (FISH) analysis of the transferred Wolbachia genes on the chromosomes of the jCAus insects. A strong signal was consistently and specifically detected on the proximal region of the basal short arm of X chromosome (Fig. 2), confirming the previous report of X-linked inheritance of the wsp gene in the jCAus insects (Kondo et al. 2002a).
Figure 2.
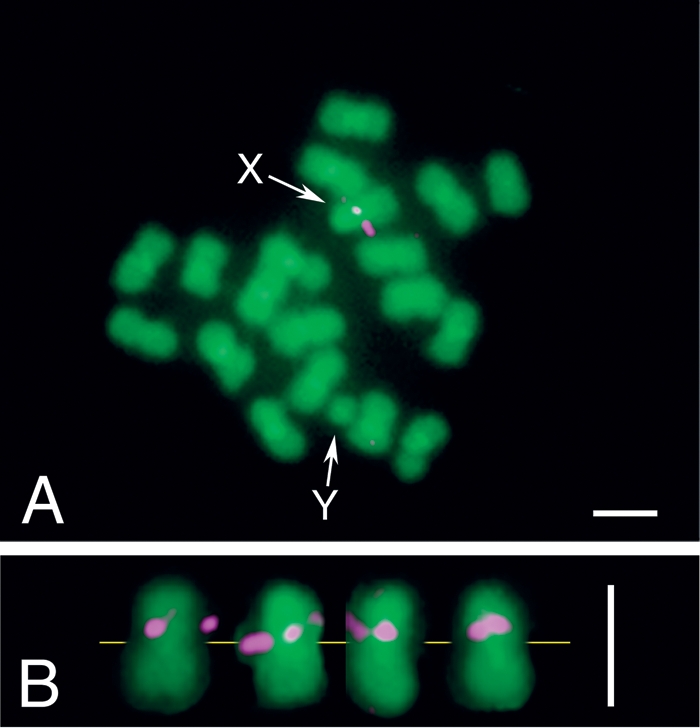
FISH detection of Wolbachia genes on the chromosomes of Callosobruchus chinensis strain jCAus. (A) Metaphase chromosomes of a male insect. X and Y indicate X chromosome and Y chromosome, respectively. Note that C. chinensis is male-heterogametic with a karyotype of 2n = 20, XY. (B) Metaphase X chromosomes from different preparations, with yellow line indicating the position of centromeres, magenta, areas indicating Wolbachia gene signals; and green indicating insect chromosome signals. Bars, 1 μm.
Phylogenetic relationship to other Wolbachia genomes
The wsp gene on the chromosome of C. chinensis was reported to belong to the Wolbachia group A (Kondo et al. 2002b). By using 56 genes commonly available for the Wolbachia strains wMel (group A) (Wu et al. 2004), wPip (group B) (Sanger Institute), and wBm (group E) (Foster et al. 2005), we analyzed phylogenetic placement of the Wolbachia genes transferred to the C. chinensis chromosome. Molecular phylogenetic analysis of the total sequences indicated that the transferred genome is closely related to the genome of wMel rather than to the genome of wPip (Supplemental Fig. S1). Distance analysis also showed that each of the transferred genes is certainly related to the gene of wMel rather than to the gene of wPip (Fig. 3A). The tendency was highly significant statistically (randomization test, P < 0.00001).
Figure 3.

(A) Distribution of genetic distances from the transferred Wolbachia genes: wMel vs. wPip. In total, 56 partial gene sequences were subjected to the analysis (see Supplemental Table S1). Filled bars show the distribution of KS values between transferred Wolbachia genes and wMel genes. Open bars indicate the distribution of KS values between transferred Wolbachia genes and wPip genes. (B) Comparison of molecular evolutionary rate between the transferred Wolbachia genes and the wMel genes. In total, 54 partial gene sequences were subjected to the analysis (see Supplemental Table S1). KA/KS values of transferred genes vs. wPip genes were respectively divided by KA/KS values of wMel genes vs. wPip genes, and the distribution of the log ratios was plotted. In the plot, values less than zero indicate that the transferred genes exhibit lower KA/KS values than the corresponding wMel genes, while values larger than zero imply that the transferred genes show higher KA/KS values than the corresponding wMel genes. Note that the transferred genes generally exhibit higher KA/KS values than the corresponding wMel genes.
Single evolutionary origin of transferred Wolbachia genes
These results, namely, the concentrated distribution of the transferred genes on the wMel genome (Fig. 1), their common physical location on the proximal region of the basal short arm of X chromosome (Fig. 2), and their consistent phylogenetic affinity to the wMel genes (Fig. 3A), suggest that the Wolbachia genes present on the C. chinensis chromosome are not acquired through multiple lateral transfer events but are of single evolutionary origin from a Wolbachia strain that belongs to group A. Of course, however, the possibility of multiple acquisitions from the same or genetically close Wolbachia strains cannot be ruled out.
Structural disruptions in transferred Wolbachia genes
It is of evolutionary interest whether the transferred Wolbachia genes on the insect chromosome are functional. Of 57 transferred genes whose sequence was determined, 27 genes contained stop codon(s) and/or frame shift(s) (Supplemental Fig. S2; Supplemental Table S1). Hence, many of the transferred Wolbachia genes have been inactivated by structural disruptions.
Accelerated molecular evolution in transferred Wolbachia genes
If, as suggested above, most of the transferred Wolbachia genes have been disrupted and lost their function, it is expected that accelerated molecular evolution should be observed in these genes due to relaxed functional constraints. In order to test this idea, we compared KA/KS values of the transferred genes with those of the corresponding wMel genes, by using the corresponding genes of wPip as outgroup. The distribution of KA/KS ratios clearly indicated that the values for the transferred genes are generally higher than the values for the wMel genes (Fig. 3B). The tendency was highly significant statistically (randomization test, P < 0.00001).
RT-PCR assay of transcriptional activity in transferred Wolbachia genes
If some of the transferred Wolbachia genes are actually functional, then such genes must be transcribed into messenger RNAs. Hence, 57 transferred Wolbachia genes were subjected to RT-PCR detection (Supplemental Table S1). While no PCR products were obtained for 23 genes, amplified products of expected size were detected for the other 34 genes (Fig. 4A). Negative controls without reverse transcription did not exhibit these products, and DNA sequencing revealed that all these RT-PCR products certainly represent the target genes (data not shown). At a glance, these results indicated that, strikingly, more than half of the transferred Wolbachia genes are transcriptionally active on the insect chromosome.
Figure 4.

Transcriptional activity of the transferred Wolbachia genes. (A) RT-PCR assay of 57 transferred Wolbachia genes. The circle is identical to the inner circle of Figure 1. Open dots indicate genes with amplified products, while filled dots show genes without amplification. (B) Quantitative RT-PCR assay of 35 transferred Wolbachia genes, of which 26 were RT-PCR positive and nine were RT-PCR negative. An exon region and an intron region of the insect ef1α gene were also quantified as positive and negative controls. Each of the green diamonds shows an RNA sample from an insect treated with reverse transcriptase, while each of the red dots indicates an RNA sample from an insect without reverse transcriptase. In the ORF line, open dots indicate structurally intact ORFs, while filled dots show disrupted ORFs. In the RT-PCR line, open dots and filled dots indicate RT-PCR–positive and RT-PCR–negative, respectively.
Quantitative RT-PCR assay of transcriptional activity in transferred Wolbachia genes
Thus, we estimated the transcriptional activity of some of these genes by a quantitative RT-PCR technique. As internal controls for the quantitative assay, exon and intron regions of elongation factor 1α (ef1α) gene of the host insect were also quantified. We measured 26 RT-PCR–positive genes and nine RT-PCR–negative genes and found that expression levels of these genes were consistently low. Most of the genes showed expression levels of 102–103 copies per microgram of total RNA; the highest level was observed with the ftsH gene at about 104 copies per microgram of total RNA, but all of these values were lower than the expression level of the ef1α intron (Fig. 4B). Since negative controls without reverse transcription always resulted in substantially zero values, transcripts of these genes may certainly exist, but their expression levels are very low. At this stage, it is obscure whether these very low expression levels of the transferred Wolbachia genes have biological significance. Considering that bacterial gene promoters usually do not work in eukaryotic cells, the possibility should be considered that these transcripts are not produced by a normal promoter-mediated transcriptional process but rather by a nonspecific leaky transcription at background levels. Future studies should focus on structure of the transcripts revealed by 5′ and 3′ RACE techniques and on spatial and temporal dynamics of the transcripts analyzed by in situ hybridization and tissue-level quantitative RT-PCR.
Are there any host insects without transferred Wolbachia genes?
In Japanese populations of C. chinensis, diagnostic PCR survey revealed that 2.9% (12/410) insects did not possess the wsp gene originating from the transferred Wolbachia genome (Kondo et al. 2002b). We established two isofemale lines from such insects—k10 and r13—of different geographic origins. By tetracycline treatment and subsequent maintenance in the laboratory, we generated two derivative lines of the insects—k10U2 and r13U1—from which no wsp gene was detectable by PCR. Recently, in 80 individuals of C. chinensis from six Taiwanese populations, we identified some insects from which wsp gene of the transferred Wolbachia type was not detected (Kondo et al., unpubl.). We established an insect line named Rm and similarly generated a tetracycline-treated derivative line Rmtet from which no wsp gene was detected by PCR (Supplemental Table S3). It was of evolutionary interest whether these insect strains are actually free of the transferred Wolbachia genes.
Variations in transferred Wolbachia genes in host insect populations
These insect strains k10U2, r13U1, and Rmtet were subjected to PCR amplification with the 205 primer sets for Wolbachia genes. It turned out that these insects certainly possessed the transferred Wolbachia genes, but the gene repertoire was remarkably different between them (Fig. 5; Supplemental Fig. S3; Supplemental Table S1). Of the 205 genes inspected, 40 genes were detected from strains k10U2 and r13U1, while only 31 genes were found from strain Rmtet. These numbers were remarkably smaller than 57 genes identified from the strain jCAus. These results indicated that the transferred Wolbachia genome fragment is universally found among natural populations of C. chinensis and that considerable variations in the transferred Wolbachia gene repertoire are found within and between the populations.
Figure 5.

PCR detection of the Wolbachia genes from different strains of Callosobruchus chinensis. (Outermost circle) Genome map of wMel as shown in Figure 1; (second circle) PCR detection of Wolbachia genes in the strain jCAus, as shown in Figure 1; (third circle) PCR detection of Wolbachia genes in the strains k10U2 and r13U1; (innermost circle) PCR detection of Wolbachia genes in the strain Rmtet. Red and black lines indicate PCR-positive and PCR-negative genes, respectively.
Parsimonious estimation of evolutionary process leading to the variations in transferred Wolbachia genes
An interesting pattern was seen in these transferred Wolbachia gene repertoires. Namely, 57 Wolbachia genes were detected in the insect strain jCAus; 40 Wolbachia genes, which comprised a subset of the 57 genes, were present in the strains k10U2 and r13U1; and 31 Wolbachia genes, 29 of which constituted a subset of the 40 genes, were found in the strain Rmtet (Supplemental Fig. S3). On the assumption that acquisition of Wolbachia genes is difficult while loss of Wolbachia genes is easy, the nested gene possession pattern suggested the following evolutionary scenario: (1) among the insect strains, jCAus diverged first; (2) 16 genes were lost in the lineage leading to the strains k10U2, r13U1, and Rmtet; (3) Rmtet diverged from the lineage of k10U2 and r13U1; (4) 11 genes were lost in the lineage of Rmtet; (5) a gene, vdcC, was lost in the lineages of jCAus and k10U2/r13U1 independently, and a gene, pyrD, was lost in the lineage of k10U2 and r13U1 (Fig. 6A).
Figure 6.

Phylogenetic relationships between the insect strains jCAus, k10U2, r13U1, and Rmtet of Callosobruchus chinensis. (A) Relationship of the insect strains deduced from their possession patterns of the transferred Wolbachia genes. (B) Relationship inferred from 522 aligned nucleotide sites of mitochondrial cytochrome oxidase I gene. (C) Relationship inferred from 590 aligned nucleotide sites of nuclear elongation factor 1α intron. (D) Relationship of the concatenated amino acid sequence of the 27 transferred Wolbachia genes based on 2929 aligned amino acid sites. NJ trees are shown, whereas ML and BA trees exhibit the same topologies. Support values >50% are indicated at the nodes.
Phylogenetic relationship between the insect strains
Indeed the parsimonious reconstruction of the evolutionary process of the transferred Wolbachia genes was simple and beautiful, but several lines of evidence raised an objection to the scenario. When we constructed phylogenetic relationships between the insect strains, both mitochondrial and nuclear genes indicated that not jCAus but Rmtet constituted the basal lineage (Fig. 6B,C). Phylogenetic analysis of the 27 transferred Wolbachia genes also supported the relationship (Fig. 6D). These results consistently refuted the above evolutionary hypothesis Fig. 6A).
Nucleotide sequence polymorphisms in transferred Wolbachia genes
Furthermore, we found an interesting pattern in the 57 transferred Wolbachia genes identified in the insect strain jCAus. When PCR products of these genes were directly sequenced, it turned out that 24 of the 57 genes contained nucleotide sequence polymorphisms. These polymorphisms were also confirmed by cloning and sequencing of the PCR products (Supplemental Table S4). Strikingly, 23 of the 24 genes, except for pyrD, were a subset of the 29 genes that were conserved among all the insect strains jCAus, k10U2, r13U1, and Rmtet (Supplemental Fig. S3).
Putative duplication of transferred Wolbachia genome fragment on the insect chromosome
These patterns suggested that (1) the transferred Wolbachia genome is, at least partially, duplicated; (2) the conserved Wolbachia genes tend to be duplicated while the other genes are likely to be single-copied; and (3) the gene duplication probably constitutes a reason why some of the transferred genes are conserved whereas others are not. The process and mechanism of the duplication are at present obscure. However, considering that the FISH signal was restricted to the proximal region of the basal short arm of X chromosome (Fig. 2), the duplicated genome fragments are likely to be located in the physical vicinity, possibly constituting tandem repeats. If so, unequal crossing over between synapsing X chromosomes is suspected as a candidate process involved in the gene duplication. To verify this idea, the genomic structure of the transferred Wolbachia genes should be determined.
Absence of transferred Wolbachia genes in bean beetles allied to C. chinensis
In Japanese populations of C. chinensis, 97.1% (398/410) insects possessed wsp gene of lateral transfer origin (Kondo et al. 2002b). In representatives of the remaining 2.9% (12 individuals)—namely, the strains k10U2 and r13U1—we found that the transferred Wolbachia genes were actually present, although wsp and some other genes were not detected. In Taiwanese populations of C. chinensis, even in the absence of wsp gene, many of the transferred Wolbachia genes were also identified (Fig. 5; Supplemental Fig. S3; Supplemental Table S1). By contrast, in Callosobruchus maculatus and Callosobruchus rhodesianus, which are congenic relatives of C. chinensis, no Wolbachia genes were detected (Supplemental Table S1). Hence, it appears likely that the Wolbachia genome fragment was transferred to the insect chromosome after speciation of C. chinensis.
When did the Wolbachia genome transfer occur?
The Wolbachia genome fragment must have been present in the ancestor of the Japanese and Taiwanese strains of C. chinensis. The evolutionary distance values between the strains jCAus and Rmtet were nearly comparable to the values between D. melanogaster and Drosophila simulans. By applying the evolutionary rates estimated from biogeography and molecular clocks of multiple loci of the fruit flies (Lachaise et al. 1988; Li et al. 1999), we estimated the divergence time of the C. chinensis strains as 2.5 MYA and 0.74 MYA on the basis of mitochondrial COI gene and nuclear ef1α intron gene sequences, respectively (Supplemental Table S5). Hence, although the estimate is quite rough, the Wolbachia genome fragment was probably transferred to the insect chromosome about one or a few million years ago in the ancestor of C. chinensis.
Possible mechanisms for prevalence of transferred Wolbachia genes in host insect populations
All of these results suggested that the Wolbachia genome fragment integrated in the X chromosome of C. chinensis is nearly fixed in the insect populations and has undergone different levels of evolution and mutational degradation in different lineages. The mechanism whereby the prevalence has been established is currently not understood. Fixation through genetic drift, possibly associated with a population bottleneck that occurred in the ancestor of C. chinensis, is a candidate mechanism. In the process of spreading in the insect populations, hitchhiking with coexisting Wolbachia endosymbionts—which are universally found in current C. chinensis populations and cause cytoplasmic incompatibility (Kondo et al. 2002b)—might have played some roles. Alternatively, the Wolbachia genome fragment might behave like Medea, a maternal-effect chromosomal factor known from the flour beetle Tribolium castaneum that is capable of spreading in host populations in a selfish manner (Beeman et al. 1992). Meaningfully, Medea phenotype and Wolbachia-induced cytoplasmic incompatibility can be explained by the same “poison-antidote” or “modification-rescue” mechanism (Poinsot et al. 2003).
Evolution and fate of laterally transferred endosymbiont genes
All of the data obtained in this study consistently indicate that the Wolbachia genes transferred to the host insect chromosome tend to be structurally disrupted (Supplemental Fig. S2), eroded and lost (Fig. 5), freed from functional constraints (Fig. 3), and transcriptionally inactive (Fig. 4). These results strongly suggest that most, if not all, of the laterally transferred endosymbiont genes have been pseudogenized and, although currently prevalent in the host insect populations, are destined to be DNA junk in the insect genome.
Similarity to organelle genomes recently transferred to host nucleus
Interestingly, these evolutionary patterns are prevalent among remnant mitochondrial and plastid genomes located on the nuclear genomes of various plants, fungi, and animals, including humans (Bensasson et al. 2001; Timmis et al. 2004). These organelles are evolutionarily of bacterial endosymbiont origins (Margulis 1981), acquired by ancestral eukaryotes very anciently (Hedges et al. 2004); most of their original gene repertoire has been lost in the evolutionary course, leading to drastically reduced organellar genomes (Lang et al. 1999; Simpson and Stern 2002), and some of the genes are transferred to the nuclear genome where such genes are transcribed, translated, and transported to the organelles for functioning (Leister 2003; Brown 2003). Aside from the ancient endosymbiotic gene transfers, a large number of organellar genomic DNA fragments of much more recent origins are present in the nuclear genomes of diverse eukaryotes, indicating that endosymbiotic gene transfer is a ubiquitous and currently ongoing process that pervades nuclear DNA dynamics (Bensasson et al. 2001; Timmis et al. 2004). The transferred organelle DNA can be amplified once inserted (Woischnik and Moraes 2002; Hazkani-Covo et al. 2003), and some of them are organized as tandem repeats (Tourmen et al. 2002). These nuclear copies of organellar genes are totally pseudogenized, like the Wolbachia genes transferred to the X chromosome of C. chinensis.
How frequent are lateral gene transfers from Wolbachia endosymbionts to their hosts?
Here it should be noted that Wolbachia endosymbionts are intimately associated with germ-line cells and are maternally transmitted, like organelles, via ovarial passage through host generations (Veneti et al. 2004). Worldwide PCR surveys of Wolbachia infection in diverse insect taxa have consistently revealed high infection frequencies, >20% at the species level (Werren and Windsor 2000). On account of such host–symbiont developmental integrity and ecological ubiquity, the following questions may arise: Why not more cases of Wolbachia–host gene transfers? Is the case of C. chinensis an orphan exception, or are other cases to be found? How frequent have such lateral gene transfers occurred?
Actually, other cases are emerging from recent genome sequencing projects of insects and nematodes. Genome fragments of Wolbachia endosymbionts, whose sizes vary from single-gene level to nearly whole-genome level, have been detected from the fruit fly Drosophila ananassae, the parasitoid wasps Nasonia giraulti, Nasonia vitripennis, and Nasonia longicornis, and the filarial nematodes Onchocerca volvulus, Onchocerca ochengi, Brugia malayi, Brugia timori, Brugia pahangi, and Dirofilaria immitis (Fenn et al. 2006; Dunning-Hotopp et al. 2007). These recent findings suggest that such symbiont–host gene transfers might have been occurring more frequently than previously envisioned, like the gene transfers from organelles to nucleus.
Conclusion and perspective
In this study, we comprehensively investigated the magnitude, process and evolution of the lateral genome transfer from Wolbachia endosymbiont to C. chinensis with reference to the wMel genome information. Currently we are making efforts to completely determine the whole genomic region of C. chinensis containing all the transferred Wolbachia genome fragments by fosmid/BAC genomic library screening.
Once all the transferred Wolbachia genes are identified, we will be able to evaluate the transcriptional activities of all the symbiont-derived genes on the insect chromosome. Certainly it is plausible that all the transferred genes will finally turn out to be molecular fossils of an evolutionary episode in the past, but if some of the genes would be identified as transcriptionally active and functional, such genes would provide novel insights into how host–symbiont gene transfers could contribute to organismal evolution.
The evolutionary origin of the transferred Wolbachia genes in natural populations of C. chinensis is still an enigma. The current worldwide distribution of the pest of stored beans is probably mediated by human activities (Borowiec 1987). Mitochondrial DNA analysis of Asian populations of C. chinensis certainly showed that insect samples collected from bean field/storage/market exhibited specific genotypes, while insect samples collected from wild leguminous plants were genetically diverse (Tuda et al. 2004). Hence, it is expected that a worldwide survey of wild C. chinensis populations not infesting agricultural beans but feeding on wild legumes would lead to key discoveries, such as insect populations without the transferred Wolbachia genes or insect populations infected with a relict bacterial Wolbachia endosymbiont closely related to the transferred Wolbachia genome.
Methods
Materials
Strains of C. chinensis and allied bean beetles used in this study are listed in Supplemental Table S3. These insects were maintained on seeds of the adzuki bean Vigna angularis at 30°C in a long-day regimen of 16 h light and 8 h dark, except for the strains Rm and Rmtet fed on seeds of the wild legume Rhynchosia minima.
DNA extraction
Female adults of C. chinensis were subjected to DNA extraction by using the QIAamp DNA Mini kit (Qiagen) for PCR amplification or by a standard proteinase K/SDS/phenol/chloroform extraction procedure for Southern blotting.
PCR detection of Wolbachia genes
Specific primer sets for amplifying each of the Wolbachia genes were designed on the basis of published and ongoing genome sequences of three Wolbachia strains, wMel (Wu et al. 2004), wBm (Foster et al. 2005), and wPip (Sanger Institute) (Supplemental Table S1). Primer pairs that failed to amplify PCR products of expected size from the positive DNA samples—the D. melanogaster strain OGS-1 infected with wMel and the Wolbachia-infected C. chinensis strain jC—were not used for further analyses. The amplified products were directly sequenced, and the sequences were subjected to BLAST search to confirm their sequence identity to Wolbachia genes.
Probe construction for Southern blotting
DNA probes for Southern blotting were synthesized by long-PCR amplification from the wMel-infected D. melanogaster DNA as template. The Wolbachia-specific gene primers used are listed in Supplemental Table S2. The amplified products were electrophoresed in agarose gels, excised and extracted from the gels, and digoxigenin-labeled by using the DIG Random Primed DNA Labeling Kit (Roche).
Southern blotting
Genomic DNA preparations of the C. chinensis strain jCAus were digested with restriction endonuclease EcoRI, BamHI, or HindIII. The digested DNA samples, 5–10 μg in each lane, were electrophoresed in agarose gels. Genomic DNA of the wMel-infected D. melanogaster was used as positive control. The separated DNA fragments were transferred to nylon membranes by a standard capillary blotting procedure, and fixed by UV-cross-linking. Hybridization and detection of each of the probes were performed by using the DIG Detection Kit (Roche) according to the manufacturer’s instructions. Specific hybridization of all the probes was confirmed by using the genomic DNA of wMel-infected D. melanogaster, wherein the banding patterns of the signals agreed with the restriction patterns inferred from the genomic sequence of wMel deposited in the DNA database.
DNA sequencing
The PCR products were sequenced by direct sequencing and/or sequencing of plasmid-cloned DNA. In the former, the PCR products were treated with Exo-SAP-IT (Amersham) and sequenced by using amplifying primers. In the latter, the PCR products were cloned into pGEM T-vector (Promega) or pCR Blunt-vector (Invitrogen). The cloned inserts were amplified by single-colony PCR with vector-specific primers and subjected to sequencing with the primers.
FISH
Testes were dissected from adult individuals of C. chinensis in a phosphate-buffered saline, swollen for 30 min in a hypotonic solution (75 mM KCl), and fixed, crushed, and suspended in methanol–acetic acid (3:1) by repeated pipetting; the suspended cells were then spread and dried on clean glass slides. Biotin-labeled DNA probes were prepared by using the Biotin Nick Translation Mix (Roche). Two sets of labeled probes were synthesized. The short-probes set was produced by labeling short PCR products (180–2500 bp) of the 36 transferred Wolbachia genes (Supplemental Table S1). The long-probes set consisted of four long PCR products (3.5, 4.0, 5.5, and 8.8 kb in size) amplified by the primer pairs as described in Supplemental Table S6. The labeled probes were dissolved in a hybridization buffer (50% formamide, 10% dextran sulfate, 2× SSC). Chromosomal DNA preparations were denatured at 76°C for 2 min in a denaturing buffer (70% formamide, 2× SSC), and were incubated overnight with the probe-containing hybridization buffer. After being thoroughly washed, the hybridized probes on the chromosomes were detected by using AlexaFluor488-labeled streptavidin (Invitrogen), and the signals were amplified by using antistreptavidin antibody biotin conjugate (Vector). The whole chromosomes were counterstained with 4,6-diamino-2-phenylidole, and the hybridization signals were observed under a fluorescent microscope (BX-50, Olympus) and recorded by a chilled CCD camera (Sensys 1400, Photometrics). Pseudocolor images were generated by IPLab (Scanalytics).
RT-PCR
Total-RNA samples were individually prepared from adult females of C. chinensis by using the RNA Mini Kit (QIAGEN). The RNA samples were treated with DNase, reverse-transcribed with SuperScriptIII (Invitrogen), and subjected to PCR using the primers listed in Supplemental Table S1. All of the RNA samples were tested for genomic DNA contamination by performing PCR without reverse transcription.
Quantitative RT-PCR
The reverse-transcribed RNA samples were subjected to real-time fluorescent quantitative RT-PCR by using ABI Prism 7300 Sequence detection system (Applied Biosystems), with SYBR Green I (Molecular Probes) as detection dye and ROX (Invitrogen) as reference dye, as described elsewhere (Narita et al. 2007). The PCR primers are listed in Supplemental Table S6.
Molecular phylogenetic and evolutionary analyses
Phylogenetic analyses were conducted by the neighbor-joining (NJ) method, the maximum-likelihood (ML) method, and Bayesian analysis (BA). NJ trees were constructed by using the program package ClustalW (Thompson et al. 1994) or MEGA3.1 (Kumar et al. 2004). Bootstrap probabilities were calculated by generating 1000 bootstrap replicates. ML trees were estimated by using the program package TREE-PUZZLE 5.2 (Schmidt et al. 2002). In BA analysis, we used the program package MrBayes 3.1.2 (Ronquist and Huelsenbeck 2003). The numbers of synonymous and nonsynonymous substitutions per site were calculated as described (Miyata and Yasunaga 1980).
Acknowledgments
We thank M. Tuda, S.-B. Horng, Y.-C. Lan, J.-F. Tsai, H.-Y. Lin, and Y. Fuyama for collection and maintenance of the insect samples; K. Goka for logistic supports; and K. Nikoh for technical assistance. This study was financially supported by the National Institute of Advanced Industrial Science and Technology (AIST) to TF, the 21st COE Program “Research Center for Integrated Science” (E-5) at the University of Tokyo to M.S., and a Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan to N.N. N.K. was supported by the Japan Society for the Promotion of Science (JSPS) Research Fellowship for Young Scientists.
Footnotes
[Supplemental material is available online at www.genome.org. The sequence data from this study have been submitted to GenBank under accession nos. EF534574–EF534700 and EF560581–EF560584.]
Article published online before print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.7144908
References
- Andersson J.O. Lateral gene transfer in eukaryotes. Cell. Mol. Life Sci. 2005;62:1182–1197. doi: 10.1007/s00018-005-4539-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beeman R.W., Friesen K.S., Denell R.E., Friesen K.S., Denell R.E., Denell R.E. Maternal-effect selfish genes in flour beetles. Science. 1992;256:89–92. doi: 10.1126/science.1566060. [DOI] [PubMed] [Google Scholar]
- Bensasson D., Zhang D., Hartl D.L., Hewitt G.M., Zhang D., Hartl D.L., Hewitt G.M., Hartl D.L., Hewitt G.M., Hewitt G.M. Mitochondrial pseudogenes: Evolution’s misplaced witnesses. Trends Ecol. Evol. 2001;16:314–321. doi: 10.1016/s0169-5347(01)02151-6. [DOI] [PubMed] [Google Scholar]
- Borowiec L. The genera of seed-beetles (Coleoptera, Bruchidae) Polsk. Pismo Entomol. 1987;57:3–207. [Google Scholar]
- Bourtzis K., Miller T.A., Miller T.A. Insect symbiosis. CRC Press; Boca Raton, FL: 2003. [Google Scholar]
- Brown J.R. Ancient horizontal gene transfer. Nat. Rev. Genet. 2003;4:121–132. doi: 10.1038/nrg1000. [DOI] [PubMed] [Google Scholar]
- Doolittle W.E. You are what you eat: A gene transfer ratchet could account for bacterial genes in eukaryotic nuclear genomes. Trends Genet. 1998;14:307–311. doi: 10.1016/s0168-9525(98)01494-2. [DOI] [PubMed] [Google Scholar]
- Dunning-Hotopp J.C., Clark M.E., Oliveira D.C.S.G., Foster J.M., Fischer P., Munoz Torres M.C., Giebel J.D., Kumar N., Ishmael N., Wang S., Clark M.E., Oliveira D.C.S.G., Foster J.M., Fischer P., Munoz Torres M.C., Giebel J.D., Kumar N., Ishmael N., Wang S., Oliveira D.C.S.G., Foster J.M., Fischer P., Munoz Torres M.C., Giebel J.D., Kumar N., Ishmael N., Wang S., Foster J.M., Fischer P., Munoz Torres M.C., Giebel J.D., Kumar N., Ishmael N., Wang S., Fischer P., Munoz Torres M.C., Giebel J.D., Kumar N., Ishmael N., Wang S., Munoz Torres M.C., Giebel J.D., Kumar N., Ishmael N., Wang S., Giebel J.D., Kumar N., Ishmael N., Wang S., Kumar N., Ishmael N., Wang S., Ishmael N., Wang S., Wang S., et al. Widespread lateral gene transfer from intracellular bacteria to multicellular eukaryotes. Science. 2007;317:1753–1756. doi: 10.1126/science.1142490. [DOI] [PubMed] [Google Scholar]
- Fenn K., Conlon C., Jones M., Quail M.A., Holroyd N.E., Parkhill J., Blaxter M., Conlon C., Jones M., Quail M.A., Holroyd N.E., Parkhill J., Blaxter M., Jones M., Quail M.A., Holroyd N.E., Parkhill J., Blaxter M., Quail M.A., Holroyd N.E., Parkhill J., Blaxter M., Holroyd N.E., Parkhill J., Blaxter M., Parkhill J., Blaxter M., Blaxter M. Phylogenetic relationships of the Wolbachia of nematodes and arthropods. PLoS Pathog. 2006;2:e94. doi: 10.1371/journal.ppat.0020094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster J., Ganatra M., Kamal I., Ware J., Makarova K., Ivanova N., Bhattacharyya A., Kapatral V., Kumar S., Posfai J., Ganatra M., Kamal I., Ware J., Makarova K., Ivanova N., Bhattacharyya A., Kapatral V., Kumar S., Posfai J., Kamal I., Ware J., Makarova K., Ivanova N., Bhattacharyya A., Kapatral V., Kumar S., Posfai J., Ware J., Makarova K., Ivanova N., Bhattacharyya A., Kapatral V., Kumar S., Posfai J., Makarova K., Ivanova N., Bhattacharyya A., Kapatral V., Kumar S., Posfai J., Ivanova N., Bhattacharyya A., Kapatral V., Kumar S., Posfai J., Bhattacharyya A., Kapatral V., Kumar S., Posfai J., Kapatral V., Kumar S., Posfai J., Kumar S., Posfai J., Posfai J., et al. The Wolbachia genome of Brugia malayi: Endosymbiont evolution within a human pathogenic nematode. PLoS Biol. 2005;3:e121. doi: 10.1371/journal.pbio.0030121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazkani-Covo E., Sorek R., Graur D., Sorek R., Graur D., Graur D. Evolutionary dynamics of large numts in the human genome: Rarity of independent insertions and abundance of post-insertion duplications. J. Mol. Evol. 2003;56:169–174. doi: 10.1007/s00239-002-2390-5. [DOI] [PubMed] [Google Scholar]
- Hedges S.B., Blair J.E., Venturi M.L., Shoe J.L., Blair J.E., Venturi M.L., Shoe J.L., Venturi M.L., Shoe J.L., Shoe J.L. A molecular timescale of eukaryote evolution and the rise of complex multicellular life. BMC Evol. Biol. 2004;4:2. doi: 10.1186/1471-2148-4-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo N., Nikoh N., Ijichi N., Shimada M., Fukatsu T., Nikoh N., Ijichi N., Shimada M., Fukatsu T., Ijichi N., Shimada M., Fukatsu T., Shimada M., Fukatsu T., Fukatsu T. Genome fragment of Wolbachia endosymbiont transferred to X chromosome of host insect. Proc. Natl. Acad. Sci. 2002a;99:14280–14285. doi: 10.1073/pnas.222228199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo N., Ijichi N., Shimada M., Fukatsu T., Ijichi N., Shimada M., Fukatsu T., Shimada M., Fukatsu T., Fukatsu T. Prevailing triple infection with Wolbachia in Callosobruchus chinensis (Coleoptera: Bruchidae) Mol. Ecol. 2002b;11:167–180. doi: 10.1046/j.0962-1083.2001.01432.x. [DOI] [PubMed] [Google Scholar]
- Koonin E.V., Makarova K.S., Aravind L., Makarova K.S., Aravind L., Aravind L. Horizontal gene transfer in prokaryotes: Quantification and classification. Annu. Rev. Microbiol. 2001;55:709–742. doi: 10.1146/annurev.micro.55.1.709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S., Tamura K., Nei M., Tamura K., Nei M., Nei M. MEGA3: Integrated software for Molecular Evolutionary Genetics Analysis and sequence alignment. Brief. Bioinform. 2004;5:150–163. doi: 10.1093/bib/5.2.150. [DOI] [PubMed] [Google Scholar]
- Lachaise D., Cariou M.L., David J.R., Lemeunier F., Tsacas L., Ashburner M., Cariou M.L., David J.R., Lemeunier F., Tsacas L., Ashburner M., David J.R., Lemeunier F., Tsacas L., Ashburner M., Lemeunier F., Tsacas L., Ashburner M., Tsacas L., Ashburner M., Ashburner M. Historical biogeography of the Drosophila melanogaster species subgroup. Evol. Biol. 1988;22:159–225. [Google Scholar]
- Lang B.F., Seif E., Gray M.W., O’Kelly C.J., Burger G., Seif E., Gray M.W., O’Kelly C.J., Burger G., Gray M.W., O’Kelly C.J., Burger G., O’Kelly C.J., Burger G., Burger G.1999. . A comparative genomics approach to the evolution of eukaryotes and their mitochondria J. Eukaryot. Microbiol. 46320–326. [DOI] [PubMed] [Google Scholar]
- Leister D. Chloroplast research in the genomic age. Trends Genet. 2003;19:47–56. doi: 10.1016/s0168-9525(02)00003-3. [DOI] [PubMed] [Google Scholar]
- Li Y.J., Satta Y., Takahata N., Satta Y., Takahata N., Takahata N. Paleo-demography of the Drosophila melanogaster subgroup: Application of the maximum likelihood method. Genes Genet. Syst. 1999;74:117–127. doi: 10.1266/ggs.74.117. [DOI] [PubMed] [Google Scholar]
- Margulis L. Symbiosis in cell evolution. Freeman; San Francisco: 1981. [Google Scholar]
- Miyata T., Yasunaga T., Yasunaga T. Molecular evolution of mRNA: A method for estimating evolutionary rates of synonymous and amino acid substitutions from homologous nucleotide sequences and its application. J. Mol. Evol. 1980;16:23–36. doi: 10.1007/BF01732067. [DOI] [PubMed] [Google Scholar]
- Narita S., Kageyama D., Nomura M., Fukatsu T., Kageyama D., Nomura M., Fukatsu T., Nomura M., Fukatsu T., Fukatsu T. Unexpected mechanism of symbiont-induced reversal of insect sex: Feminizing Wolbachia continuously acts on the butterfly Eurema hecabe during larval development. Appl. Environ. Microbiol. 2007;73:4332–4341. doi: 10.1128/AEM.00145-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochman H., Lawrence J.G., Groisman E.A., Lawrence J.G., Groisman E.A., Groisman E.A. Lateral gene transfer and the nature of bacterial innovation. Nature. 2000;405:299– 304. doi: 10.1038/35012500. [DOI] [PubMed] [Google Scholar]
- O’Neill S.L., Hoffmann A.A., Werren J.H., Hoffmann A.A., Werren J.H., Werren J.H. Influential passengers: Inherited microorganisms and arthropod reproduction. Oxford University Press; New York: 1997. [Google Scholar]
- Poinsot D., Charlat S., Mercot H., Charlat S., Mercot H., Mercot H. On the mechanism of Wolbachia-induced cytoplasmic incompatibility: Confronting the models with the facts. Bioessays. 2003;25:259–265. doi: 10.1002/bies.10234. [DOI] [PubMed] [Google Scholar]
- Ronquist F., Huelsenbeck J.P., Huelsenbeck J.P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003;19:1572–1574. doi: 10.1093/bioinformatics/btg180. [DOI] [PubMed] [Google Scholar]
- Schmidt H.A., Strimmer K., Vingron M., von Haeseler A., Strimmer K., Vingron M., von Haeseler A., Vingron M., von Haeseler A., von Haeseler A. TREE-PUZZLE: Maximum likelihood phylogenetic analysis using quartets and parallel computing. Bioinformatics. 2002;18:502–504. doi: 10.1093/bioinformatics/18.3.502. [DOI] [PubMed] [Google Scholar]
- Simpson C.L., Stern D.B., Stern D.B. The treasure trove of algal chloroplast genomes. Surprises in architecture and gene content, and their functional implications. Plant Physiol. 2002;129:957–966. doi: 10.1104/pp.010908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson J.D., Higgins D.G., Gibson T.J., Higgins D.G., Gibson T.J., Gibson T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmis J.N., Ayliffe M.A., Huang C.Y., Martin W., Ayliffe M.A., Huang C.Y., Martin W., Huang C.Y., Martin W., Martin W. Endosymbiotic gene transfer: Organelle genomes forge eukaryotic chromosomes. Nat. Rev. Genet. 2004;5:123–135. doi: 10.1038/nrg1271. [DOI] [PubMed] [Google Scholar]
- Tourmen Y., Baris O., Dessen P., Jacques C., Malthièry Y., Reynier P, Baris O., Dessen P., Jacques C., Malthièry Y., Reynier P, Dessen P., Jacques C., Malthièry Y., Reynier P, Jacques C., Malthièry Y., Reynier P, Malthièry Y., Reynier P, Reynier P. Structure and chromosomal distribution of human mitochondrial pseudogenes. Genomics. 2002;80:71–77. doi: 10.1006/geno.2002.6798. [DOI] [PubMed] [Google Scholar]
- Tuda M., Wasano N., Kondo N., Horng S.B., Chou L.Y., Tateishi Y., Wasano N., Kondo N., Horng S.B., Chou L.Y., Tateishi Y., Kondo N., Horng S.B., Chou L.Y., Tateishi Y., Horng S.B., Chou L.Y., Tateishi Y., Chou L.Y., Tateishi Y., Tateishi Y. Habitat-related mtDNA polymorphism in the stored-bean pest Callosobruchus chinensis (Coleoptera: Bruchidae) Bull. Entomol. Res. 2004;94:75–80. doi: 10.1079/ber2003277. [DOI] [PubMed] [Google Scholar]
- Veneti Z., Clark M.E., Karr T.L., Savakis C., Bourtzis K., Clark M.E., Karr T.L., Savakis C., Bourtzis K., Karr T.L., Savakis C., Bourtzis K., Savakis C., Bourtzis K., Bourtzis K. Heads or tails: Host–parasite interactions in the Drosophila– Wolbachia system. Appl. Environ. Microbiol. 2004;70:5366–5372. doi: 10.1128/AEM.70.9.5366-5372.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werren J.H., Windsor D.M., Windsor D.M. Wolbachia infection frequencies in insects: Evidence of a global equilibrium? Proc. R. Soc. Lond. B Biol. Sci. 2000;267:1277–1285. doi: 10.1098/rspb.2000.1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woischnik M., Moraes C.T., Moraes C.T. Pattern of organization of human mitochondrial pseudogenes in the nuclear genome. Genome Res. 2002;12:885–893. doi: 10.1101/gr.227202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu M., Sun L.V., Vamathevan J., Riegler M., Deboy R., Brownlie J.C., McGraw E.A., Martin W., Esser C., Ahmadinejad N., Sun L.V., Vamathevan J., Riegler M., Deboy R., Brownlie J.C., McGraw E.A., Martin W., Esser C., Ahmadinejad N., Vamathevan J., Riegler M., Deboy R., Brownlie J.C., McGraw E.A., Martin W., Esser C., Ahmadinejad N., Riegler M., Deboy R., Brownlie J.C., McGraw E.A., Martin W., Esser C., Ahmadinejad N., Deboy R., Brownlie J.C., McGraw E.A., Martin W., Esser C., Ahmadinejad N., Brownlie J.C., McGraw E.A., Martin W., Esser C., Ahmadinejad N., McGraw E.A., Martin W., Esser C., Ahmadinejad N., Martin W., Esser C., Ahmadinejad N., Esser C., Ahmadinejad N., Ahmadinejad N., et al. Phylogenomics of the reproductive parasite Wolbachia pipientis wMel: A streamlined genome overrun by mobile genetic elements. PLoS Biol. 2004;2:e69. doi: 10.1371/journal.pbio.0020069. [DOI] [PMC free article] [PubMed] [Google Scholar]