

Abstract
Previous studies identified several single-point mutants of the prion protein that displayed dominant-negative effects on prion replication. The dominant-negative effect was assumed to be mediated by protein X, an as-yet-unknown cellular cofactor that is believed to be essential for prion replication. To gain insight into the mechanism that underlies the dominant-negative phenomena, we evaluated the effect of the Q218K variant of full-length recombinant prion protein (Q218K rPrP), one of the dominant-negative mutants, on cell-free polymerization of wild-type rPrP into amyloid fibrils. We found that both Q218K and wild-type (WT) rPrPs were incorporated into fibrils when incubated as a mixture; however, the yield of polymerization was substantially decreased in the presence of Q218K rPrP. Furthermore, in contrast to fibrils produced from WT rPrP, the fibrils generated in the mixture of WT and Q218K rPrPs did not acquire the proteinase K-resistant core of 16 kDa that was shown previously to encompass residues 97–230 and was similar to that of PrPSc. Our studies demonstrate that the Q218K variant exhibits the dominant-negative effect in cell-free conversion in the absence of protein X, and that this effect is, presumably, mediated by physical interaction between Q218K and WT rPrP during the polymerization process.
Keywords: prion protein, dominant-negative effect, prion diseases, amyloid fibrils, cell-free conversion
Prion diseases are a group of fatal neurodegenerative maladies that can arise spontaneously or be inherited, and that can also be infectious (Prusiner 1998). All three forms of the prion diseases, sporadic, familial, and infectious, are induced by misfolding and aggregation of the prion protein (PrP). Single-point mutations at several positions within the PrP open reading frame were linked to familial prion diseases. Remarkably, the polymorphisms at other codons of PrP gene were found to protect against scrapie infection or development of sporadic forms of prion diseases. For example, sheep with Q/R polymorphisms at the codon 171 (equivalent to the codon 167 in mouse PrP) were resistant to scrapie, while sheep carrying the Q/Q alleles developed prion disease (Ikeda et al. 1995; Hunter et al. 1997). Human genetic studies revealed that 12% of the Japanese population carries the polymorphism E/K at the codon 219 (equivalent to the codon 218 of mouse PrP) (Shibuya et al. 1998b). No sporadic CJD cases were reported among this subpopulation (Shibuya et al. 1998a).
Because heterozygosity at certain positions was found to be protective against human prion diseases, it can be exploited for developing effective therapeutic strategies. While several low-molecular-weight compounds including porphyrins and phthalocyanines (Caughey et al. 1998), quinacrine (Korth et al. 2001), branched polyamines (Supattapone et al. 1999), or compound Cp-60 (Perrier et al. 2000) have been reported to inhibit PrPSc formation in cultured cells, no therapeutic strategy has been shown to be successful so far for animals or humans. The natural polymorphisms in the amino acid sequence of PrPC could be used for designing an alternative therapeutic strategy against sporadic and infectious forms of prion diseases.
Previous studies revealed that substitution of Gln at positions 167 or 218 (mouse classification) to Arg or Lys, respectively, prevented PrPSc formation in transgenic mice and cultured cells that coexpressed wild-type (WT) and Q167R or Q218K PrPC (Kaneko et al. 1997; Perrier et al. 2002). Because these PrP variants were able to block prion replication, they were referred to as “dominant-negative” mutants. The potential practical value of the dominant-negative effect for anti-prion gene therapy was demonstrated in recent studies in which the prion infection was cured in cultured cells after transferring lentiviral vectors carrying Q167R or Q218K PrP (Crozet et al. 2004).
In the previous studies, the dominant-negative effect was proposed to be mediated by the protein X, an as-yet-unidentified cellular cofactor that is believed to be essential for prion propagation (Kaneko et al. 1997). It has been speculated that dominant-negative PrPC variants block prion replication by binding to protein X and depleting it from mediating the conversion of wild-type PrPC into PrPSc (Kaneko et al. 1997). In this study, we tested whether an alternative mechanism that does not require protein X may account for the dominant-negative effect. To test whether dominant-negative phenomena could be observed in the absence of protein X, we evaluated the effect of the recombinant Q218K PrP variant (Q218K rPrP) on conversion of WT rPrP into amyloid fibrils in the absence of a cellular environment. We found that in the mixture of Q218K and WT rPrPs, the yield of polymerization was substantially decreased, while both Q218K and WT rPrPs were incorporated into fibrils. Furthermore, in contrast to the WT fibrils, the fibrils generated in the mixture of WT and Q218K rPrP failed to produce the 16-kDa proteinase K-resistant fragment in the PK-digestion assay. Our studies suggest that the dominant-negative effect is mediated by physical interaction between Q218K and WT rPrP within the fibrillar form and may not require protein X.
Results
Q218K and WT rPrP display similar secondary structure and conformational stability
Both Q218K and WT rPrP variants were expressed and purified as described in our previous studies (Bocharova et al. 2005a; Breydo et al. 2005). To examine whether Q218K substitution affected the secondary structure and conformational stability of the α-helical monomeric form of rPrP, we collected CD spectra and performed thermal denaturation for both WT and Q218K variants (Fig. 1). CD spectra revealed that both proteins have identical, predominantly α-helices secondary structure (Fig. 1A). As judged from the temperature denaturation experiments, both WT and Q218K rPrPs displayed cooperative unfolding and showed very similar conformational stability, with the Q218K variant being slightly less stable than WT rPrP (Fig. 1B). Because the cell-free conversion into the fibrillar form was carried out under partially denaturing conditions and, specifically, in the presence of GdnHCl, we were interested in knowing whether GdnHCl had a similar effect on the conformational stability of both proteins. We found that in the presence of 0.5 M GdnHCl, the stability of WT and Q218K variants was reduced to a similar extent (Fig. 1B). Under these partially denaturing conditions, both rPrP variants displayed cooperative unfolding and showed similar conformation stability. These data illustrate that Q218K substitution did not significantly affect the α-helical content or the conformational stability of the native α-helical form of rPrP.
Figure 1.
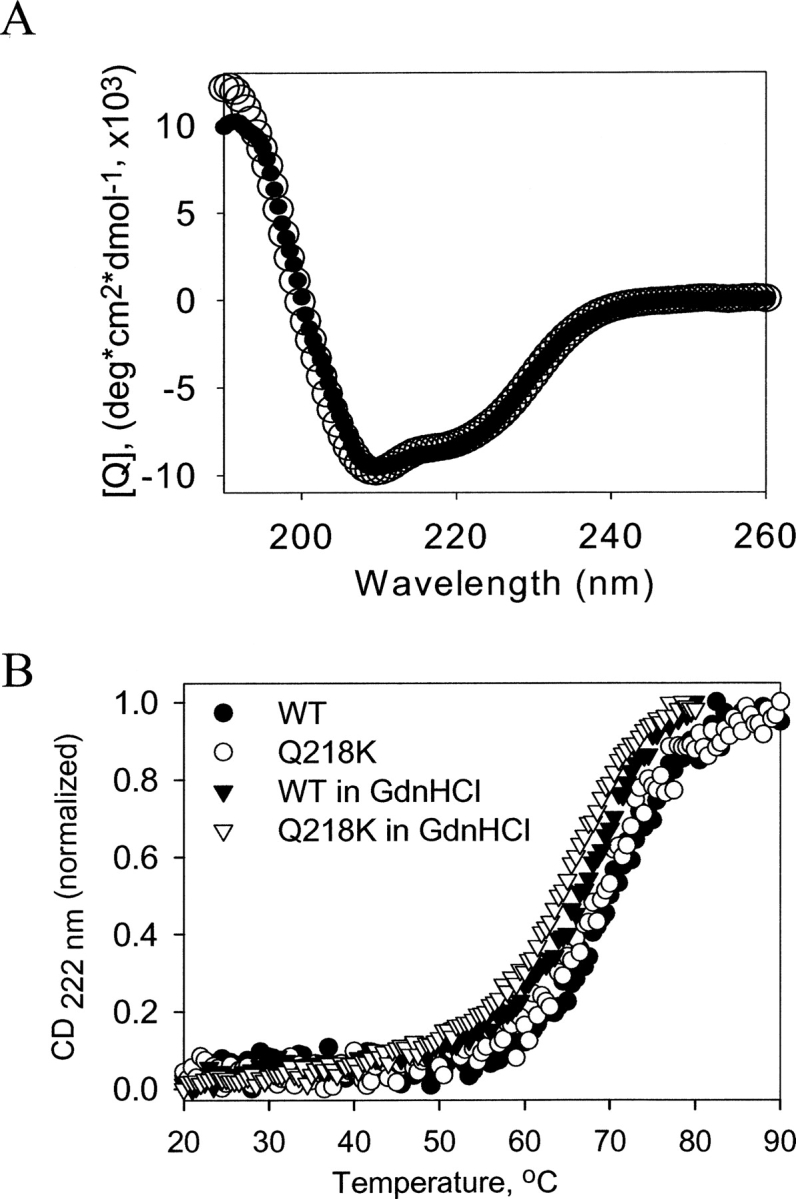
Thermal denaturation of wild-type (WT) and Q218K α-rPrPs. (A) Far-UV CD spectra of (•) WT and (○) Q218K α-rPrPs (0.2 mg/mL) collected in 1 mM HEPES (pH 7). (B) The thermal denaturation curves for WT (•, ▾) and Q218K (○, ▽) α-rPrPs collected in the absence (circles) or presence of 0.5 M GndHCl (triangles) in 50 mM MES (pH 6.0).
Q218K inhibits fibril formation of WT rPrP
To examine whether Q218K has any effect on fibrillation of WT rPrP, we monitored the kinetics of polymerization for two rPrP variants separately or in a mixture using the in vitro conversion reaction developed in our earlier studies (Fig. 2; Baskakov and Bocharova 2005; Bocharova et al. 2005a). As judged from the ThT-fluorescence assay, under optimal solvent conditions (in 2 M GdnHCl at pH 6.0), WT rPrP formed fibrils after a very short lag phase and with high yield. In contrast, the polymerization of Q218K rPrP alone displayed a prolonged lag phase and a substantially reduced yield. Mixing of WT with Q218K at a 1:1 ratio increased the lag phase and reduced the yield of fibrillization (Fig. 2). These data demonstrated that the Q218K variant had a low propensity to form fibrils alone and imposed a negative effect on the polymerization of WT rPrP.
Figure 2.

The kinetics of amyloid fibril formation. WT rPrP (10 μM) (▪), Q218K rPrP (10 μM) (▾), or a 1:1 WT:Q218K rPrPs mixture (total protein concentration of 20 μM) (•) was incubated in 50 mM MES at pH 6.0, 2 M GdnHCl at 37°C upon continuous shaking at 600 rpm and analyzed using ThT fluorescence assay. The ThT fluorescence reading was normalized per 0.3 μM/mL rPrP present in the ThT assay mixture.
Q218K copolymerizes with WT rPrP
The experiments on the kinetics of polymerization revealed that Q218K partially inhibited fibril formation. The inhibitory effect could be due to binding to and “poisoning” of the growing fibrillar ends or due to incorporation of Q218K into fibrils and slowing the rate of polymerization. The first mechanism predicts that the fibrils produced in the mixture of WT and Q218K rPrPs would consist of predominantly WT rPrP, whereas the second mechanism assumes that both WT and Q218K rPrPs would be incorporated into fibrils. To gain insight into the mechanism of Q218K-dependent inhibition, we analyzed the composition of fibrils obtained in the mixture of WT and Q218K rPrPs. Fibrils taken at the endpoint of the conversion reaction were separated from the soluble rPrP isoform by centrifugation and then denatured in 8 M GdnHCl (see Materials and Methods). The amounts of Q218K and WT rPrP variants were analyzed using analytical reverse-phase HPLC. Because Q218K and WT rPrPs elute at different times (Fig. 3A), reverse-phase HPLC can be used for quantitative assessment of the amounts of Q218K and WT rPrPs present in mixtures. To estimate the ratio of Q218K:WT rPrPs in dissociated fibrils, we first built a calibration curve using mixtures of monomeric WT and Q218K rPrPs of known concentrations (Fig. 3B). Based on the calibration curve, we found that the actual ratio of WT:Q218K rPrPs that were incorporated in fibrils was very similar to the ratio of two PrP variants used for the conversion reaction (Table 1). These results were consistent with the model that postulates that Q218K rPrP copolymerizes with WT rPrP and slows down the fibrillization rate.
Figure 3.
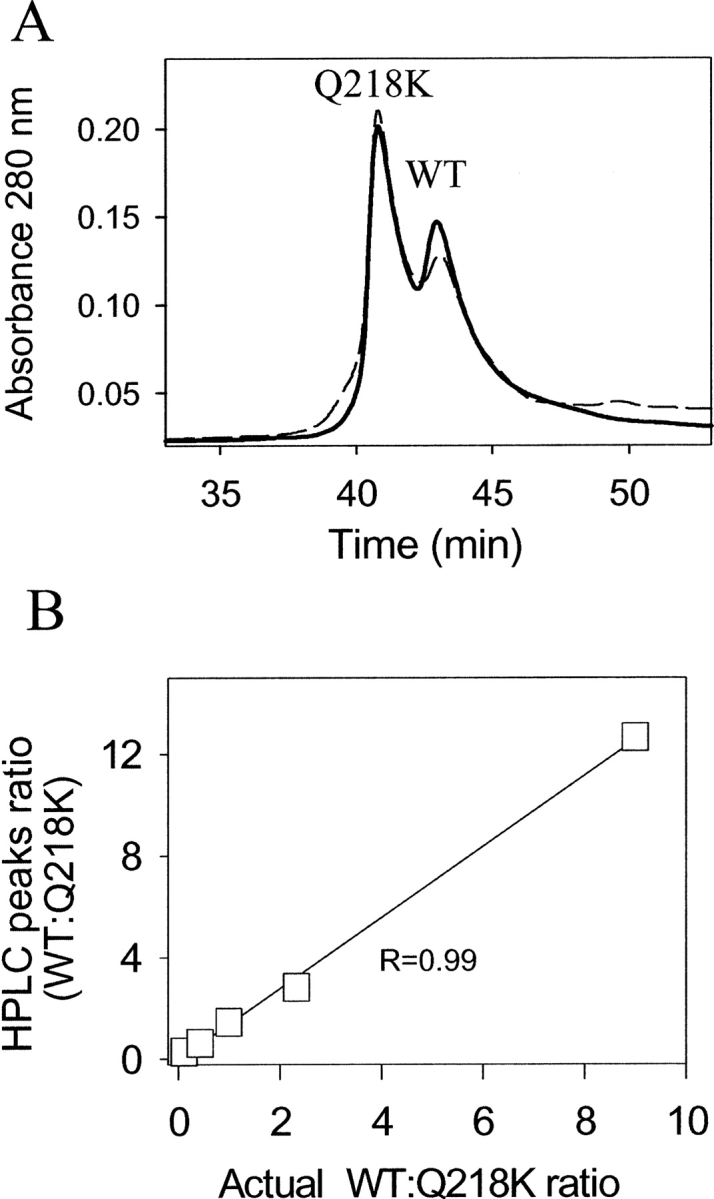
HPLC analysis of fibril composition. (A) Examples of HPLC profiles obtained for monomeric WT and Q218K rPrPs mixed at the ratio of 3:7 (solid line) and for fibrils generated in the reaction mixtures containing WT and Q218K rPrPs at a ratio of 1:2 and denatured with GdnHCl (dashed line). Q218K was eluted in the first peak and WT in the second peak. (B) Calibration curve representing the ratio of HPLC peaks recorded for the mixtures of monomeric WT and Q218K rPrPs of known concentrations plotted versus actual ratio of WT and Q218K rPrPs.
Table 1.
HPLC analysis of fibrillar composition

Fibrils formed from WT, Q218K, or in a mixture of WT and Q218K rPrPs show similar substructure
Next, we were interested in knowing whether incorporation of the Q218K variant had any effect on fibrillar substructure. To address this question, we evaluated fibrillar secondary structure using FTIR, fibrillar morphology using AFM, and conformational stability in GdnSCN-induced denaturation experiments. The FTIR spectra revealed that the fibrils produced from WT, Q218K, or WT:Q218K rPrP mixtures had very similar secondary structure (Fig. 4). As judged from the secondary derivatives of FTIR spectra, the positions of the two major peaks that accounted for the intermolecular β-sheets (at 1626 cm−1) and for β-turns (at 1662 cm−1) was identical regardless of the composition of fibrils. The minor peak at 1613 cm−1 that corresponded to the β-sheets slightly shifted by 1 or 2 cm−1 in fibrils prepared from mixtures of WT and Q218K rPrPs or from Q218K rPrP alone. This slight shift could be due to the presence of nonfibrillar or protofibrillar species in samples containing the Q218K variant. Overall, the FTIR spectroscopy revealed that the amyloid fibrils prepared from WT, Q218K, or from WT:Q218K rPrP mixtures had very similar, if not identical, secondary structure.
Figure 4.
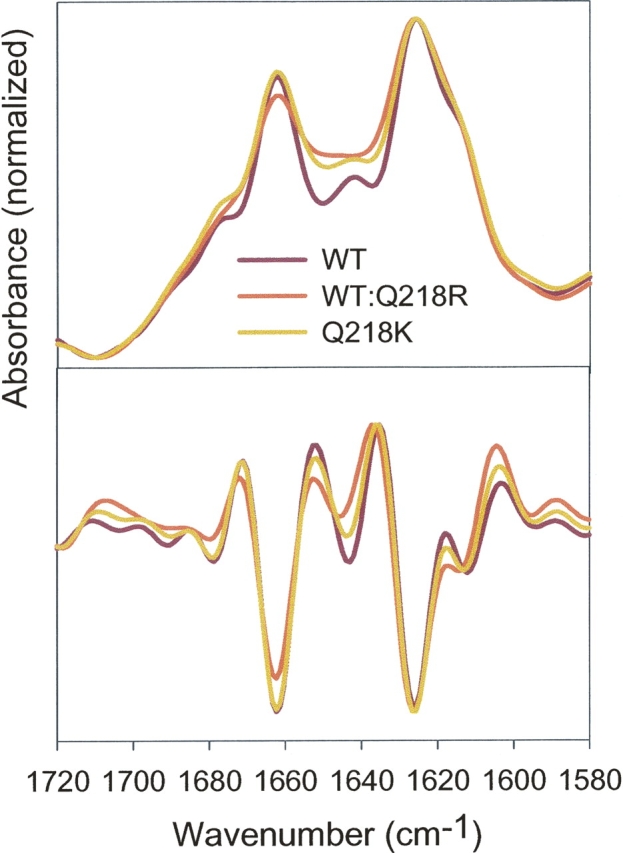
FTIR spectra (top panel) and their secondary derivatives (bottom panel) obtained for fibrils produced from WT, Q218K, or a 1:1 mixture of WT and Q218K rPrPs.
AFM images collected at the late stages of fibrillization showed the presence of fibrils in samples produced from WT, Q218K, or WT:Q218K rPrP mixtures (Fig. 5). The fibrils were morphologically similar regardless of whether the Q218K variant was present in the conversion reaction or not. The Q218K variant alone formed fibrils that were morphologically similar to those of WT rPrP.
Figure 5.
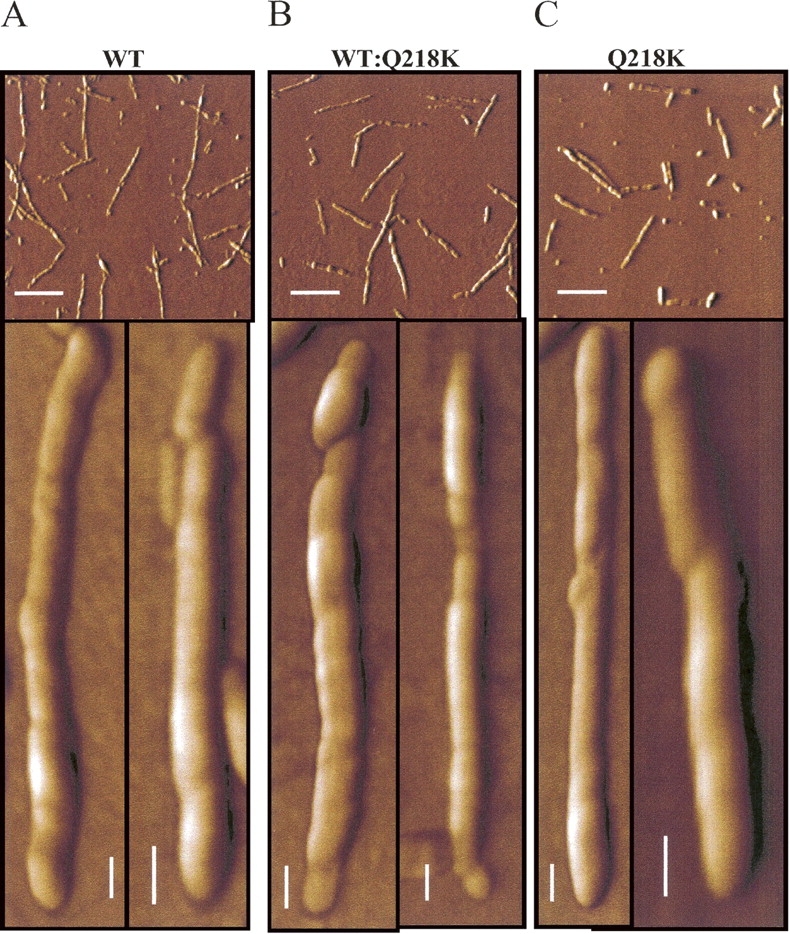
AFM images of fibrils produced from WT rPrP (A), Q218K rPrP (C), and from the 1:1 mixture of WT and Q218K rPrPs (B). To compensate for the different yield of fibrillation observed for different variants, the fibrils were diluted to the final concentrations of 2.5, 10, and 5 μg/mL for WT rPrP, Q218K rPrP, and the WT:Q218K rPrPs mixture, respectively, prior to deposition. Scale bars = 1 μm on the top panels and 0.1 μm on the bottom panels.
To evaluate the conformational stability, fibrils were incubated in the presence of increasing concentrations of GdnSCN (from 0 to 3.5 M) for 1 h, and the amount of intact amyloid structure was analyzed using the ThT-binding assay (see Materials and Methods). The fibrils produced from WT rPrP or from the WT:Q218K rPrP mixture showed very similar denaturation profiles with a C 1/2 of ∼2 M (Fig. 6). This result indicated that incorporation of Q218K rPrP did not substantially change the conformational stability of amyloid fibrils. Owing to the very low yield of fibrillation, we were not able to collect the denaturation profile for Q218K fibrils. Taken together, these studies demonstrated that incorporation of Q218K rPrP into amyloid fibrils did not change the fibrillar substructure; however, it did decrease the yield and the rate of polymerization.
Figure 6.

GndSCN-induced denaturation profiles of the amyloid fibrils produced from WT rPrP (•) and from the 1:1 mixture of WT and Q218K rPrPs (○). Amyloid fibrils were incubated for 1 h at 23°C in the presence of different concentrations of GndSCN. The GndSCN concentration was then adjusted to 0.35 M, followed by a ThT fluorescence assay. The slight increase in ThT fluorescence observed at low concentrations of denaturant was presumably due to GndSCN-induced dissociation of coaggregated amyloid fibrils.
Incorporation of Q218K rPrP blocks maturation of fibrils into PrPSc-like form
Our previous studies revealed that the amyloid fibrils produced in vitro show three PK-resistant products that encompassed residues 138–230, 152–230, and 162–230 and appeared on SDS-PAGE as 12-, 10-, and 8-kDa bands, respectively (Bocharova et al. 2005b). Upon brief heating in the presence of low concentrations of Triton X-100 or brain homogenate (the procedure referred to as “maturation”), amyloid fibrils showed an additional PK-resistant band of 16 kDa that encompassed residues 97–230 (Bocharova et al. 2006). While we do not know the physical mechanisms that account for the extension of the PK-resistant core during maturation, our previously published data indicated that this process may be an important step in the conversion of PrPC into PrPSc. First, we showed that maturation was strictly limited to self-propagating amyloid fibrils; β-sheet-rich oligomers and prefibrillar oligomers failed to undergo maturation (Breydo et al. 2005; Bocharova et al. 2006). Second, upon maturation, rPrP fibrils acquired biochemical properties typical for PrPSc. Therefore, we were interested in testing whether fibrils produced in the mixtures of WT and Q218K variants were capable of undergoing maturation.
To monitor maturation, fibrils were heated for 5 min at 80°C, digested with PK for 1 h at 37°C, and subjected to SDS-PAGE as described in Materials and Methods. While the fibrils produced from the individual solutions of WT or Q218K rPrPs displayed a 16-kDa PK-resistant band, the WT:Q218K fibrils failed to show the 16-kDa band upon maturation (Fig. 7). This result illustrates the differences in the substructure of fibrils produced from the mixtures of rPrP variants and from individual rPrPs.
Figure 7.

PK-digestion assay of amyloid fibrils produced from WT rPrP (lanes 1–3), a 1:1 mixture of WT and Q218K rPrP (lanes 4–6), and Q218K rPrP (lanes 7–9). Fibrils in lanes 3, 6, and 9 were subjected to a maturation procedure (heating for 5 min at 80°C in the presence of 1% Triton X-100). Fibrils in lanes 2, 3, 5, 6, 8, and 9 were treated with PK for 1 h at 37°C at a PK-to-rPrP ratio of 1:50.
Discussion
A previous study identified several single-point PrP mutants that displayed dominant-negative effects in prion replication when coexpressed with WT PrPC (Kaneko et al. 1997). The same study suggested that the dominant-negative effect is mediated by protein X, an as-yet-unidentified cellular cofactor that is essential for conversion of PrPC into PrPSc, and that certain PrP mutants act as “dominant negative” by the binding and sequestering of protein X (Kaneko et al. 1997).
In the present study, we showed that the dominant-negative rPrP variant Q218K inhibited conversion of WT rPrP into the fibrillar form in the absence of a cellular environment, that is, in the absence of protein X. Specifically, we found that Q218K rPrP decreased the yield and slightly delayed the lag phase of polymerization. Q218K rPrP was shown to copolymerize together with WT PrP, and the amounts of WT and Q218K rPrPs incorporated into fibrils were found to be proportional to the amounts used in the reaction mixtures. Therefore, the negative effect of Q218K rPrP on polymerization of WT rPrP seemed to occur via the slowing down of the polymerization rate rather than binding to and blocking the growing fibrillar edges. AFM, FTIR, and conformational stability studies did not reveal substantial differences in the substructure of fibrils formed from WT rPrP, Q218K rPrP, or from their mixtures. While the aforementioned techniques indicated similar physical properties regardless of fibrillar composition, the maturation assay revealed fundamental differences in the substructure of fibrils formed in the mixtures of WT and Q218K rPrPs. Upon maturation, WT:Q218K fibrils did not acquire the PK-resistant core of 16 kDa that was typically observed for WT fibrils. The 16-kDa PK-resistant core was shown previously to encompass residues 97–230 and, therefore, is similar to that of PrPSc (Bocharova et al. 2006). Surprisingly, the Q218K substitution interfered with the ability of the central region of ∼97–140 to acquire the PK-resistant conformation in the fibrils composed of WT and Q218K rPrPs. It is noteworthy that fibrils made of Q218K rPrP were able to produce a 16-kDa band in the maturation assay. These data illustrate that the substructure of “hybrid” fibrils produced in the mixtures of WT and Q218K rPrPs was fundamentally different.
Our observation that Q218K rPrP inhibited fibril formation from WT rPrP was consistent with previous studies, where Q218K PrPC was found to inhibit formation of WT PrPSc and delay the prion disease in transgenic mice infected with prions (Perrier et al. 2002). Q218K PrPC retained the ability to block formation of PrPSc when coexpressed together with WT PrPC in cultured cells (Kaneko et al. 1997; Crozet et al. 2004). Remarkably, recent studies demonstrated that recombinant Q218K PrP inhibited PrPSc replication in a highly efficient manner, when supplemented in cultured cells, despite the lack of glycosylphosphatidylinositol and carbohydrates (50% effective concentration, EC50 = 0.2 μM) (Kishida et al. 2004). Previous and current studies indicate that Q218K PrPC displayed the dominant-negative effect regardless of whether it was expressed in animals or in cultured cells, supplemented in cultured cells in the form of recombinant PrP, or used in cell-free conversion reactions. When considered together, these studies suggest that the single-point mutation Q218K is essential and sufficient for the dominant-negative effect.
What is the possible mechanism that accounts for the dominant-negative role of Q218K rPrP and its low amyloidogenic propensity? As judged from the temperature denaturation experiments, WT and Q218K α-PrPs displayed very similar conformational stability (Fig. 1). Therefore, the low propensity of Q218K rPrP to form fibrils cannot be explained by the hypothesis that predicts low amyloidogenic propensity for the variants with a highly stable native state. The residue 218 is located within helix C in the folded, α-helical domain of PrP. As suggested by the molecular dynamics simulations, the residues 184–223 have a high tendency to undergo a transition from α-helical to a β− or random-coil state and, therefore, might be involved in the initial stages of the conversion from PrPC to PrPSc (Dima and Thirumalai 2004). Consistent with the simulation results, our recent kinetics studies showed that the C-terminal part encompassing residues 196–230 was involved in the early stages of polymerization (Sun et al. 2007). Furthermore, in amyloid fibrils produced in vitro, residue 218 is located within the PK-resistant cross β-sheet core. The fact that Q218K significantly decreased the yield and delayed the lag phase of fibrillization suggests that the additional positive charge in the C-terminal domain at position 218 interfered with the early stages of the transition from the α-helical conformation to a β-sheet-rich form.
The study on transgenic mice overexpressing Q218K PrPC on an ablated background revealed that spontaneous neurologic dysfunction develops at the late stage of the animal's life even in the absence of WT PrPC. Consistent with these results, we observed that in the absence of WT rPrP, Q218K rPrP formed amyloid fibrils, however, with much lower yield and only after a prolonged incubation time (Fig. 2). These parallels observed between prion polymerization in vitro and in transgenic animals emphasize that biological outcomes are determined to a large extent by the intrinsic physical properties of PrP variants involved in prion replication. Taken together, our results were consistent with previous observations and support a dominant-negative role of Q218K in prion replication. However, our study suggests that the underlying mechanism of the dominant-negative effect does not require protein X.
Materials and Methods
Expression and purification of wild-type and Q218K rPrP
Full-length mouse WT and Q218K rPrP variants encompassing residues 23–230 were expressed and purified as described earlier (Bocharova et al. 2005a; Breydo et al. 2005). The purified rPrPs were confirmed by SDS-PAGE and electrospray mass spectrometry to be a single species with an intact disulfide bond and correct molecular weight.
CD spectra and thermal denaturation scans
CD studies were performed using a J-810 CD spectrometer (Jasco) equipped with a temperature-controlled water-circulated quartz cell (1 mm pathlength). CD spectra of WT or Q218K rPrP (0.2 mg/mL) were collected in 1 mM HEPES (pH 7). Each spectrum represents the average of five individual scans. Thermal denaturation was performed using rPrP variants (0.25 mg/mL) prepared freshly in 50 mM MES buffer (pH 6) in the presence or absence of 0.5 M GdnHCl by monitoring the ellipticity at 222 nm with a scan rate of 1.0°C/min.
Conversion of rPrP into amyloid fibrils
Upon purification, WT and Q218K were stored in lyophilized form at −20°C. To prepare stock solutions, rPrP variants were dissolved in 50 mM MES (pH 6) prior to experiments. To form amyloid fibrils, rPrP variants were incubated individually at a concentration of 10 μM or as mixtures at a total protein concentration of 20 μM in 50 mM MES (pH 6) at 37°C in the presence of 2 M GdnHCl upon continuous shaking at 600 rpm as described previously (Baskakov and Bocharova 2005). The aliquots collected during the time course of fibril formation were diluted with 10 mM Na-acetate (pH 5), then Thioflavine T was added to the total concentration of 10 μM, and ThT fluorescence was measured as described previously (Baskakov 2004).
Maturation assay and PK-digestion assay
Tris-HCl (pH 7.5) and Triton X-100 were added to fibrils to final concentrations of 100 mM and 1%, respectively. Samples were heated in a water bath for 5 min at 80°C, cooled down, and incubated with PK at a PK-to-rPrP ratio of 1:50 for 1 h at 37°C. After PK digestion, samples were analyzed in pre-cast 12% SDS-PAGE (Invitrogen).
Atomic force microscopy (AFM)
The samples were imaged with a PicoSPM LE AFM (Molecular Imaging), operating in the AAC (acoustic alternative current) AFM mode and using a silicon cantilever PPP-NCH (Nanoscience) with a tip radius <7 nm and a spring constant of ∼42 N/m. rPrP fibrils (10 μL) were deposited onto 25 mm × 25-mm glass coverslips (Fisher). The coverslips were cleaned with 30% HNO3 and rinsed extensively with distilled H2O before use. After deposition, samples were washed with distilled H2O for 10 min and dried with nitrogen. The images (512 × 512 pixel scans) were collected at a scan rate of 1–2 lines/sec.
FTIR spectroscopy
FTIR spectra were measured with a Bruker Tensor 27 FTIR instrument (Bruker Optics) equipped with an MCT detector cooled with liquid nitrogen. Fibrils were dialyzed against 10 mM Na-acetate (pH 5.0); 20 μL of each sample was loaded into a BioATR II cell. A total of 1024 scans at 2 cm−1 resolution were collected for each sample under constant purging with nitrogen. Spectra were corrected for water vapor, and background spectra of the same buffer were subtracted. The bands were resolved by Fourier self-deconvolution in the Opus 4.2 software package using a Lorentzian line shape and parameters equivalent to 30 cm−1 bandwidth at half height and a noise suppression factor of 0.2.
HPLC analysis of fibril composition
The fibrils produced in the mixtures of WT and Q218K rPrPs were spun down at 15,400g for 1.5 h, supernatant was removed, and the pellets were incubated overnight with 8 M GdnHCl to denature fibrils. After fibril denaturation, GdnHCl was adjusted to the final concentration of 6 M, and protein composition was analyzed by HPLC (Symmetry 300 C4, 4.6 × 250 mm; Waters Corp.). The eluant gradient consisted of 0%–25% acetonitrile in water for 15 min, followed by 25%–35% acetonitrile in water for >65 min; the flow rate was 0.8 mL/min, and the eluant contained 0.1% TFA. To build a calibration curve, WT and Q218K rPrPs were mixed at different ratios in 6 M GdnHCl and analyzed by HPLC. The heights of the HPLC peaks corresponding to WT and Q218K rPrPs were measured, and their ratio was plotted versus the ratio of actual amounts of WT and Q218K used for the calibration experiment.
GdnSCN-induced denaturation of amyloid fibrils
rPrP fibrils (10 μL) were mixed with 100 mM MES (pH 6.0; 30 μL) containing various concentrations of GdnSCN (0–3.5 M), incubated for 1 h at 23°C, and diluted with 100 mM MES (pH 6.0) and GdnSCN to the final volume of 300 μL and final GdnSCN concentration of 0.35 M. ThT was added to a final concentration of 10 μM, and ThT fluorescence spectra were collected as described previously.
Acknowledgments
We thank Pamela Wright for editing the manuscript. This work was supported by the Program in Prion Diseases at the Medical Biotechnology Center, University of Maryland Biotechnology Institute.
Footnotes
Reprint requests to: Ilia V. Baskakov, 725 West Lombard Street, Baltimore, MD 21201, USA; e-mail: Baskakov@umbi.umd.edu; fax: (410) 706-8184.
Abbreviations: PrP, prion protein; PrPC, normal cellular isoform of the prion protein; PrPSc, abnormal pathological isoform of the prion protein; rPrP, full-length recombinant prion protein; WT, wild type; FTIR, Fourier transform infrared spectroscopy; ThT, Thioflavin T; GdnHCl, guanidine hydrochloride; GdnSCN, guanidine thiocyanate.
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.072954607.
References
- Baskakov I.V. 2004. Autocatalytic conversion of recombinant prion proteins displays a species barrier. J. Biol. Chem. 279: 586–595. [DOI] [PubMed] [Google Scholar]
- Baskakov I.V. and Bocharova, O.V. 2005. In vitro conversion of mammalian prion protein into amyloid fibrils displays unusual features. Biochemistry 44: 2339–2348. [DOI] [PubMed] [Google Scholar]
- Bocharova O.V., Breydo, L., Parfenov, A.S., Salnikov, V.V., and Baskakov, I.V. 2005a. In vitro conversion of full length mammalian prion protein produces amyloid form with physical property of PrPSc. J. Mol. Biol. 346: 645–659. [DOI] [PubMed] [Google Scholar]
- Bocharova O.V., Breydo, L., Salnikov, V.V., Gill, A.C., and Baskakov, I.V. 2005b. Synthetic prions generated in vitro are similar to a newly identified subpopulation of PrPSc from sporadic Creutzfeldt-Jakob Disease PrPSc . Protein Sci. 14: 1222–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bocharova O.V., Makarava, N., Breydo, L., Anderson, M., Salnikov, V.V., and Baskakov, I.V. 2006. Annealing PrP amyloid firbils at high temperature results in extension of a proteinase K-resistant core. J. Biol. Chem. 281: 2373–2379. [DOI] [PubMed] [Google Scholar]
- Breydo L., Bocharova, O.V., Makarava, N., Salnikov, V.V., Anderson, M., and Baskakov, I.V. 2005. Methionine oxidation interferes with conversion of the prion protein into the fibrillar proteinase K-resistant conformation. Biochemistry 44: 15534–15543. [DOI] [PubMed] [Google Scholar]
- Caughey W.S., Raymond, L.D., Horiuchi, M., and Caughey, B. 1998. Inhibition of protease-resistant prion protein formation by porphyrins and phthalocyanines. Proc. Natl. Acad. Sci. 95: 12117–12122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crozet C., Lin, Y.L., Metting, C., Mourton-Gilles, C., Corbeau, P., Lehmann, S., and Perrier, V. 2004. Inhibition of PrPSc formation by lentiviral gene transfer of PrP containing dominant negative mutations. J. Cell Sci. 117: 5591–5597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dima R.I. and Thirumalai, D. 2004. Probing the instabilities in the dynamics of helical fragments from mouse PrPC. Proc. Natl. Acad. Sci. 101: 15335–15340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter N., Moore, L., Hosie, B.D., Dingwall, W.S., and Greig, A. 1997. Association between natural scrapie and PrP genotype in a flock of Suffolk sheep in Scotland. Vet. Rec. 140: 59–63. [DOI] [PubMed] [Google Scholar]
- Ikeda T., Horiuchi, M., Ishiguro, N., Muramatsu, Y., Kai-Uwe, G.D., and Shinagawa, M. 1995. Amino acid polymorphisms of PrP with reference to onset of scrapie in Suffolk and Corriedale sheep in Japan. J. Gen. Virol. 76: 2577–2581. [DOI] [PubMed] [Google Scholar]
- Kaneko K., Zulianello, L., Scott, M., Cooper, C.M., Wallace, A.C., James, T.L., Cohen, F.E., and Prusiner, S.B. 1997. Evidence for protein X binding to a discontinuous epitope on the cellular prion protein during scrapie prion propagation. Proc. Natl. Acad. Sci. 94: 10069–10074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishida H., Sakasegawa, Y., Watanabe, K., Yamakawa, Y., Nishijima, M., Kuroiwa, Y., Hachiya, N.S., and Kaneko, K. 2004. Non-glycosylphosphatidylinositol (GPI)-anchored recombinant prion protein with dominant-negative mutation inhibits PrPSc replication in vitro. Amyloid 11: 14–20. [DOI] [PubMed] [Google Scholar]
- Korth C., May, B.C., Cohen, F.E., and Prusiner, S.B. 2001. Acridine and phenothiazine derivatives as pharmacotherapeutics for prion disease. Proc. Natl. Acad. Sci. 98: 9836–9841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrier V., Wallace, A.C., Kaneko, K., Safar, J., Prusiner, S.B., and Cohen, F.E. 2000. Mimicking dominant negative inhibition of prion replication through structure-based drug design. Proc. Natl. Acad. Sci. 97: 6073–6078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrier V., Kaneko, K., Safar, J., Vergara, J., Tremblay, P., DeArmond, S.J., Cohen, F.E., Prusiner, S.B., and Wallace, A.C. 2002. Dominant-negative inhibition of prion replication in transgenic mice. Proc. Acad. Natl. Sci. 99: 13079–13084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prusiner S.B. 1998. Prions (Les Prix Nobel Lecture). In Les Prix Nobel (ed. T. Frängsmyr), pp. 268–323. Almqvist and Wiksell International, Stockholm, Sweden.
- Shibuya S., Higuchi, J., Shin, R.-W., Tateishi, J., and Kitamoto, T. 1998a. Codon 219 Lys allele of PRNP is not found in sporadic Creutzfeldt-Jakob disease. Ann. Neurol. 43: 826–828. [DOI] [PubMed] [Google Scholar]
- Shibuya S., Higuchi, J., Shin, R.-W., Tateishi, J., and Kitamoto, T. 1998b. Protective prion protein polymorphisms against sporadic Creutzfeldt-Jakob disease. Lancet 351: 419. doi: 10.1016/S0140-6736(05)78358-6. [DOI] [PubMed] [Google Scholar]
- Sun Y., Breydo, L., Makarava, N., Yang, Q., Bocharova, O.V., and Baskakov, I.V. 2007. Site-specific conformational studies of PrP amyloid fibrils revealed two cooperative folding domain within amyloid structure. J. Biol. Chem. 282: 9090–9097. [DOI] [PubMed] [Google Scholar]
- Supattapone S., Nguyen, H.-O.B., Cohen, F.E., Prusiner, S.B., and Scott, M.R. 1999. Elimination of prions by branched polyamines and implications for therapeutics. Proc. Natl. Acad. Sci. 96: 14529–14534. [DOI] [PMC free article] [PubMed] [Google Scholar]