

Summary
Recent studies of resected hippocampus from patients with intractable temporal lobe epilepsy (TLE) have yielded biochemical evidence of signalling pathways associated with apoptosis. The tumor suppressor and transcription factor p53 regulates expression of several genes involved in apoptosis. Cellular levels of p53 are regulated in part by murine double minute 2 (MDM2) via ubiquitination and degradation through the proteasome. Presently, we compared expression of p53 and MDM2 in resected hippocampus from patients with intractable TLE to matched autopsy control samples. Western blotting detected significantly higher levels of p53 within TLE samples than controls. MDM2 levels were significantly lower in patient brain and its cleaved form was more abundant. Immunohistochemistry localized elevated p53 to a mainly nuclear distribution in neurons and glia in sections from TLE hippocampus. These data extend other findings on altered expression of genes regulating cell death and survival decisions in human intractable TLE.
Keywords: Apoptosis, Bcl-2, DNA damage, Epileptogenesis, Mesial temporal sclerosis
Introduction
Temporal lobe epilepsy (TLE) is the most common and intractable seizure disorder in adults. Up to 70% of refractory TLE patients display neuronal loss within the hippocampus and repeated seizures have been implicated as additive causal factors in progressive hippocampal changes and cognitive decline in some patients (Kalviainen et al., 1998; Bernasconi et al., 2002; Mathern et al., 2002; Liu et al., 2003). Experimental modelling has established repeated brief seizures can cause neuronal death within hippocampus and proportional cognitive deficits (Kotloski et al., 2002).
Biochemical and molecular features associated with apoptosis signalling have been identified within damaged neuronal populations in animal models of seizure-induced brain injury (Henshall and Simon, 2005). Studies of resected hippocampi from patients with intractable TLE have also found regulation of these pathways (Shinoda et al., 2004; Yamamoto et al., 2006a,b). The tumor suppressor and nuclear transcription factor p53 is one of the most intensely studied apoptosis-regulatory molecules (Hofseth et al., 2004). A major mechanism regulating p53 levels involves a reciprocal relationship with murine double minute 2 (MDM2) (Brooks and Gu, 2003). Under normal conditions MDM2 regulates p53 stability, blocking its activity and promoting transport for ubiquitin-proteasome degradation. Cellular stressors lead to p53 stabilization, MDM2 degradation and transcription of p53’s target genes, which include death receptors and pro-apoptotic Bcl-2 family proteins (Hofseth et al., 2004).
Seizures in experimental models induce and activate p53 and mice deficient in p53 or mice treated with p53 inhibitors show protection against seizure damage (Morrison et al., 1996; Culmsee et al., 2001; Araki et al., 2004). Recent immunohistochemical findings suggest p53 expression may also be elevated in human epilepsy brain (Xu et al., 2007). To investigate this further we examined p53 and MDM2 expression in resected hippocampus from patients with intractable TLE.
Methods
Human hippocampal samples
This study was approved by the Legacy Health System Institutional Review board and informed consent was obtained from all patients. Patient and control demographics have been reported elsewhere (Shinoda et al., 2004; Yamamoto et al., 2006a) and are summarized in Table 1. Briefly, all patients were determined to have medically intractable TLE and had been referred for surgical resection by an epileptologist following neurological assessment, video-EEG recording and neuroimaging (magnetic resonance imaging, MRI). Hippocampal sclerosis was present in all cases as assessed by MRI and/or pathology and all patients were taking antiepileptic drugs prior to surgery. No patient had experienced status epilepticus during the year of their surgery.
Table 1.
Clinical data
| Variable | Autopsy controls | TLE patients |
|---|---|---|
| Group (n) | 6 | 10 |
| Age | 29 ± 18 years (range 13–53 years) | 37 ± 15 years (range 15–57 years) |
| Gender | Male, 5; female, 1 | Male, 3; female 7 |
| Post mortem interval | 8 ± 3 h (range 5–12 h) | n.a. |
| Side of resection | n.a. | 5 left, 5 right |
Data are mean ± S.D. for controls (n = 6) and TLE (n = 10). n.a., not applicable/available.
The hippocampus was separated from any adjacent temporal cortex, flash-frozen in liquid nitrogen and stored at −70 °C until use. Specimen quantity and orientation precluded comparative analysis of specific hippocampal subfields (e.g. CA1 versus hilus). Hippocampi were sectioned on a cryostat (12 μm), with the remaining bloc prepared for biochemical analysis.
Human control (C) tissue (n = 6) was similar fresh-frozen, en bloc hippocampi from people who died of causes not related to known neurological disease (Brain and Tissue Bank for Developmental Disorders at the University of Maryland, Baltimore, Maryland, USA). The causes of death were cardiac arrest (C1), pulmonary thromboembolus (C2), traffic accident (C3, C6), suicide (hanging) (C4) and gunshot wound (chest) (C5).
Western blotting
Western blotting was performed as previously described with modifications (Schindler et al., 2006; Yamamoto et al., 2006a). Due to sample numbers, separate gels were organized with TLE samples E1–E8 run against controls C1–C4 and TLE samples E9 and E10 run against controls C5 and C6. Whole cell protein lysates (50 μg per lane) were run on 12–15% SDS-PAGE gels, transferred to polyvinylidene difluoride membranes and then incubated with polyclonal antibodies against p53, MDM2 or α-tubulin (Santa Cruz Biotechnology, Santa Cruz, CA, USA). Membranes were next incubated with HRP-conjugated secondary antibodies followed by chemiluminescence detection. Protein bands were semi-quantified using gel-scanning integrated optical density (O.D.) software (AlphaEaseFC 4.0). Band O.D. values from control samples were expressed as fold relative to the group mean and O.D. values from each concurrently run TLE sample expressed as fold difference from the control mean. Immunoblotting for α-tubulin was used as a guide to equal protein loading but expression data were not corrected to α-tubulin levels. Data are presented as mean ± S.D. and comparisons made using a t-test with significance accepted at p < 0.05.
Immunohistochemistry
Immunohistochemistry was performed as previously described with modifications (Schindler et al., 2006; Yamamoto et al., 2006a). Briefly, hippocampal sections from controls (C2, C5 and C6) and TLE patients (E7, E8 and E9) were post-fixed, blocked in goat serum and incubated with antibodies against either p53 and NeuN (Chemicon, Temecula, CA, USA) or p53 and glial fibrillary acidic protein (GFAP) (Sigma–Aldrich, St. Louis, MO, USA). Sections were then washed and incubated with secondary antibodies conjugated to fluorescein isothiocyante (FITC) and Rhodamine, mounted and imaged using a Nikon 2000s epifluorescence microscope equipped with an Orca 285 camera. Additional sections where the primary antibody was omitted were included to assess non-specific staining. Attempts to visualize MDM2 were not successful.
Simulated post mortem delay
Simulated post mortem delay experiments were undertaken as previously described (Yamamoto et al., 2006b). Animal procedures were approved by the Research Ethics Committee of the Royal College of Surgeons in Ireland. Hippocampi were extracted from mice (adult C57Bl/6, n = 12) following deep anesthesia with pentobarbital and decapitation. Hippocampi were either frozen immediately (“surgical” control) or frozen 4 or 8 h after being left at room temperature (simulated post mortem interval). Samples were then processed for Western blotting as described above.
Results
Higher hippocampal levels of p53 and lower MDM2 levels in TLE
Western blotting detected constitutive expression of p53 at its predicted molecular weight in all control human hippocampal samples (Fig. 1a). Expression of p53 was significantly higher in hippocampus from patients with intractable TLE (Fig. 1a and b).
Fig. 1.
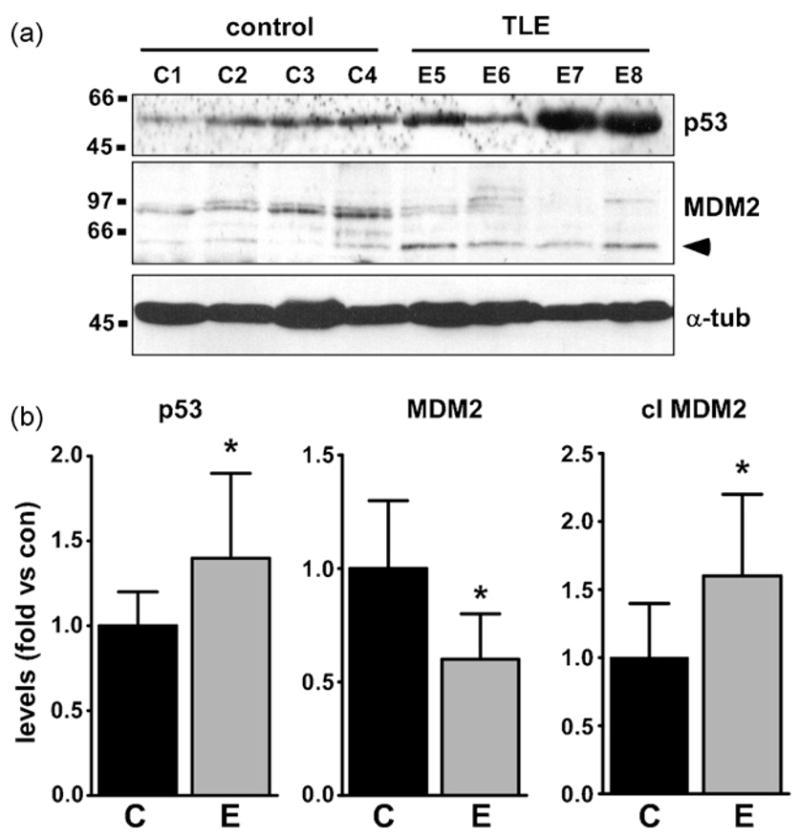
Elevated p53 and downregulated MDM2 in TLE hippocampus. (a) Representative Western blots (n = 1 per lane) showing (top panel) higher levels of p53 in TLE patient hippocampus (E) compared to controls (C) and (bottom panels) higher levels of MDM2 in controls compared to patients along with higher levels of the cleaved form of MDM2 (arrowhead) in patients. The ages of the controls for the presented data examples (33 ± 22 years) were similar to TLE patients (34 ± 14 years). Loading control shown for α-tubulin (α-tub). Protein weight markers (in kDa) depicted to left. (b) Graphs showing semi-quantification of Western data differences in p53, MDM2 and cleaved (cl) MDM2 levels between patient and control brain. *p < 0.05 vs. control.
MDM2 was present at low levels in control hippocampus at its predicted weight of ~95 kDa (Fig. 1a). Western blotting determined MDM2 levels were significantly lower in TLE patient hippocampi (Fig. 1a and b). Additionally, levels of a second band corresponding to the MDM2 cleavage product (~60 kDa) were significantly higher in TLE samples than controls (Fig. 1a and b).
To rule out expressional differences arising as a result of autopsy delay in human controls we examined p53 and MDM2 levels in mouse hippocampus left at room temperature to simulate post mortem interval. Western blotting of hippocampal samples left for up to 8 h (average autopsy delay, see Table 1) showed no significant changes to p53 or MDM2 levels (data not shown).
Immunohistochemical localization of p53 in human hippocampus
To further examine p53 expression and identify the cell types involved we undertook double-label fluorescence microscopy experiments. Expression of p53 was very limited in NeuN-immunoreactive cells in hippocampal sections from controls (n = 3) (Fig. 2a). In contrast, p53 immunostaining was more widespread in sections of TLE patient hippocampus (n = 3) (Fig. 2a). Here, p53 appeared in a mainly nuclear distribution in cells co-labeled for NeuN, although some extra-nuclear staining was also present (Fig. 2a).
Fig. 2.

p53 immunohistochemistry. (a) Photomicrographs (40× lens) showing NeuN (red, Rhodamine) and p53 (green, FITC) immunostaining in control (top panels) and TLE patient (middle, bottom panels) hippocampal sections. Note more widespread, mainly nuclear p53 immunostaining in TLE sections. Arrows label neurons expressing p53 while neurons not expressing p53 are identified by arrowheads. (b) Double-staining of the astrocyte marker GFAP (red) with p53 (green) in control (top panels) and TLE patient (middle, bottom panels) hippocampal sections. GFAP-p53 double staining can be seen in sections from both groups (arrowheads) while GFAP-negative/p53-positive cells are also present in TLE sections (arrows). Scale bar in top left panels in a and b is 20 μm, in bottom left in a is 10 μm.
GFAP-positive cells in control hippocampal sections showed very little p53 immunoreactivity (Fig. 2b). In TLE sections, GFAP staining was more prominent and small numbers of GFAP-positive cells exhibited light nuclear p53 immunostaining (Fig. 2b).
Discussion
Identifying determinants of seizure-induced neuronal death may improve our understanding of molecular mechanisms contributing to long-term changes in intractable epilepsy and whether recurrent seizures damage brain in TLE patients. The present data suggest the p53 transcription factor may be induced in the hippocampus of patients with intractable epilepsy. The lower levels of its negative regulator MDM2 are also compatible with the known relationship of these two proteins whereby lowered MDM2 enables p53 to accumulate and function. Our data extend a recent immunohistochemical report of increased p53 in TLE patient hippocampus (Xu et al., 2007) and are reflective of p53 responses in rat hippocampus during seizure-induced neuronal death (Sakhi et al., 1994; Tan et al., 2001; Araki et al., 2004). Of additional note, activity of forkhead in rhabdomyosarcoma (FKHR) appears suppressed in TLE hippocampus (Shinoda et al., 2004) suggesting selectivity in the regulation of apoptosis-associated transcription factors. The data are also compatible with other, potentially downstream pro-apoptotic signalling reported in TLE hippocampus. This includes caspase processing and nuclear localization of the caspase-activated DNAase (Schindler et al., 2006; Yamamoto et al., 2006a). Nevertheless, neuroimaging and neuropathology suggest any seizure-induced cell death in TLE patients must be very limited in scale (Kalviainen et al., 1998; Henshall et al., 2000; Bernasconi et al., 2002; Mathern et al., 2002; Liu et al., 2003). Thus, pro-apoptotic functions of p53 may be blunted. Indeed, several anti-apoptotic signalling activities may be functional in the same tissue including X-linked inhibitor of apoptosis protein, Akt and endoplasmic reticulum stress chaperones (Shinoda et al., 2004; Yamamoto et al., 2006a).
Whether p53 is a potential target for neuroprotection in epilepsy is uncertain and requires proof of the therapeutic value of targeting apoptosis to block seizure-induced neuronal death. Certainly, a variety of apoptotic triggers relevant to seizures converge on p53 (Toledo and Wahl, 2006) and p53 coordinates a multitude of pro-apoptotic transcriptional events (Hofseth et al., 2004; Toledo and Wahl, 2006). Potent neuroprotection is also achieved when p53 is targeted during experimental status epilepticus (Culmsee et al., 2001). However, the large number of p53-regulated genes means specific, relevant targets would need to be carefully evaluated.
Since post-translational modifications can alter p53 activity the expression differences seen here do not necessarily translate to a direct measure of function (Toledo and Wahl, 2006). Additionally, p53 has been implicated in direct promotion of pro-apoptotic Bcl-2 protein activities at the mitochondrial membrane (Toledo and Wahl, 2006) so non-transcriptional functions might also be relevant. Finally, p53 may also have roles in signalling unrelated to cell death. Indeed, while only a subset of p53 target genes have been studied to date in human TLE, expressional differences were not found for apoptosis-associated death receptor 5 or Bid (Shinoda et al., 2004; Yamamoto et al., 2006b). Of particular interest in the setting of intractable epilepsy might be p53-mediated regulation of neurite growth and expression of genes involved in axon guidance (Arakawa, 2005).
Several limitations and caveats should be considered in the interpretation of the present findings. Principally, it is not possible to attribute p53/MDM2 differences just to effects of repeated seizures because of the presence of structural damage (hippocampal sclerosis) in our material. Second, the group sizes in this study were small: the extent to which differences would match other TLE patient cohorts is unknown, although our p53 data reflect other findings (Xu et al., 2007). Third, we are comparing material obtained at surgery to that at autopsy, although post mortem simulation using mouse hippocampus did not change p53 or MDM2 levels. Fourth, our patients had long histories of exposure to a variety of antiepileptic drugs, the effects of which were not controlled for presently.
Further insights into this system could be gained by visualizing p53 and the cleaved form of MDM2 in the same TLE sections, not possible presently due to technical limitations. Additional aspects of p53 and MDM2 expression not resolved presently include their localization within specific hippocampal subfields and extra-hippocampal material, regulation within non-TLE epilepsy patient brain and correlative studies on expression-seizure frequency relationships. In summary, the present study provides evidence that the transcription factor p53 and its regulator MDM2 are altered in hippocampus from TLE patients with intractable seizures.
Acknowledgments
The authors thank Ronan Conroy for statistical support, Ina Koegel and Jing-Quan Lan for technical support. We express our great thanks to Drs So, Rosenban and Abtin for surgical collection of the specimens and the University of Maryland Brain and Tissue Bank for autopsy specimens. This research was supported in part by grants from the Health Research Board Ireland (RP/2005/24 and PD/2005/35), Science Foundation Ireland (04/IN3/B466) and NIH/NINDS (NS39016 and NS41935).
References
- Arakawa H. p53, apoptosis and axon–guidance molecules. Cell Death Differ. 2005;12:1057–1065. doi: 10.1038/sj.cdd.4401601. [DOI] [PubMed] [Google Scholar]
- Araki T, Shinoda S, Schindler CK, Quan-Lan J, Meller R, Taki W, Simon RP, Henshall DC. Expression, interaction, and proteolysis of death-associated protein kinase and p53 within vulnerable and resistant hippocampal subfields following seizures. Hippocampus. 2004;14:326–336. doi: 10.1002/hipo.10184. [DOI] [PubMed] [Google Scholar]
- Bernasconi A, Tasch E, Cendes F, Li LM, Arnold DL. Proton magnetic resonance spectroscopic imaging suggests progressive neuronal damage in human temporal lobe epilepsy. Prog Brain Res. 2002;135:297–304. doi: 10.1016/S0079-6123(02)35027-1. [DOI] [PubMed] [Google Scholar]
- Brooks CL, Gu W. Ubiquitination, phosphorylation and acetylation: the molecular basis for p53 regulation. Curr Opin Cell Biol. 2003;15:164–171. doi: 10.1016/s0955-0674(03)00003-6. [DOI] [PubMed] [Google Scholar]
- Culmsee C, Zhu X, exci-totoxic Yu QS, Chan SL, Camandola S, Guo Z, Greig NH, Mattson MP. A synthetic inhibitor of p53 protects neurons against death induced by ischemic and insults, and amyloid beta-peptide. J Neurochem. 2001;77:220–228. doi: 10.1046/j.1471-4159.2001.t01-1-00220.x. [DOI] [PubMed] [Google Scholar]
- Henshall DC, Clark RS, Adelson PD, Chen M, Watkins SC, Simon RP. Alterations in bcl-2 and caspase gene family protein expression in human temporal lobe epilepsy. Neurology. 2000;55:250–257. doi: 10.1212/wnl.55.2.250. [DOI] [PubMed] [Google Scholar]
- Henshall DC, Simon RP. Epilepsy and apoptosis pathways. J Cereb Blood Flow Metab. 2005;25:1557–1572. doi: 10.1038/sj.jcbfm.9600149. [DOI] [PubMed] [Google Scholar]
- Hofseth LJ, Hussain SP, Harris CC. p53: 25 years after its discovery. Trends Pharmacol Sci. 2004;25:177–181. doi: 10.1016/j.tips.2004.02.009. [DOI] [PubMed] [Google Scholar]
- Kalviainen R, Salmenpera T, Partanen K, Vainio P, Riekkinen P, Pitkanen A. Recurrent seizures may cause hippocampal damage in temporal lobe epilepsy. Neurology. 1998;50:1377–1382. doi: 10.1212/wnl.50.5.1377. [DOI] [PubMed] [Google Scholar]
- Kotloski R, Lynch M, Lauersdorf S, Sutula T. Repeated brief seizures induce progressive hippocampal neuron loss and memory deficits. Prog Brain Res. 2002;135:95–110. doi: 10.1016/S0079-6123(02)35010-6. [DOI] [PubMed] [Google Scholar]
- Liu RS, Lemieux L, Bell GS, Hammers A, Sisodiya SM, Bartlett PA, Shorvon SD, Sander JW, Duncan JS. Progressive neocortical damage in epilepsy. Ann Neurol. 2003;53:312–324. doi: 10.1002/ana.10463. [DOI] [PubMed] [Google Scholar]
- Mathern GW, Adelson PD, Cahan LD, Leite JP. Hippocampal neuron damage in human epilepsy: Meyer’s hypothesis revisited. Prog Brain Res. 2002;135:237–251. doi: 10.1016/s0079-6123(02)35023-4. [DOI] [PubMed] [Google Scholar]
- Morrison RS, Wenzel HJ, Kinoshita Y, Robbins CA, Done-hower LA, Schwartzkroin PA. Loss of the p53 tumor suppressor gene protects neurons from kainate-induced cell death. J Neurosci. 1996;16:1337–1345. doi: 10.1523/JNEUROSCI.16-04-01337.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakhi S, Bruce A, Sun N, Tocco G, Baudry M, Schreiber SS. p53 induction is associated with neuronal damage in the central nervous system. Proc Natl Acad Sci USA. 1994;91:7525–7529. doi: 10.1073/pnas.91.16.7525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindler CK, Pearson EG, Bonner HP, So NK, Simon RP, Prehn JH, Henshall DC. Caspase-3 cleavage and nuclear localization of caspase-activated DNase in human temporal lobe epilepsy. J Cereb Blood Flow Metab. 2006;26:583–589. doi: 10.1038/sj.jcbfm.9600219. [DOI] [PubMed] [Google Scholar]
- Shinoda S, Schindler CK, Meller R, So NK, Araki T, Yamamoto A, Lan JQ, Taki W, Simon RP, Henshall DC. Bim regulation may determine hippocampal vulnerability after injurious seizures and in temporal lobe epilepsy. J Clin Invest. 2004;113:1059–1068. doi: 10.1172/JCI19971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan Z, Levid J, Schreiber SS. Increased expression of Fas (CD95/APO-1) in adult rat brain after kainate-induced seizures. Neuroreport. 2001;12:1979–1982. doi: 10.1097/00001756-200107030-00040. [DOI] [PubMed] [Google Scholar]
- Toledo F, Wahl GM. Regulating the p53 pathway: in vitro hypotheses, in vivo veritas. Nat Rev Cancer. 2006;6:909–923. doi: 10.1038/nrc2012. [DOI] [PubMed] [Google Scholar]
- Xu S, Pang Q, Liu Y, Shang W, Zhai G, Ge M. Neuronal apoptosis in the resected sclerotic hippocampus in patients with mesial temporal lobe epilepsy. J Clin Neurosci. 2007;14:835–840. doi: 10.1016/j.jocn.2006.08.002. [DOI] [PubMed] [Google Scholar]
- Yamamoto A, Murphy N, Schindler CK, So NK, Stohr S, Taki W, Prehn JH, Henshall DC. Endoplasmic reticulum stress and apoptosis signaling in human temporal lobe epilepsy. J Neuropathol Exp Neurol. 2006a;65:217–225. doi: 10.1097/01.jnen.0000202886.22082.2a. [DOI] [PubMed] [Google Scholar]
- Yamamoto A, Schindler CK, Murphy BM, Bellver-Estelles C, So NK, Taki W, Meller R, Simon RP, Henshall DC. Evidence of tumor necrosis factor receptor 1 signaling in human temporal lobe epilepsy. Exp Neurol. 2006b;202:410–420. doi: 10.1016/j.expneurol.2006.07.003. [DOI] [PubMed] [Google Scholar]