

Abstract
Polyketides are a medicinally important class of natural products. The architecture of modular polyketide synthases (PKSs), composed of multiple covalently linked domains grouped into modules, provides an attractive framework for engineering novel polyketide-producing assemblies. However, impaired domain–domain interactions can compromise the efficiency of engineered polyketide biosynthesis. To facilitate the study of these domain–domain interactions, we have used nuclear magnetic resonance (NMR) spectroscopy to determine the first solution structure of an acyl carrier protein (ACP) domain from a modular PKS, 6-deoxyerythronolide B synthase (DEBS). The tertiary fold of this 10-kD domain is a three-helical bundle; an additional short helix in the second loop also contributes to the core helical packing. Superposition of residues 14–94 of the ensemble on the mean structure yields an average atomic RMSD of 0.64 ± 0.09 Å for the backbone atoms (1.21 ± 0.13 Å for all non-hydrogen atoms). The three major helices superimpose with a backbone RMSD of 0.48 ± 0.10 Å (0.99 ± 0.11 Å for non-hydrogen atoms). Based on this solution structure, homology models were constructed for five other DEBS ACP domains. Comparison of their steric and electrostatic surfaces at the putative interaction interface (centered on helix II) suggests a model for protein–protein recognition of ACP domains, consistent with the previously observed specificity. Site-directed mutagenesis experiments indicate that two of the identified residues influence the specificity of ACP recognition.
Keywords: polyketide, acyl carrier protein, NMR, structure, protein interaction, electrostatic, homology modeling, mutagenesis
Modular polyketide synthases (PKSs) are multidomain enzymes responsible for biosynthesis of polyketides, a class of natural products that includes a number of medicinally important compounds. This family of PKS enzymes (also known as type I PKS) is distinguished by its modular organization, where each polypeptide corresponds to a polyfunctional protein containing several catalytic domains grouped into discrete modules, each responsible for a round of polyketide chain extension and functional group modification (Cane et al. 1998; Khosla et al. 1999; Staunton and Weissman 2001; Staunton and Wilkinson 2001). Related modular megasynthases are also responsible for the biosynthesis of nonribosomal peptides (Cane et al. 1998; Walsh 2002; Finking and Marahiel 2004; Sieber and Marahiel 2005).
6-Deoxyerythronolide B synthase (DEBS) is a prototypical modular polyketide synthase that produces the macrolide core of the antibiotic erythromycin. DEBS consists of three large polypeptides, 330–370 kD in size, (Cortes et al. 1990; Donadio et al. 1991; Donadio and Katz 1992), each harboring two modules. Each module includes all the catalytic domains responsible for one round of chain extension and modification on the growing polyketide intermediate (Fig. 1). While the number and composition of domains in a PKS module can vary, each of the six DEBS modules contains at least three types of domains: a ketosynthase (KS), which catalyzes carbon–carbon bond formation in the condensation reaction that extends the growing polyketide chain; an acyl transferase (AT), which transfers the appropriate building block for chain elongation from the corresponding acyl-CoA metabolite to the polyketide synthase; and an acyl carrier protein (ACP), which first accepts the extender unit from the AT, then collaborates with the KS domain in chain elongation, and finally anchors the newly elongated chain as it undergoes modification at the β-keto position. In order to carry out their function, the ACP domains require post-translational addition of a phosphopantetheine group to a conserved serine residue of the ACP. The terminal sulfhydryl group of the phosphopantetheine is the site of attachment of the growing polyketide chain. Additional ketoreductase (KR), dehydratase (DH), and enoyl reductase (ER) domains can also be part of a given module and their combined catalytic actions determine whether a keto group, a hydroxy group, an olefinic group, or a fully reduced methylene group will be present at the β-carbon of the newly synthesized polyketide product. A dedicated cyclization domain, thioesterase (TE), located at the C terminus of DEBS module 6, releases the final heptaketide product as the macrolactone, 6-deoxyerythronolide B (6-dEB).
Figure 1.

Schematic diagram of the modular organization of 6-deoxyerythronolide B synthase (DEBS). Three polypeptides (DEBS1, DEBS2, and DEBS3) each contain two modules that are, in turn, composed of distinct catalytic domains. KS indicates ketosynthase; AT, acyl transferase; KR, ketoreductase; ACP, acyl carrier protein; DH, dehydratase; ER, enoyl reductase; and TE, thioesterase. DEBS1 also contains a loading didomain, consisting of an AT and an ACP. KR0 denotes an inactive KR domain. Inter-protein linker (docking) domains are shown as matching tabs.
In modular polyketide synthases such as DEBS, specific intermodular transfer of intermediates between two different polypeptides (for example, between the ACP domain of module 2 [ACP2] and the KS domain of module 3 [KS3] in DEBS) is mediated by intermodular docking domains or linker sequences located at the N and C termini of the acceptor and donor modules of adjacent PKS polypeptides (shown as complementary tabs in Fig. 1). The C-terminal (∼100 residues) and N-terminal (∼40 residues) linkers (or docking domains) have been demonstrated to form compatible pairs that play a role in the specificity of intermodular interactions between the modules located on different polypeptides (Gokhale et al. 1999; Tsuji et al. 1999; Wu et al. 2001, 2002; Broadhurst et al. 2003; Kumar et al. 2003). This linker–linker recognition is accomplished by coiled coil interactions between the helical regions of these linker (or docking) domains (Broadhurst et al. 2003; Kumar et al. 2003). However, in addition to linker-mediated interactions, ACP–KS interactions and enzyme–substrate interactions have been shown to influence the efficiency and specificity of the polyketide chain transfer between the modules (Wu et al. 2001, 2002).
An understanding of the basis for the specificity of protein–protein interactions between PKS domains would be valuable, as it could assist in engineering synthases with novel productive combinations of catalytic domains, leading to new biosynthetic products. Until recently, the absence of high-resolution structures for most of the constituent domains of modular polyketide synthases posed a major limitation for more detailed studies of the determinants of specificity in domain–domain interactions. The recently solved 2.7 Å X-ray crystal structure of a portion of module 5 of DEBS has provided the first atomic resolution view of the KS5 and AT5 domains, along with the flanking interdomain linkers (Tang et al. 2006). In addition, the crystal structure of a KR domain from module 1 of DEBS has recently been reported (Keatinge-Clay and Stroud 2006).
The core (or minimal) set of catalytic domains of a PKS module consists of a KS, an AT, and an ACP. Although relatively small (∼10 kD), the ACP domain is especially important in that it interacts with all other domains in the course of polyketide biosynthesis. Presently, no structures have been reported for ACP domains from type I PKSs. A recent study of the interactions between an ACP domain of DEBS and two phosphopantetheinyl transferases relied on homology modeling of the ACP structure (Weissman et al. 2006).
The ACP domain from module 2 of DEBS was chosen as a target for structural characterization by nuclear magnetic resonance (NMR) spectroscopy, because it is involved in a number of well-characterized domain–domain interactions and because its size (∼10 kD) is amenable to structure determination by NMR. The solution structure of ACP2 from DEBS serves as a prototype for this class of domains from modular polyketide synthase assemblies, analogous to the previously reported structure of the PCP domain from the tyrocidine nonribosomal peptide synthase (Weber et al. 2000). To demonstrate this potential, we have constructed homology models for the other ACP domains in DEBS. A comparison of steric and electrostatic surface properties of these domains reveals plausible mechanistic explanations for several earlier observations with regard to ACP domain specificity in the context of polyketide chain transfer and chain elongation. In addition, three single-site mutants of the ACP2 domain were constructed and evaluated for their ability to support polyketide chain elongation in reactions with KS–AT didomain fragments from module 3 and from module 6 of DEBS. Two of the three mutated residues appear to have an effect on the specificity of ACP interactions.
Results
As a representative ACP domain from a modular PKS, the ACP2 domain of DEBS was chosen for structure determination. Cloning of this construct (ACP2 without the C-terminal linker) has been described previously as ACP2(Ø) (Wu et al. 2002). Additionally, for the NMR sample preparation, the His6-tag sequence was removed by thrombin cleavage (as described in Materials and Methods). Thus, in the final protein sample, only the first four residues originate from the expression vector, while residues 5–95 are identical to the DEBS residues 3318–3408 (EryA, EntrezProtein accession no. AAA26493). Protein sample purity was verified using SDS-PAGE and electrospray ionization mass spectrometry (ESI-MS) (Supplemental Fig. S1A,B, supporting information), as well as by the presence of the expected number of peaks in the two-dimensional 1H-15N HSQC spectrum (Supplemental Fig. S1C, supporting information). The protein was not coexpressed with a phosphopantetheinyl transferase and was confirmed to be more than 95% apo-ACP by ESI-MS. The phosphopantetheine arm has typically been shown to be flexible and to interact only transiently, if at all, with the polypeptide portion of the ACP (Holak et al. 1988a; Weber et al. 2000; Xu et al. 2001; Li et al. 2003), at least in the absence of the attached substrate. (One exception has been reported [Sharma et al. 2006] in the case of the ACP from the fatty acid synthase [FAS] of Plasmodium falciparum.) In this work, we proceeded to determine the solution structure of the apo-ACP2(Ø).
Resonance assignments
The assigned 1H-15N HSQC spectrum of DEBS ACP2(Ø) is shown in Figure 2. The spectrum is well resolved, with sufficient chemical-shift dispersion in both the 1H and the 15N dimension and only a few instances of overlap. Nearly complete sequence-specific backbone and side-chain assignments were achieved (96.8% of the assignable atoms from residues 3–95, including 99% for 13C [292 assigned carbons], 99% for 15N [101 assigned nitrogens], and more than 96% for 1H [485 assigned protons]) using the data from a series of triple-resonance NMR experiments described in Materials and Methods. Side-chain amide protons of the three asparagine residues were assigned based on the intraresidue (1Hδ to 1Hα and 1Hβ) and short-range NOEs in the 13C/15N NOESY-HSQC spectra, whereas side-chain amide nitrogens and protons (Nɛ and Hɛ) of arginine residues were assigned using the NOEs to 1Hδ resonances in 13C/15N NOESY-HSQC and 15N-edited NOESY-HSQC spectra.
Figure 2.

Assigned two-dimensional 1H-15N HSQC spectrum of apo-ACP2(Ø) from DEBS. Backbone amide resonances are labeled with the one-letter amino acid code and residue number. Pairs of peaks from asparagine side-chain amide groups are connected by horizontal lines and labeled.
Secondary structure
Observed patterns of short- and medium-range NOEs are summarized in the secondary structure plot (Fig. 3). Judging from these indicators, three major helical regions, connected by two loops, are present. These are described in more detail below (in the Tertiary Structure section). The first 13 N-terminal residues of this construct do not exhibit any medium- or long-range NOEs to the rest of the protein, and therefore, their relative orientation with respect to the helical bundle could not be defined. However, a segment starting with residue 5 showed some evidence of helical propensity, as indicated by several αHi − NHi+3 NOEs and phi (φ) torsion angle values (deduced from the 3JHNHα scalar coupling constants; see Materials and Methods) consistent with a helical structure. Local superposition of residues 6–11 in the calculated structure ensemble reveals a short, relatively structured (backbone RMSD 0.73 Å) helical region. Interestingly, the R8-G11 portion of this segment matches the first four residues of a previously identified conserved junction sequence RLAGL, located just upstream of many ACP domains, as observed in sequences of several modular polyketide synthases (Kim et al. 2004).
Figure 3.

Summary of sequential and medium-range NOE patterns for DEBS ACP2(Ø). The relative intensities of sequential NOEs are indicated by the heights of the connecting boxes. Medium-range NOEs observed between residue pairs are indicated by horizontal lines. Helical regions are indicated above the amino acid sequence.
Quality of the calculated structure
Structure calculations, performed by the DYANA software package (Guntert et al. 1997), utilized 1791 distance constraints and 50 φ backbone torsion angle constraints, which are summarized in Table 1. Out of the calculated set of 50 structures, an ensemble of 30 structures with the lowest target function values was used to obtain the mean structure (see Materials and Methods). As shown in Table 1B, the structural statistics demonstrate good agreement of the structures with the experimental data, giving rise to no upper limit violations greater than 0.40 Å, no van der Waals violations greater than 0.39 Å, and no torsion angle violations greater than 5°.
Table 1.
Structural statistics for 30 calculated conformers of the apo-ACP2(Ø)

A measure of precision for this family of 30 structures is given by the superposition of residues 14–94 of the ensemble on the mean structure, with a backbone RMSD value of 0.64 ± 0.09 Å and non-hydrogen atoms RMSD value of 1.21 ± 0.13 Å. Ensemble convergence for the helical regions is illustrated by superimposing the three major helices, yielding a backbone RMSD value of 0.48 ± 0.10 Å and a non-hydrogen atoms RMSD value of 0.99 ± 0.11 Å.
Ramachandran statistics (evaluated with PROCHECK-NMR software v3.5.4 for residues 14–94 of the ensemble of 30 structures) (Laskowski et al. 1996) show 79.6% of the (φ,ψ) backbone torsion angle pairs occupying the most favored regions, 17.5% in the additionally allowed regions, 1.5% in the generously allowed regions, and 1.4% in the disallowed regions.
Tertiary structure
The overall structure of this ACP domain consists of a right-hand twisted bundle formed by three major α-helices connected by two loops. An additional short helix in the second loop lies nearly perpendicular to the other three helices and contributes to the helical packing of the bundle. Figure 4A shows the superposition of the 30 conformers by overlaying the three major helical regions, and Figure 4B shows the ribbon diagram of the minimized mean structure. The observed right-twisted helical bundle topology matches the conserved fold for the ACP and PCP family (Holak et al. 1988a,b; Kim and Prestegard 1989; Crump et al. 1997; Weber et al. 2000; Volkman et al. 2001; Xu et al. 2001; Wong et al. 2002; Findlow et al. 2003; Li et al. 2003; Drake et al. 2006; Sharma et al. 2006), with helix I (residues 14–32) running antiparallel to helices II (residues 55–68) and III (residues 83–94). The first helix is longer than the other helices and is positioned at an angle relative to the helices II and III. In turn, helices II and III are also tilted relative to each other. The first half of the long loop between helices I and II is structured, but less well defined, compared with the second half of the same loop (starting near residue 43). The latter is more tightly constrained and exhibits not only short- and medium-range NOEs, but also long-range signals to helix I (for instance, NOEs from V22 to A43, T44, from R26 to V41, A43, and from T29, A30 to L50) and helix III (NOEs from A43, T44 to A83, S84, and from T44, F47 to N82).
Figure 4.

NMR solution structure of DEBS ACP2(Ø). (A) Superposition of residues 14–95 for the 30 conformers from torsion angle dynamics calculation. (B) Ribbon diagram of the minimized mean structure of ACP2(Ø). (C) Electrostatic potential surface of ACP2(Ø). Positively charged areas are colored in blue, negatively charged areas are in red, and uncharged areas are in gray. Secondary structure elements are labeled as follows: helices—HI, HII′, HII, HIII′, HIII; loops—LI, LII.
Hydrophobic interactions stabilize the helical bundle, as evidenced by the NOE signals between core leucine, valine, and alanine residues. For example, helix I and helix III are aligned by the NOEs between L21 and V86, A87, L90, between V22 and A83, A87, as well as between V25 and A83, V86, A87, L90. The connection between the buried residues of helix I and helix II is established by the NOE signals between L24 and A66, A67, between V25 and L64, as well as between T29, V32 and L60, while the C-terminal end of helix II is additionally aligned with helix III by the observed NOEs between T68 and L90, L94. Likewise, L60, L64 (from helix II) and L70, L72 (from the loop LII between helices II and III) also participate in the hydrophobic interactions, as evidenced by the corresponding NOEs. Additional signals are observed between V58 (helix II) and S74 (loop LII), as well as between R61 (helix II) and L76 (helix III′). Furthermore, NOEs between A57, L60 from helix II and V77 from the short helix III′ (residues 76–79) in the second loop indicate that the latter helix contributes to the packing of the bundle.
The distribution of surface charge for the ACP2 domain is shown in Figure 4C. It is notable that, overall, the protein surface appears less charged for this ACP domain (which belongs to a modular PKS), compared with the discrete ACPs from type II PKS (Li et al. 2003) or from FAS (Roujeinikova et al. 2002), which are mostly acidic proteins. A similar observation has been made with regard to the structure of a PCP from a modular nonribosomal peptide synthetase (Weber et al. 2000), as the PCP was also found to have a larger uncharged surface area than a FAS ACP or a type II PKS ACP used for comparison.
Homology models and electrostatic potential surfaces of other ACP domains from DEBS
Each module of DEBS contains an ACP domain (Fig. 1). Amino acid sequences of these domains have a high degree of similarity (45%–55% sequence identity based on pairwise amino acid sequence alignments of ACP2 with other ACP domains) (also see multiple sequence alignment shown in Fig. 5). Therefore, the overall structures of these domains are also expected to be quite similar. Previous studies have demonstrated, however, that the ACP domains are not always functionally interchangeable, for example, in their ability to transfer substrates to the KS domain of the downstream PKS module (Wu et al. 2002) or to participate in chain elongation catalyzed by the KS domain of the same module (Kim et al. 2004). Computational docking and mutagenesis studies on the analogous structurally characterized subunits (ACP and KSIII) of the Escherichia coli FAS suggested that helix II of that ACP participates in an extensive binding interface with the KS, and highlighted the potential role of electrostatic interactions at this protein–protein interface (Zhang et al. 2001). Moreover, a residue on helix II of ACP subunits from aromatic (type II) polyketide synthases was shown to contribute to the interactions with the initiation KS domain (Tang et al. 2003). A recently described homology model of a DEBS ACP (Weissman et al. 2006), as well as the results from a number of other studies on carrier proteins, has also pointed to the involvement of helix II in protein–protein recognition (Linne et al. 2001; Mofid et al. 2002; Worsham et al. 2003; Zhang et al. 2003; Finking et al. 2004; Lai et al. 2006). Therefore, to decode the functional differences between the ACP domains of DEBS, this proposed interface (centered on helix II) was evaluated.
Figure 5.

Sequence alignment of the DEBS ACP domains. The protein sequences were obtained from the PKSDB (a Database of Modular Polyketide Synthases, www.nii.res.in/pksdb.html). Secondary structure elements from ACP2 structure are shown above the sequences. Several residues discussed in the text are numbered, including the conserved serine (Ser54) that serves as the phosphopantetheine attachment site.
Homology models for the ACP domains of DEBS were generated based on the solution structure of DEBS ACP2(Ø) as a template. Multiple sequence alignment of the six ACP domains from DEBS, generated using PileUp program (GCG SeqWeb sequence analysis software package, Accelrys Inc.), is shown in Figure 5. Models for ACP1, ACP3, ACP4, ACP5, and ACP6 domains were generated using the WHATIF homology model server (Rodriguez et al. 1998). Electrostatic potential surfaces were calculated using MOLMOL (Koradi et al. 1996). The resulting models are shown in Figure 6, with blue and red colors on the surface representing positively and negatively charged residues, respectively. In ACP2, helix II spans residues 55–68, but the interface under consideration also includes adjacent regions, such as the loop LII following helix II and some residues at the C terminus of helix I.
Figure 6.
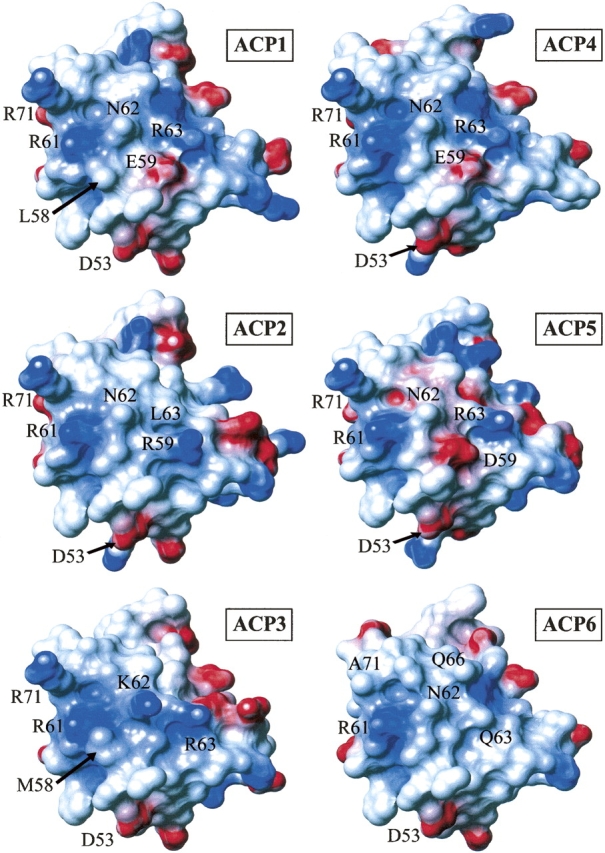
Electrostatic potential surface diagrams for six DEBS ACP domains. Positively charged residues are colored blue and negatively charged residues are colored red. Residue numbers for the labeled residues correspond to ACP2 numbering.
Comparison of the homology models (Fig. 6) shows that the overall topology of the surface encompassing helix II is similar among the ACP domains of DEBS, as might be expected based on their high sequence similarity. However, several variations can be noted, mostly due to the absence of a few bulky side chains. For example, R71 (here and throughout, the numbering is based on ACP2; as a point of reference, the conserved serine is S54) in the loop LII immediately following helix II is conserved in five of the six ACPs but is replaced with an alanine at the corresponding position in ACP6. Similarly, ACP6 has a glycine as residue 59, while four of the remaining five ACPs have bulkier side chains in that position. Only ACP3 has an alanine at position 59, but this is partially compensated by a lysine (K62) located one helical turn away, in place of an asparagine residue in the other ACPs. In addition, there is a methionine residue at position 58 in ACP3, rather than a smaller valine present in ACP2, ACP4, ACP5, and ACP6, or a leucine in ACP1. Although this difference is not dramatic, it might be functionally relevant, as residue 58 corresponds to the position previously shown to influence the ACP recognition in an aromatic (type II) PKS (Tang et al. 2003), as well as the aryl carrier protein (ArCP) recognition in a nonribosomal peptide synthetase (Lai et al. 2006). Two additional steric differences are apparent from a comparison of the homology models shown in Figure 6. ACP6 has the bulkier residue (glutamine) at position 66, whereas the other ACP domains have a smaller alanine residue (or threonine in the case of ACP4) in that position. Lastly, position 63 is occupied by an arginine in four of the ACP domains, but ACP6 and ACP2 have somewhat smaller side chains (Gln and Leu, respectively) at this location.
The differences in the electrostatic potential surfaces of these ACPs are more pronounced. The models show distinct patterns of charge for different ACP domains. For example, ACP6 is the only domain that presents an essentially neutral surface around helix II, except for the negative charge from D53 (ACP2 numbering) at the N terminus of helix II and the positive charge from R61, extending into the space between helices II and III′ (Fig. 6). Both D53 and R61 are conserved among the DEBS ACP domains examined here (Fig. 5) and might be involved in conserved domain–domain binding interactions, but are unlikely to serve as specificity determinants. In contrast to the largely neutral surface of the ACP6 domain, ACP3 has (in addition to the conserved D53 and R61 residues mentioned above) a string of positively charged side chains (R63, K62, and R71) starting near the N-terminal end of helix II and traversing diagonally across the surface to the top of the loop that connects helix II to helix III′. The remaining ACP domains have three of the four positive charges that are present in ACP3, corresponding to residues R63 (or R59 in ACP2), R71, and the conserved R61. At the same time, ACP1, ACP5, and ACP4 are distinguished from the remaining ACP domains of DEBS by the presence of a second negatively charged side chain (in addition to the conserved D53 residue) in the N-terminal half of helix II, namely, E59 in ACP1 and ACP4, or D59 in ACP5 (Fig. 6).
In summary, examination of the models suggests that ACP6 is the least similar, both sterically and electrostatically, to the ACPs from other elongation modules of DEBS. Several distinguishing features could also be discerned for ACP3.
Interestingly, these trends parallel earlier observations from experiments comparing several ACP domains from DEBS as interacting partners with a given KS domain for chain transfer or for condensation reactions. Specifically, ACP4(Ø) (i.e., ACP4 without the C-terminal linker) has been shown to transfer substrates to module 2 and module 6 (thereby substituting for ACP1 and ACP5, respectively) but not to module 3 (being unable to substitute for ACP2) (Wu et al. 2002). This is in agreement with the charge pattern noted above, where ACP4, ACP1, and ACP5, but not ACP2, display a negative charge at residue 59 in the N-terminal half of helix II. Moreover, in experiments that probed the ability of several methylmalonyl-ACP domains to support chain elongation with the KS–AT didomain fragment from module 3 of DEBS, it was observed that ACP6 was the poorest substitute for ACP3 (the natural interaction partner of the KS–AT fragment from module 3), whereas ACP2 and ACP4 were both able to participate in the condensation reaction (Kim et al. 2004). However, a lower amount of product was observed in the reaction mediated by ACP4 than in reactions mediated by ACP2 or ACP3 (Kim et al. 2004), as judged by phosphorimaging. These observations agree well with the conclusions from steric and electrostatic surface comparisons of the ACPs, as ACP6 domain is most different from ACP3 while ACP2 and ACP4 are less dissimilar from ACP3, possessing three of the four positive charges that the ACP3 presents on the interaction surface. However, ACP2 and ACP4 differ in position 59, which is occupied by a positively charged arginine (R59) in ACP2 and by a negatively charged glutamate (E59) in ACP4.
Interaction of ACP2 point mutants with KS–AT didomains in a triketide lactone formation assay
Site-directed mutagenesis experiments in combination with a functional assay were used to test the influence of several residues on the specificity of ACP-KS interactions. Three single-point mutants of ACP2—R59G, R71A, and L63Q—were cloned, expressed, and purified. Each mutation converts an ACP2 residue to the corresponding ACP6 residue (see alignment in Fig. 5). All three residues are located within the proposed interaction interface (helix II and the subsequent loop), as discussed above. The resulting ACP2 mutants were evaluated (alongside the wild-type ACP2 and ACP6) for their ability to participate in the triketide lactone formation reaction together with the KS–AT didomains from module 3 and module 6 of DEBS. In each reaction (see Fig. 7A), the KS domain of KS–AT catalyzed the condensation between the [14C]-methylmalonyl extender unit, presented by the ACP, and the N-acetylcysteamine (NAc)-activated diketide, (2S,3R)-2-methyl-3-hydroxypentanoyl-N-acetylcysteamine thioester, or NDK-SNAc, previously shown to be a substrate for modules 3 and 6 of DEBS (Gokhale et al. 1999; Wu et al. 2000). The reaction was quenched with base, and the amount of radioactively labeled triketide lactone product was subsequently visualized and quantified using thin layer chromatography (TLC) and phosphorimaging.
Figure 7.
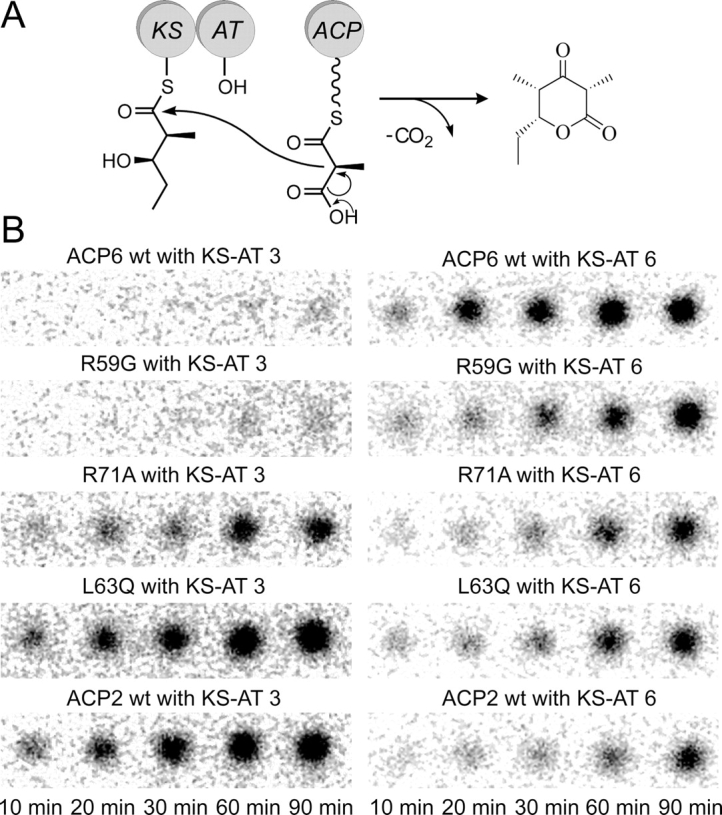
Triketide lactone formation assay. (A) Chain elongation reaction catalyzed by a KS–AT didomain interacting with a methylmalonyl-ACP. The KS domain is preacylated with the electrophilic diketide substrate (NDK). (B) TLC phosphorimaging results, showing the amount of 14C-labeled triketide lactone product in each reaction at the indicated time points.
The phosphorimaging results are presented in Figure 7B. For each of the 10 reactions, five time points were analyzed. The spot intensities were converted to triketide lactone amounts using a set of standards containing known quantities of radioactive material, and the initial rate of triketide lactone product formation was determined in each case (ranging from 10–200 nM/min) (see Supplemental Table ST2, supporting information). The rate of product formation in this assay is expected to reflect the rate of the condensation reaction involving KS and ACP domains, since the KS domain is preacylated by incubation with the diketide substrate for one hour and the AT-catalyzed reaction (transfer of the methylmalonyl extender unit from methylmalonyl-CoA onto the ACP domain) is faster than the condensation reaction (Hans et al. 2003; Liou et al. 2003).
As previously observed (Kim et al. 2004), the KS–AT didomain from module 3 interacts more efficiently with the wild-type ACP2 than with the wild-type ACP6, judging from the amount of the triketide lactone produced in the condensation reactions (see the left-hand column of Fig. 7B, where the bottom panel is the ACP2-mediated reaction and the top panel is the ACP6-mediated reaction). On the other hand, the KS–AT didomain from module 6 displays the opposite preference: Although the wild-type ACP2 can support a detectable level of product formation, the wild-type ACP6 is clearly a better interaction partner in this case (see the right-hand column of Fig. 7B).
Remarkably, when the R59G mutant of ACP2 is evaluated in this product formation assay, the observed interaction specificity mirrors that of the wild-type ACP6, rather than the wild-type ACP2. Specifically, a very low level of product formation is seen in the chain elongation reaction involving the R59G ACP2 and the KS–AT didomain from module 3, whereas the reaction mediated by the R59G ACP2 and the KS–AT from module 6 proceeds efficiently (see second row from the top in Fig. 7B). Hence, a single substitution is sufficient in this case to change the specificity for ACP2, turning it into a poor interaction partner for the KS–AT from module 3 and a good interaction partner for the KS–AT from module 6. This result suggests that the residue at position 59 might be involved in the KS–ACP interactions required for KS-catalyzed condensation reactions.
Another single-site substitution, R71A, also appears to alter the interaction specificity relative to the wild-type ACP2, but the change is less dramatic than for the R59G mutant discussed above. When the R71A mutant of ACP2 interacts with the KS–AT didomain from module 3, the triketide lactone product formation is less efficient compared with the corresponding reaction involving the wild-type ACP2 (see middle and bottom panels in the left-hand column of Fig. 7B). At the same time, the ability of the R71A mutant to interact with the KS–AT from module 6 is not compromised relative to the wild-type ACP2, although the observed triketide lactone formation levels are modest (see the right-hand column of Fig. 7B). Thus, in contrast to the wild-type ACP2, the R71A mutant no longer shows a preference for the KS–AT from module 3 over the KS–AT from module 6.
Lastly, the L63Q mutant of ACP2 is able to support product formation in combination with either of the two KS–AT didomains used in this assay; however, the reaction involving the KS–AT from module 3 is more efficient (see Fig. 7B). Thus, the interaction specificity of this mutant is similar to that of the wild-type ACP2, although the ability of L63Q ACP2 to interact with the KS-AT from module 3 appears to be slightly lower, while the ability to interact with the KS-AT from module 6 shows a modest increase relative to the wild-type ACP2 (see Supplemental Table ST2, supporting information).
Taken together, the results from these experiments indicate that the interaction specificity of the ACP2 domain can be altered by amino acid substitutions within the proposed interaction interface. It is notable that each of the ACP2 mutant proteins used in these assays is active in at least one chain elongation reaction (Fig. 7B), suggesting that the amino acid substitutions did not cause major structural perturbations to the ACP domain.
Discussion
The biosynthesis of 6-dEB and other polyketides assembled by modular polyketide synthases involves sequential transfer of intermediates between the modules of the PKS. Within modules, distinct protein domains are responsible for elongating and chemically modifying the intermediates. The modular architecture of these multifunctional enzymes can be exploited to engineer synthases that contain desired combinations of catalytic domains and modules, generating novel biosynthetic products (Cane et al. 1998; McDaniel et al. 1999; Staunton and Weissman 2001; Staunton and Wilkinson 2001; Menzella et al. 2005). However, it has been observed that not all combinations of modules or domains enable efficient formation of polyketide products (Ranganathan et al. 1999; Wu et al. 2001, 2002; Hans et al. 2003; Watanabe et al. 2003; Kim et al. 2004), and impaired interactions between the PKS domains have been implicated in at least some of these cases. Thus, engineered biosynthesis of polyketides would be facilitated by an improved understanding of the factors governing the specificity of domain–domain recognition.
The ACP domains of DEBS are involved in a number of interactions with the other domains in the course of polyketide biosynthesis; therefore, interchanging the ACP domains can have a negative impact on the efficiency of substrate transfer (Wu et al. 2002) or condensation reactions (Kim et al. 2004). Structure-based studies could suggest explanations for the experimentally observed preferences, but no structure of an ACP domain from a modular polyketide synthase has previously been reported. The structures of several related ACPs from bacterial FASs (Holak et al. 1988a; Kim and Prestegard 1989; Xu et al. 2001; Roujeinikova et al. 2002; Wong et al. 2002; Park et al. 2004) and from aromatic (type II) polyketide synthases (Crump et al. 1997; Findlow et al. 2003; Li et al. 2003) have been published, as well as the structures of some functionally analogous carrier proteins (Weber et al. 2000; Volkman et al. 2001; Drake et al. 2006). The available structures show that the core structural fold (a three-helical bundle with an additional short helix in one of the intervening loops) is conserved among this family of carrier proteins (Weber et al. 2000; Findlow et al. 2003; Li et al. 2003). However, the structure of an ACP domain from a modular PKS provides a better starting point for a detailed analysis of the specificity in ACP recognition within modular polyketide synthases, because the amino acid sequences of the ACP domains from DEBS are 45%–55% identical to each other in pairwise sequence alignments, but the level of identity drops to ∼25%, when, for instance, the sequence of ACP2 from DEBS is compared to the sequence of an ACP from an aromatic PKS (frenolicin or oxytetracycline). Amino acid sequence alignment, followed by analysis using CLUSTAL W software (Thompson et al. 1994), illustrates that ACP domains cluster into subfamilies by type (Donadio and Katz 1992): The modular (type I) polyketide ACP domains form a different subgroup than do the ACPs from aromatic (type II) PKSs, and both of these polyketide ACP subfamilies are distinct from the ACP domains that belong to bacterial FASs (Fig. 8).
Figure 8.

A phylogram illustrating the clustering of ACP domains into subfamilies. The first 10 ACPs are from modular polyketide synthases: either erythromycin PKS (labeled DebsACP) or rifamycin PKS (labeled RifACP). The ACP domains from fatty acid synthases (E. coli ACP and B. subtilis ACP) form a distinct subgroup. In turn, the actinorhodin (act), frenolicin (fren), and oxytetracycline (otc) ACPs, which originate from type II (aromatic) polyketide synthases, also form a subfamily distinct both from the ACP domains of the fatty acid synthase origin and from the ACPs that belong to modular polyketide synthases. The sequences were aligned using PileUp program (GCG SeqWeb software package, Accelrys Inc.) and the phylogram was produced using the CLUSTAL W server at the European Bioinformatics Institute (www.ebi.ac.uk/clustalw/).
We have determined the solution structure of the ACP domain from module 2 of DEBS, and have used this structure to build homology models for ACP domains from the other five DEBS elongation modules. As also seen in other carrier proteins whose structures have been reported to date, ACP2 from DEBS is composed of a scaffold consisting of three main helices (residues 14–32, 55–68, and 83–94) packed in a bundle, with an additional contribution from a short helix (residues 76–79) in the intervening loop LII between helices II and III. Hydrophobic interactions stabilize this helical bundle fold.
The structure of ACP2 allowed us to define unambiguously the boundaries of an ACP domain in a PKS module. Specifically, we have shown that the structured core of the ACP2 domain spans ∼40 residues on each side of the conserved serine (S54) that serves as the attachment site for the phosphopantetheine arm, or 81 residues total (from R14 at the N terminus of helix I to L94 at the C terminus of helix III) versus 75 residues for the tyrocidine PCP (Weber et al. 2000) or 73–78 residues for the ACP domains from the aromatic PKSs (act ACP, fren ACP, and otc ACP) (Crump et al. 1997; Findlow et al. 2003; Li et al. 2003). In addition, a short N-terminal region with some helical propensity (residues 6–11) has been identified in the ACP2 of DEBS. This segment partially coincides with the RLAGL motif (residues 8–12 in ACP2), which is conserved among many PKS modules, and has been proposed as a domain boundary based on limited proteolysis of an entire PKS module (Kim et al. 2004).
Comparison of the structure of DEBS ACP2 to the structures of ACPs from aromatic PKSs (act ACP, fren ACP, and otc ACP) shows that helix I is two turns longer in the former protein (19 residues in the ACP2 vs. 12 residues in the ACPs from type II PKSs), whereas the loop between helices I and II is one or two residues shorter. Similar observations were reported when the structure of the PCP domain was compared to the structure of the FAS ACP from E. coli and to the act ACP (Weber et al. 2000): Relative to the latter ACPs, the PCP domain from the tyrocidine synthetase has a longer first helix but a shortened subsequent loop. Helix II of the ACP2 is approximately the same length (14 residues) as in the ACPs from type II PKSs, but the loop LII is a residue or two shorter in the DEBS ACP2. As a result, the short helix (helix III′) within the LII loop is held closer to the core three-helical bundle in the ACP2 domain structure than, for instance, in the fren ACP. The final helix (helix III) is almost one turn longer in ACP2 than in either the fren ACP or the otc ACP.
Superposition of the backbone atoms of the three major helices of ACP2 and fren ACP (ACP2 residues: 21–32, 54–67, 84–92; fren ACP residues: 5–16, 39–52, 69–77) yields the RMSD value of 2.43 Å. This is less than the RMSD value of ∼3 Å obtained by superposition of ACP from E. coli to fren ACP (Li et al. 2003) but greater than the values from pairwise comparisons of ACPs from type II PKSs; RMSD of 1.36 Å for superposition of fren ACP and act ACP (Li et al. 2003) and RMSD of 0.86 Å for superposition of fren ACP and otc ACP. This suggests that ACPs from a type II PKS (fren ACP, act ACP, and otc ACP) are more closely related to each other than to the ACP2 domain from DEBS (a modular PKS).
Surprisingly, superposition of the same helical regions of ACP2 and the corresponding helical regions of two representative ACPs from type II FASs suggests a closer structural similarity. Superimposing ACP2 and ACP from Bacillus subtilis (ACP2 residues: 21–32, 54–67, 84–92; B. subtilis ACP residues: 3–14, 36–49, 66–74) yields the RMSD value of 1.75 Å, while the superposition of ACP2 with ACP from E. coli (ACP2 residues: 21–32, 54–67, 84–92; E. coli ACP residues: 3–14, 36–49, 66–74) gives the RMSD of 1.31 Å. Additional structures of ACP domains from modular PKSs would be necessary to evaluate the generality of this observation.
Previous studies have shown that the KS–AT didomain from module 3 of DEBS did not catalyze chain elongation when ACP6 from DEBS was presented as the partner domain, whereas ACP2 and ACP4 were able to substitute for ACP3 in the same assay system (Kim et al. 2004). A model has been reported for the analogous interaction between a condensing enzyme (KSIII) and an ACP in a bacterial FAS, based on computational structure docking and mutagenesis (Zhang et al. 2001). It suggests that KS–ACP specificity is likely to be influenced by electrostatic interactions at the protein–protein interface, and that helix II of the ACP participates in that interface. A number of other reports point to the role of helix II (and the adjacent loop) of ACP in the domain–domain interactions (Worsham et al. 2003; Zhang et al. 2003) and studies on modular nonribosomal peptide synthetases have also demonstrated that helix II of the peptidyl carrier protein (PCP) is involved in protein–protein recognition (Linne et al. 2001; Mofid et al. 2002; Finking et al. 2004; Lai et al. 2006). Furthermore, in type II polyketide synthases, a residue on helix II (corresponding to position 58 in the ACP2 structure described here) has been found to influence the specificity of ACP recognition by the initiation versus elongation KS (Tang et al. 2003). In light of the precedents summarized above, the topology and electrostatic potential of the putative protein interaction interface (centered on helix II and encompassing the adjacent loop LII) was compared among the six ACP domains of DEBS for clues as to the specificity determinants of the ACP to KS interaction.
Based on this comparison, ACP6 stood out as the domain that differs the most from the other DEBS ACPs. ACP6 displays very little charge on the proposed interaction face and lacks several bulky residues that are present in most of the other DEBS ACP domains, as described in the Results section (e.g., residues at position 59 and position 71) (see Figs 5, 6). At the other end of the spectrum, helix II of the ACP3 domain is predicted to have the most positively charged surface (three positive charges on this interface, in addition to the one contributed by the conserved R61 residue), including a unique, bulky lysine residue (K62). Moreover, ACP3 has a methionine at the position 58, rather than a smaller valine that is present in the other DEBS ACPs (with the exception of ACP1, which has a leucine at that position). These differences could potentially explain the inability of ACP6 to substitute for ACP3 in a chain elongation assay (Kim et al. 2004), as summarized above. At the same time, as seen in Figure 7B, a KS–AT didomain from module 6 of DEBS can interact with ACP2 to catalyze some level of product formation, possibly because both ACP2 and ACP6 lack the positively charged residue at position 63. However, an evaluation of the role of other ACP residues, as well as an examination of the complementary binding interface of the KS domains would be necessary to elucidate the origin of this flexibility.
It has also been shown that ACP2 was unable to substitute for ACP4, and vice versa, in intermodular chain transfer reactions (to module 5 and module 3, respectively), but ACP4 could substitute for ACP1 and ACP5 in analogous assays (Wu et al. 2001, 2002). Consistent with these earlier observations, our homology models reveal that the electrostatic potential surfaces of ACP1, ACP4, and ACP5 of DEBS have two negatively charged residues (rather than just one) in the N-terminal half of helix II. In contrast, ACP2 differs from these three domains at position 59, due to a positively charged arginine residue. Thus, electrostatic interactions appear likely to play a role in the KS–ACP recognition specificity in modular PKSs.
Results of the initial mutagenesis experiments indicate that, at least for the ACP2 domain, the specificity of ACP interactions with KS domains can be influenced by the amino acids located at the proposed interaction interface. The R59G substitution has the most pronounced effect, changing the ACP2 specificity to resemble that of the ACP6. The R71A substitution also leads to a noticeable, but less dramatic change, possibly because residue R59 still prevents efficient interaction with the KS–AT6 didomain. Both the R59G and the R71A mutations remove positively charged residues potentially involved in electrostatic interactions, as noted in the discussion presented above. Additional insight into the exact role of these residues might be gained from examining the complementary interface of the KS domains. To generalize these results and to evaluate possible contributions from other ACP residues to the specificity of domain–domain interactions, more extensive experiments on this and other ACP domains from DEBS (and from other modular polyketide synthases) are warranted. For instance, monitoring ACP chemical shift changes upon titration of the interacting KS domain could point out the interacting residues and their role can be further verified by mutagenesis and functional interaction assays. The structure and models presented here provide a foundation for more comprehensive studies to map the specificity determinants of protein–protein interactions involving the ACP domains in modular polyketide synthases.
Materials and Methods
Sample preparation
The DEBS ACP2(Ø) domain (without the C-terminal linker) was expressed as a uniformly 13C/15N- or 15N-labeled protein in BL21(DE3) E. coli cells that have been transformed with pET28(a)-derived plasmid pNW7 (Wu et al. 2002). The cells were grown at 37°C in minimal medium M9 (final concentrations: 6 g/L Na2HPO4 [anhydrous], 3 g/L KH2PO4 [crystal], 0.5 g/L NaCl, 0.014 g/L CaCl2·2H2O, 0.25 g/L MgSO4 7H2O, 0.005 g/L FeCl3·6H2O, 0.001 g/L thiamine), supplemented with trace metals and vitamins (Tsai et al. 1987; Pfeifer et al. 2002) (final concentrations of trace metals: 0.002 g/L ZnCl2, 0.0025 g/L Na2MoO4, 0.0028 g/L CuSO4·5H2O, 0.00075 g/L H3BO3, 0.015% [v/v] conc. HCl; final concentrations of vitamins: 0.0065 g/L pantothenic acid, 0.00007 g/L biotin, 0.0072 g/L niacin [nicotinic acid], 0.00168 g/L pyridoxine, 0.00005 g/L folic acid, 0.0005 g/L riboflavin—note that the limited solubility of riboflavin in water [on the order of 0.1 g/L] makes it more convenient not to add it as part of the common vitamin stock solution but to prepare a separate, more dilute riboflavin stock solution instead). The medium also contained 4 g/L [U-13C6 (99%)]-glucose and/or 1 g/L [15N (98%+)]-ammonium chloride (Cambridge Isotope Laboratories). The culture was induced at an OD600 of 0.95–1.1 by addition of IPTG (Invitrogen) to the final concentration of 0.5 mM and incubated in a shaker incubator for 16–18 h at 22°C. Cells were harvested by centrifugation and lysed using a homogenizer, and the protein was purified on a Ni-NTA affinity column (Ni-NTA resin from Qiagen), utilizing the N-terminal hexa-histidine tag present in the plasmid construct. After this step, the His6-tag was no longer necessary and was removed by proteolytic cleavage using biotinylated thrombin (Novagen), which was subsequently captured on streptavidin-conjugated resin (Novagen). Separation of cleaved and noncleaved protein was accomplished by batch loading the reaction mixture onto Ni-NTA resin and collecting the desired cleavage product (lacking the His6-tag) in the flow-through. This cleavage step removed 17 of the 20 nonnative N-terminal residues, which originate from the pET28(a) plasmid vector and are not part of the ACP2 sequence.
After several rounds of buffer exchange and concentration using 3 kD cut-off Centriprep and Centricon units (Millipore), which were prerinsed overnight in the appropriate buffer to reduce glycerol contamination in the NMR samples, final NMR samples contained ∼0.8–1 mM of ACP2 in 30 mM sodium phosphate buffer (pH 5.5), 0.05% sodium azide (NaN3), 90% H2O/10%D2O mixture. Protein concentration was estimated by UV absorbance at 205 nm (Scopes 1974).
Data acquisition and analysis
All NMR experiments were acquired at the Stanford Magnetic Resonance Laboratory (SMRL) at 15°C on Varian Inova 500-, 600-, and 800-MHz spectrometers running VNMR v6.1B or v6.1C, equipped with 5-mm inverse-detect, triple-resonance 1H{13C/15N}, pulse-field gradient probes. All data were processed using VNMR and analyzed with SPARKY (Goddard and Kneller 2002). Data from the following two- and three-dimensional isotope edited NMR experiments contained within the ProteinPack suite of pulse sequences (Varian, Inc.) have been collected: 15N HSQC, 13C HSQC, 15N NOESY-HSQC (mixing time, 80 ms), 13C HCCH-TOCSY (mixing time, 14 ms), 13C/15N NOESY-HSQC (mixing time, 100 ms), three-dimensional HNCACB, three-dimensional C(CO)NH, three-dimensional 15N HNHA, three-dimensional 15N HNHB. The 3JHNHα scalar coupling constants were obtained from the ratio of intensities of the cross-peak and the diagonal peak in the HNHA experiment (Grzesiek et al. 1995) and used to derive φ torsion angle restraints according to a previously described method (Zhang et al. 1997), setting the φ constraint to −120° ± 30° for 3JHNHα > 9 Hz, to −120° ± 50° for 9 Hz > 3JHNHα > 8 Hz, or to 60° ± 30° for 3JHNHα < 6 Hz in the α-helical regions.
Structure determination
Structures were calculated using the torsion angle dynamics protocol implemented in DYANA v1.5 software (Guntert et al. 1997) with 4000 steps per structure. Distance constraint upper bounds were generated from assigned NOE data (divided into separate peak lists for aromatic and nonaromatic protons) with the CALIBA function of DYANA (nonaromatic protons: avedis = 3.85 Å, dmax = 7.5 Å; aromatic protons: bb = 5 × 109, dmax = 9 Å). The φ torsion angle restraints were derived from the 3JHNHα scalar coupling constants, as described in the preceding paragraph. The standard simulated annealing protocol included as part of DYANA software was used to calculate a total of 50 structures, of which 30 structures with the lowest target function values were chosen for the solution structure ensemble. The helical regions of these 30 structures were superimposed, and the mean structure was obtained using MOLMOL software (Koradi et al. 1996) and then subjected to the standard variable target function minimization implemented in DYANA. The MOLMOL (v2K.1 and v2K.2.0) (Koradi et al. 1996), DYANA v1.5 (Guntert et al. 1997), and PROCHECK-NMR v3.5.4 (Laskowski et al. 1996) software packages were used to analyze the structures. The coordinates of the ensemble of 30 structures have been deposited in the Protein Data Bank (PDB ID 2JU1), along with the coordinates of the minimized mean structure (PDB ID 2JU2).
Sequence alignment
Multiple sequence alignment of the ACP domains from DEBS was performed using the PileUp program from the GCG SeqWeb sequence analysis software package (Accelrys Inc.), with the following parameters: blosum65 matrix, gap creation penalty 11, gap extension penalty 5. The sequences were obtained from PKSDB: A Database of Modular Polyketide Synthases (www.nii.res.in/pksdb.html) (Yadav et al. 2003).
Homology models of other ACP domains from DEBS
Automated pairwise amino acid sequence alignments of ACP2 to each of the other five ACP domains from DEBS were performed using the GAP program from the GCG SeqWeb sequence analysis software package (Accelrys Inc.). The alignments were then used to build homology models for the ACP domains of DEBS, based on the solution structure of ACP2 as a template. Residues 12–95 (numbering according to ACP2) were taken for homology model building. The homology models for ACP1, ACP3, ACP4, ACP5, and ACP6 domains of DEBS were generated using the WHATIF homology model server (Rodriguez et al. 1998), available at http://swift.cmbi.kun.nl/swift/servers/modserver-submit.html. Electrostatic potential surfaces were calculated using MOLMOL (Koradi et al. 1996), with blue and red colors on the surface representing positively and negatively charged residues, respectively.
Phylogram of ACP domain subfamilies
Sequences of the 16 carrier proteins used to construct the phylogram were obtained either from PKSDB (in case of the 10 ACP domains originating from modular polyketide synthases, i.e., ACPs from DEBS and rifamycin PKSs) (Yadav et al. 2003) or from the following sources: The sequence of the frenolicin (fren) ACP was obtained from a published reference (Li et al. 2003), the sequence of the structurally characterized PCP domain from tyrocidine NRPS from a published reference (Weber et al. 2000), the sequence of the oxytetracycline (otc) ACP from EntrezProtein (accession no. P43677), the sequence of the actinorhodin (act) ACP from EntrezProtein (accession no. Q02054), the sequence of the E. coli FAS ACP from a published reference (also see EntrezProtein, accession no. 1L0HA) (Fig. 3 in Xu et al. 2001), the sequence of the B. subtilis FAS ACP from a published reference (also see EntrezProtein, accession no. P80643) (Xu et al. 2001) Multiple sequence alignment for these 16 carrier proteins was performed using default parameters (blosum62, gap creation penalty 8, gap extension penalty 2) of the PileUp program (GCG SeqWeb sequence analysis software package, Accelrys Inc.), and the aligned sequences were submitted to the CLUSTAL W server (Thompson et al. 1994) at the European Bioinformatics Institute (www.ebi.ac.uk/clustalw/) to generate the phylogram shown in Figure 8.
Construction, expression, and purification of ACP point mutants
Starting with the native ACP2 sequence (plasmid pNW7) (Wu et al. 2002) as a template, the QuikChange kit (Stratagene) was used to introduce point mutations according to the manufacturer's instructions. The following primers were used (the bold underlined bases correspond to the introduced changes): eryACP2R71Aforward (5′-GCGGCCACCGGGCTCGCCCTGCCGTCG-3′) and eryACP2R71Areverse (5′-CGACGGCAGGGCGAGCCCGGTGGCCGC-3′) for the construction of ACP2 R71A mutant, eryACP2R59Gforward (5′-CTGGCGGCCGTCGGACTGCGCAACCTGCTC-3′) and eryACP2R59Greverse (5′-GAGCAGGTTGCGCAGTCCGACGGCCGCCAG-3′) for the construction of the ACP2 R59G mutant, and eryACP2L63Q forward (5′-GTCCGGCTGCGCAACCAGCTGAACGCGGCCACC-3′) and eryACP2L63Qreverse (5′-GGTGGCCGCGTTCAGCTGGTTGCGCAGCCGGAC-3′) for the construction of the ACP2 L63Q mutant. The cloning steps and the screening for the desired mutants were done in E. coli XL1-Blue cells (Stratagene). The incorporation of the desired base changes and the absence of other mutations in the final construct were confirmed by DNA sequencing. The resulting plasmids (pACP2R71A, pACP2R59G and pACP2L63Q, encoding three single-point mutants of ACP2—R71A, R59G, and L63Q, respectively) were transformed into E. coli BAP1 electrocompetent cells. The E. coli BAP1 cell line (Pfeifer et al. 2001) harbors a plasmid encoding the Sfp phosphopantetheinyl transferase, which was used to effect pantetheinylation of ACP. The three ACP2 mutant proteins were expressed and purified according to a previously described procedure (Kim et al. 2004), utilizing Ni-NTA affinity purification and an additional FPLC step involving application of the protein to a HiTrap-Q anion exchange column—the protein was recovered in flow through, while impurities remained on the column and were removed by a washing step using a high salt buffer. After these steps, each of the three ACP2 mutant proteins was found to be pure by SDS-PAGE and was concentrated using Vivaspin 20 centrifugal concentrators with the polyethersulfone membrane, molecular weight cutoff 5000 (Sartorius AG) to ∼1 mM concentration (as determined by UV absorbance at 205 nm) (Scopes 1974) in a phosphate buffer (pH 7.2). The wild-type ACP2 and ACP6 proteins were also expressed and purified according to the above procedure. The expression and purification procedure for KS–AT didomains has been previously published (Kim et al. 2004).
Triketide lactone formation assay using ACP point mutants
The experiments evaluating the ability of each of the three ACP2 mutants to support chain elongation in reactions with KS–AT didomains from DEBS module 3 and from DEBS module 6 were performed as described previously (Kim et al. 2004), with the exception that the radioactive tracer originated from the [14C]-labeled extender unit (D,L-[2-methyl-14C]-methylmalonyl-CoA, American Radiolabeled Chemicals), rather than from the electrophilic NDK-SNAc ((2S,3R)-2-methyl-3-hydroxypentanoyl-N-acetylcysteamine (NAC) thioester) substrate that acylates the KS domain. In addition to the ACP2 R71A, ACP2 R59G, and ACP2 L63Q mutants, wild-type ACP2 and wild-type ACP6 proteins were also evaluated in these assays for comparison. The final reaction volumes were 10 μL. Each KS–AT didomain (10 μM) was incubated for 1 h with 5 mM nonlabeled NDK-SNAc ((2S,3R)-2-methyl-3-hydroxypentanoyl-SNAc, a known substrate for DEBS module 3 and DEBS module 6) (Gokhale et al. 1999; Wu et al. 2000), to acylate KS to completion. The incubation buffer contained 100 mM phosphate (pH 7.2) and 5 mM TCEP (tris-[2-carboxyethyl]phosphine hydrochloride). The appropriate ACP (200 μM) and 200 μM D,L-[2-methyl-14C]methylmalonyl-CoA were then added and allowed to react at room temperature for 10, 20, 30, 60, or 90 min. At each time point, the reaction was quenched by adding 20 μL of 0.5 M potassium hydroxide and heating the mixture for 20 min at 65°C. Hydrochloric acid (10 μL of 1.5 M) was then added, and the mixture was dried by centrifugation under reduced pressure for 2 h. The pellet was resuspended in 10 μL of ethyl acetate and spotted onto a TLC plate (J.T. Baker). A 60:40 mixture of ethyl acetate/hexane was used for the TLC and the plates were then visualized and quantified using a Packard phosphorimager.
Electronic supplemental material
Supplemental Table ST1 contains NMR resonance assignments for DEBS ACP2(Ø). Supplemental Table ST2 contains initial rates of triketide lactone product formation in condensation reactions catalyzed by KS–AT didomains interacting with methylmalonyl-ACPs. Supplemental Figure S1 shows characterization of the ACP2(Ø) protein sample (electrophoresis, mass spectrometry, and NMR data used to evaluate protein sample homogeneity and purity).
Acknowledgments
We thank Dr. Pawan Kumar for advice on protein expression and purification, Dr. Stephen R. Lynch for valuable assistance with the preliminary HSQC experiments, and Dr. Qing Li for many helpful suggestions throughout the structure determination process, as well as Alice Chen for the KS–AT didomain proteins and Dr. Nathan Schnarr for kindly providing the NDK-SNAc. This research was supported by a grant from the National Institutes of Health (CA 66736 to C. K.). V.Y.A was a Howard Hughes Medical Institute pre-doctoral fellow. Stanford Magnetic Resonance Laboratory is supported by the Stanford University School of Medicine.
Footnotes
Supplemental material: see www.proteinscience.org
Reprint requests to: Chaitan Khosla, Department of Chemistry, Stanford University, MC5080, Stanford CA 94305, USA; e-mail: khosla@stanford.edu; fax: (650) 723-6538.
Abbreviations ACP, acyl carrier protein; ArCP, aryl carrier protein; AT, acyltransferase; CoA, coenzyme A; DEBS, 6-deoxyerythronolide B synthase; DH, dehydratase; ER, enoyl reductase; ESI-MS, electrospray ionization mass spectrometry; FAS, fatty acid synthase; HSNAc, N-acetylcysteamine; HSQC, heteronuclear single-quantum coherence; IPTG, isopropyl-β-D-thiogalactopyranoside; kD, kilodalton; KR, ketoreductase; KS, ketosynthase; NDK-SNAc, the natural diketide substrate, 2-methyl-3-hydroxypentanoyl-N-acetylcysteamine thioester; NMR, nuclear magnetic resonance; NOE, nuclear Overhauser effect; Ni-NTA, nickel-nitrilotriacetic acid; NRPS, nonribosomal peptide synthetase; PCP, peptidyl carrier protein; PKS, polyketide synthase; RMSD, root-mean-square deviation; SDS-PAGE, sodium dodecyl sulfate polyacrylamide gel electrophoresis; TCEP, tris-(2-carboxyethyl)phosphine hydrochloride; TE, thioesterase; TLC, thin layer chromatography.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.073011407.
References
- Broadhurst R.W., Nietlispach, D., Wheatcroft, M.P., Leadlay, P.F., and Weissman, K.J. 2003. The structure of docking domains in modular polyketide synthases. Chem. Biol. 10: 723–731. [DOI] [PubMed] [Google Scholar]
- Cane D.E., Walsh, C.T., and Khosla, C. 1998. Harnessing the biosynthetic code: Combinations, permutations, and mutations. Science 282: 63–68. [DOI] [PubMed] [Google Scholar]
- Cortes J., Haydock, S.F., Roberts, G.A., Bevitt, D.J., and Leadlay, P.F. 1990. An unusually large multifunctional polypeptide in the erythromycin-producing polyketide synthase of Saccharopolyspora erythraea . Nature 348: 176–178. [DOI] [PubMed] [Google Scholar]
- Crump M.P., Crosby, J., Dempsey, C.E., Parkinson, J.A., Murray, M., Hopwood, D.A., and Simpson, T.J. 1997. Solution structure of the actinorhodin polyketide synthase acyl carrier protein from Streptomyces coelicolor A3(2) . Biochemistry 36: 6000–6008. [DOI] [PubMed] [Google Scholar]
- Donadio S. and Katz, L. 1992. Organization of the enzymatic domains in the multifunctional polyketide synthase involved in erythromycin formation in Saccharopolyspora erythraea . Gene 111: 51–60. [DOI] [PubMed] [Google Scholar]
- Donadio S., Staver, M.J., McAlpine, J.B., Swanson, S.J., and Katz, L. 1991. Modular organization of genes required for complex polyketide biosynthesis. Science 252: 675–679. [DOI] [PubMed] [Google Scholar]
- Drake E.J., Nicolai, D.A., and Gulick, A.M. 2006. Structure of the EntB multidomain nonribosomal peptide synthetase and functional analysis of its interaction with the EntE adenylation domain. Chem. Biol. 13: 409–419. [DOI] [PubMed] [Google Scholar]
- Findlow S.C., Winsor, C., Simpson, T.J., Crosby, J., and Crump, M.P. 2003. Solution structure and dynamics of oxytetracycline polyketide synthase acyl carrier protein from Streptomyces rimosus . Biochemistry 42: 8423–8433. [DOI] [PubMed] [Google Scholar]
- Finking R. and Marahiel, M.A. 2004. Biosynthesis of nonribosomal peptides. Annu. Rev. Microbiol. 58: 453–488. [DOI] [PubMed] [Google Scholar]
- Finking R., Mofid, M.R., and Marahiel, M.A. 2004. Mutational analysis of peptidyl carrier protein and acyl carrier protein synthase unveils residues involved in protein–protein recognition. Biochemistry 43: 8946–8956. [DOI] [PubMed] [Google Scholar]
- Goddard T.D. and Kneller, D.G. 2002. SPARKY 3. University of California, San Francisco, CA.
- Gokhale R.S., Tsuji, S.Y., Cane, D.E., and Khosla, C. 1999. Dissecting and exploiting intermodular communication in polyketide synthases. Science 284: 482–485. [DOI] [PubMed] [Google Scholar]
- Grzesiek S., Kuboniwa, H., Hinck, A.P., and Bax, A. 1995. Multiple-quantum line narrowing for measurement of Hα-Hβ J couplings in isotopically enriched proteins. J. Am. Chem. Soc. 117: 5312–5315. [Google Scholar]
- Guntert P., Mumenthaler, C., and Wuthrich, K. 1997. Torsion angle dynamics for NMR structure calculation with the new program DYANA. J. Mol. Biol. 173: 283–298. [DOI] [PubMed] [Google Scholar]
- Hans M., Hornung, A., Dziarnowski, A., Cane, D.E., and Khosla, C. 2003. Mechanistic analysis of acyl transferase domain exchange in polyketide synthase modules. J. Am. Chem. Soc. 125: 5366–5374. [DOI] [PubMed] [Google Scholar]
- Holak T.A., Kearsley, S.K., Kim, Y., and Prestegard, J.H. 1988a. Three-dimensional structure of acyl carrier protein determined by NMR pseudoenergy and distance geometry calculations. Biochemistry 27: 6135–6142. [DOI] [PubMed] [Google Scholar]
- Holak T.A., Nilges, N., Prestegard, J.H., Gronenborn, A.M., and Clore, G.M. 1988b. Three-dimensional structure of acyl carrier protein in solution determined by nuclear magnetic resonance and the combined use of dynamical simulated annealing and distance geometry. Eur. J. Biochem. 175: 9–15. [DOI] [PubMed] [Google Scholar]
- Keatinge-Clay A.T. and Stroud, R.M. 2006. The structure of a ketoreductase determines the organization of the β-carbon processing enzymes of modular polyketide synthases. Structure 14: 737–748. [DOI] [PubMed] [Google Scholar]
- Khosla C., Gokhale, R.S., Jacobsen, J.R., and Cane, D.E. 1999. Tolerance and specificity of polyketide synthases. Annu. Rev. Biochem. 68: 219–253. [DOI] [PubMed] [Google Scholar]
- Kim C.-Y., Alekseyev, V.Y., Chen, A.Y., Tang, Y., Cane, D.E., and Khosla, C. 2004. Reconstituting modular activity from separated domains of 6-deoxyerythronolide B synthase. Biochemistry 43: 13892–13898. [DOI] [PubMed] [Google Scholar]
- Kim Y. and Prestegard, J.H. 1989. A dynamic model for the structure of acyl carrier protein in solution. Biochemistry 28: 8792–8797. [DOI] [PubMed] [Google Scholar]
- Koradi R., Billeter, M., and Wuthrich, K. 1996. MOLMOL: A program for display and analysis of macromolecular structures. J. Mol. Graph. 14: 51–55. [DOI] [PubMed] [Google Scholar]
- Kumar P., Li, Q., Cane, D.E., and Khosla, C. 2003. Intermodular communication in modular polyketide synthases: Structural and mutational analysis of linker mediated protein–protein recognition. J. Am. Chem. Soc. 125: 4097–4102. [DOI] [PubMed] [Google Scholar]
- Lai J.R., Fischbach, M.A., Liu, D.R., and Walsh, C.T. 2006. A protein interaction surface in nonribosomal peptide synthesis mapped by combinatorial mutagenesis and selection. Proc. Natl. Acad. Sci. 103: 5314–5319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski R.A., Rullmann, J.A., MacArthur, M.W., Kaptein, R., and Thornton, J.M. 1996. AQUA and PROCHECK-NMR: Programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR 8: 477–486. [DOI] [PubMed] [Google Scholar]
- Li Q., Khosla, C., Puglisi, J.D., and Liu, C.W. 2003. Solution structure and backbone dynamics of the holo form of the frenolicin acyl carrier protein. Biochemistry 42: 4648–4657. [DOI] [PubMed] [Google Scholar]
- Linne U., Doekel, S., and Marahiel, M.A. 2001. Portability of epimerization domain and role of peptidyl carrier protein on epimerization activity in nonribosomal peptide synthetases. Biochemistry 40: 15824–15834. [DOI] [PubMed] [Google Scholar]
- Liou G.F., Lau, J., Cane, D.E., and Khosla, C. 2003. Quantitative analysis of loading and extender acyltransferases of modular polyketide synthases. Biochemistry 42: 200–207. [DOI] [PubMed] [Google Scholar]
- McDaniel R., Thamchaipenet, A., Gustafsson, C., Fu, H., Betlach, M., Betlach, M., and Ashley, G. 1999. Multiple genetic modifications of the erythromycin polyketide synthase to produce a library of novel “unnatural” natural products. Proc. Natl. Acad. Sci. 96: 1846–1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menzella H.G., Reid, R., Carney, J.R., Chandran, S.S., Reisinger, S.J., Patel, K.G., Hopwood, D.A., and Santi, D.V. 2005. Combinatorial polyketide biosynthesis by de novo design and rearrangement of modular polyketide synthase genes. Nat. Biotechnol. 23: 1171–1176. [DOI] [PubMed] [Google Scholar]
- Mofid M.R., Finking, R., and Marahiel, M.A. 2002. Recognition of hybrid peptidyl carrier proteins/acyl carrier proteins in nonribosomal peptide synthetase modules by the 4′-phosphopantetheinyl transferases AcpS and Sfp. J. Biol. Chem. 277: 17023–17031. [DOI] [PubMed] [Google Scholar]
- Park S.J., Kim, J.S., Son, W.S., and Lee, B.J. 2004. pH-Induced conformational transition of H. pylori acyl carrier protein: Insight into the unfolding of local structure. J. Biochem. 135: 337–346. [DOI] [PubMed] [Google Scholar]
- Pfeifer B.A., Admiraal, S.J., Ness, H., Ness, G., Cane, D.E., and Khosla, C. 2001. Biosynthesis of complex polyketides in a metabolically engineered strain of E. coli . Science 291: 1790–1792. [DOI] [PubMed] [Google Scholar]
- Pfeifer B.A., Hu, Z., Licari, P., and Khosla, C. 2002. Process and metabolic strategies for improved production of Escherichia coli-derived 6-deoxyerythronolide B. Appl. Environ. Microbiol. 68: 3287–3292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranganathan A., Timoney, M., Bycroft, M., Cortes, J., Thomas, I.P., Wilkinson, B., Kellenberger, L., Hanefeld, U., Galloway, I.S., Staunton, J., et al. 1999. Knowledge-based design of bimodular and trimodular polyketide synthases based on domain and module swaps: A route to simple statin analogues. Chem. Biol. 6: 731–741. [DOI] [PubMed] [Google Scholar]
- Rodriguez R., Chinea, G., Lopez, N., Pons, T., and Vriend, G. 1998. Homology modeling, model and software evaluation: Three related resources. Bioinformatics 14: 523–528. [DOI] [PubMed] [Google Scholar]
- Roujeinikova A., Baldock, C., Simon, W.J., Gilroy, J., Baker, P.J., Stuitje, A.R., Rice, D.W., Slabas, A.R., and Rafferty, J.B. 2002. X-ray crystallographic studies on butyryl-ACP reveal flexibility of the structure around a putative acyl chain binding site. Structure 10: 825–835. [DOI] [PubMed] [Google Scholar]
- Scopes R.K. 1974. Measurement of protein by spectrophotometry at 205 nm. Anal. Biochem. 59: 277–282. [DOI] [PubMed] [Google Scholar]
- Sharma A.K., Sharma, S.K., Surolia, A., Surolia, N., and Sarma, S.P. 2006. Solution structures of conformationally equilibrium forms of holo-acyl carrier protein (PfACP) from Plasmodium falciparum provides insight into the mechanism of activation of ACPs. Biochemistry 45: 6904–6916. [DOI] [PubMed] [Google Scholar]
- Sieber S.A. and Marahiel, M.A. 2005. Molecular mechanisms underlying nonribosomal peptide synthesis: Approaches to new antibiotics. Chem. Rev. 105: 715–738. [DOI] [PubMed] [Google Scholar]
- Staunton J. and Weissman, K.J. 2001. Polyketide biosynthesis: A millennium review. Nat. Prod. Rep. 18: 380–416. [DOI] [PubMed] [Google Scholar]
- Staunton J. and Wilkinson, B. 2001. Combinatorial biosynthesis of polyketides and nonribosomal peptides. Curr. Opin. Chem. Biol. 5: 159–164. [DOI] [PubMed] [Google Scholar]
- Tang Y., Lee, T.S., Kobayashi, S., and Khosla, C. 2003. Ketosynthases in the initiation and elongation modules of aromatic polyketide synthases have orthogonal acyl carrier protein specificity. Biochemistry 42: 6588–6595. [DOI] [PubMed] [Google Scholar]
- Tang Y., Kim, C.Y., Mathews, I.I., Cane, D.E., and Khosla, C. 2006. The 2.7 Å crystal structure of a 194-kDa homodimeric fragment of the 6-deoxyerythronolide B synthase. Proc. Natl. Acad. Sci. 103: 11124–11129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson J.D., Higgins, D.G., and Gibson, T.J. 1994. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22: 4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai L.B., Mann, M., Morris, F., Rotgers, C., and Fenton, D. 1987. The effect of organic nitrogen and glucose on the production of recombinant human insulin-like growth factor in high cell density Escherichia coli fermentations. J. Ind. Microbiol. 2: 181–187. [Google Scholar]
- Tsuji S.Y., Wu, N., and Khosla, C. 1999. Intermodular communication in polyketide synthases: Comparing the role of protein–protein interactions to those in other multidomain proteins. Biochemistry 40: 2317–2325. [DOI] [PubMed] [Google Scholar]
- Volkman B.F., Zhang, Q., Debabov, D.V., Rivera, E., Kresheck, G.C., and Neuhaus, F.C. 2001. Biosynthesis of D-alanyl-lipoteichoic acid: The tertiary structure of apo-D-alanyl carrier protein. Biochemistry 40: 7964–7972. [PubMed] [Google Scholar]
- Walsh C.T. 2002. Combinatorial biosynthesis of antibiotics: Challenges and opportunities. ChemBioChem 3: 124–134. [DOI] [PubMed] [Google Scholar]
- Watanabe K., Wang, C.C.C., Boddy, C.N., Cane, D.E., and Khosla, C. 2003. Understanding substrate specificity of polyketide synthase modules by generating hybrid multimodular synthases. J. Biol. Chem. 278: 42020–42026. [DOI] [PubMed] [Google Scholar]
- Weber T., Baumgartner, R., Renner, C., Marahiel, M.A., and Holak, T.A. 2000. Solution structure of PCP, a prototype for the peptidyl carrier domains of modular peptide synthetases. Structure 8: 407–418. [DOI] [PubMed] [Google Scholar]
- Weissman K.J., Hong, H., Popovic, B., and Meersman, F. 2006. Evidence for a protein–protein interaction motif on an acyl carrier protein domain from a modular polyketide synthase. Chem. Biol. 13: 625–636. [DOI] [PubMed] [Google Scholar]
- Wong H.C., Liu, G.H., Zhang, Y.M., Rock, C.O., and Zheng, J. 2002. The solution structure of acyl carrier protein from Mycobacterium tuberculosis . J. Biol. Chem. 277: 15874–15880. [DOI] [PubMed] [Google Scholar]
- Worsham L.M., Earls, L., Jolly, C., Langston, K.G., Trent, M.S., and Ernst-Fonberg, M.L. 2003. Amino acid residues of Escherichia coli acyl carrier protein involved in heterologous protein interactions. Biochemistry 42: 167–176. [DOI] [PubMed] [Google Scholar]
- Wu N., Kudo, F., Cane, D.E., and Khosla, C. 2000. Analysis of the molecular recognition features of individual modules derived from the erythromycin polyketide synthase. J. Am. Chem. Soc. 122: 4847–4852. [Google Scholar]
- Wu N., Tsuji, S.Y., Cane, D.E., and Khosla, C. 2001. Assessing the balance between protein–protein interactions and enzyme-substrate interactions in the channeling of intermediates between polyketide synthase modules. J. Am. Chem. Soc. 123: 6465–6474. [DOI] [PubMed] [Google Scholar]
- Wu N., Cane, D.E., and Khosla, C. 2002. Quantitative analysis of the relative contributions of donor acyl carrier proteins, acceptor ketosynthases, and linker regions to intermodular transfer of intermediates in hybrid polyketide synthases. Biochemistry 41: 5056–5066. [DOI] [PubMed] [Google Scholar]
- Xu G.Y., Tam, A., Lin, L., Hixon, J., Fritz, C.C., and Powers, R. 2001. Solution structure of B. subtilis acyl carrier protein. Structure 9: 277–287. [DOI] [PubMed] [Google Scholar]
- Yadav G., Gokhale, R.S., and Mohanty, D. 2003. SEARCHPKS: A program for detection and analysis of polyketide synthase domains. Nucleic Acids Res. 31: 3654–3658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W., Smithgall, T.E., and Gmeiner, W.H. 1997. Three-dimensional structure of the Hck SH2 domain in solution. J. Biomol. NMR 10: 263–272. [DOI] [PubMed] [Google Scholar]
- Zhang Y.-M., Rao, M.S., Heath, R.J., Price, A.C., Olson, A.J., Rock, C.O., and White, S.W. 2001. Identification and analysis of the acyl carrier protein (ACP) docking site on β-ketoacyl-ACP synthase III. J. Biol. Chem. 276: 8231–8238. [DOI] [PubMed] [Google Scholar]
- Zhang Y.M., Wu, B., Zheng, J., and Rock, C.O. 2003. Key residues responsible for acyl carrier protein and β-ketoacyl-acyl carrier protein reductase (FabG) interaction. J. Biol. Chem. 278: 52935–52943. [DOI] [PubMed] [Google Scholar]