

Abstract
Many human proteins contain consecutive amino acid repeats, known as homopolymeric amino acid (HPAA) tracts. Some inherited diseases are caused by proteins in which HPAAs are expanded to an excessive length. To this day, nine polyglutamine-related diseases and nine polyalanine-related diseases have been reported, including Huntington's disease and oculopharyngeal muscular dystrophy. In this study, potential HPAA–HPAA interactions were examined by yeast two-hybrid assays using HPAAs of ∼30 residues in length. The results indicate that hydrophobic HPAAs interact with themselves and with other hydrophobic HPAAs. Previously, we reported that hydrophobic HPAAs formed large aggregates in COS-7 cells. Here, those HPAAs were shown to have significant interactions with each other, suggesting that hydrophobicity plays an important role in aggregation. Among the observed HPAA–HPAA interactions, the Ala28–Ala29 interaction was notable because polyalanine tracts of these lengths have been established to be pathogenic in several polyalanine-related diseases. By testing several constructs of different lengths, we clarified that polyalanine self-interacts at longer lengths (>23 residues) but not at shorter lengths (six to ∼23 residues) in a yeast two-hybrid assay and a GST pulldown assay. This self-interaction was found to be SDS sensitive in SDS-PAGE and native-PAGE assays. Moreover, the intracellular localization of these long polyalanine tracts was also observed to be disturbed. Our results suggest that long tracts of polyalanine acquire SDS-sensitive self-association properties, which may be a prerequisite event for their abnormal folding. The misfolding of these tracts is thought to be a common molecular aspect underlying the pathogenesis of polyalanine-related diseases.
Keywords: polyalanine, polyglutamine, triplet repeat, aggregate
Many human proteins contain homopolymeric amino acid (HPAA) repeats, and different HPAA species are identified in an increasing number of proteins (Karlin et al. 2002a; Alba and Guigo 2004; Faux et al. 2005). Each HPAA possesses characteristic properties inherently attributable to the nature of the repeated amino acid. Based on an analysis of 20 different HPAA types, we previously reported that different HPAAs present wide-ranging characteristic intracellular localizations (Oma et al. 2004). Hydrophobic HPAAs exhibit a strong tendency toward aggregation and produce severe cytotoxic effects when expressed in mammalian cells (Oma et al. 2005); consequently, hydrophobic HPAA tracts are rarely observed in natural proteins (Dorsman et al. 2002; Oma et al. 2004, 2005). In contrast, the less toxic HPAA tracts such as polyalanine, polyglutamine, polyproline, polyserine, and polyglycine are relatively abundant, especially among transcription factors (Karlin et al. 2002a; Cocquet et al. 2003; Lavoie et al. 2003; Alba and Guigo 2004; Faux et al. 2005).
Some HPAA-containing proteins become pathological when the HPAA tract expands beyond a certain length. Nine polyglutamine-related diseases and nine polyalanine-related diseases have been reported (Amiel et al. 2004; Brown and Brown 2004; Albrecht et al. 2004; Gatchel and Zoghbi 2005). Although these two HPAA-expanded diseases are different in several ways, the expanded polyalanine and polyglutamine tracts both result in protein misfolding. The pathological length threshold differs between polyglutamine (38–40 residues) and polyalanine tracts (about 20 residues). Polyglutamine-related disorders, including Huntington's disease, are all neurodegenerative conditions caused by the expansion of a polyglutamine tract, but the expansions are found in functionally unrelated proteins (Gatchel and Zoghbi 2005). Proteins containing expanded polyglutamine tracts lead to the appearance of intracellular aggregates in the central nervous system of patients and disease-model animals (Davies et al. 1997; Michalik and Van Broeckhoven 2003). The misfolding of the expanded polyalanine tract of poly(A)-binding protein nuclear 1 (PABPN1) was shown to cause oculopharyngeal muscular dystrophy (OPMD), which is the only polyalanine disorder caused by an expansion in a protein that is not a transcription factor (Brais 2003). Intranuclear protein aggregates were observed in skeletal muscle fibers of patients with OPMD (Becher et al. 2000; Calado et al. 2000; Shanmugam et al. 2000; Uyama et al. 2000), and the expression of an expanded-polyalanine version of PABPN1 resulted in aggregate formation in cultured cells (Fan et al. 2001; Bao et al. 2002; Abu-Baker et al. 2003; Wang et al. 2005; Verheesen et al. 2006) and in animal models (Hino et al. 2004; Davies et al. 2005; Dion et al. 2005; Chartier et al. 2006). Polyalanine tracts containing 15–20 residues, but not those with three to eight residues, spontaneously form β-sheets in vitro (Shinchuk et al. 2005), and polyglutamine-containing peptides have also been shown to form such structures in vitro and in vivo (Scherzinger et al. 1997; Huang et al. 1998). Except for OPMD, all polyalanine-related conditions involve abnormal development caused by the expansion of a polyalanine tract in a transcription factor (Amiel et al. 2004; Brown and Brown 2004; Albrecht and Mundlos 2005). It was recently reported that expression of the different disease-related polyalanine-containing transcription factors in mammalian cells results in their cytoplasmic and/or nuclear aggregation (Albrecht et al. 2004; Caburet et al. 2004; Nasrallah et al. 2004; Bachetti et al. 2005; Brown et al. 2005), which disrupts their normal function and may cause the transcriptional dysregulation. However, the molecular mechanisms causing the mislocalization of the polyalanine protein is thus far unknown. In addition, it was reported by us and others that long polyalanine tracts are toxic to cells, which may also contribute to pathogenesis in polyalanine-related diseases (Rankin et al. 2000; Oma et al. 2005).
HPAA tracts have been used in protein engineering to construct functional peptides. For example, polyarginine-containing peptides serve as a drug-delivery system owing to their ability to enter cells (Tung and Weissleder 2003), six-residue polyhistidine tags exhibit a metal-binding activity widely exploited for purifying recombinant proteins, and polylysine provides a hydrophilic coating for the surfaces of culture dishes, facilitating cell adhesion and proliferation. Therefore, the HPAAs are useful peptides that can be used in multiple applications. However, no study so far has investigated all of the possible interactions that might occur between HPAAs of the same or of different amino acids.
As repetitive stretches of identical amino acids, HPAAs can adopt characteristic conformations with significant effects on protein–protein interactions. An examination of HPAA–HPAA interactions may reveal novel physiological interactions as well as interactions between rare HPAAs. Here, we examined HPAA–HPAA interactions using yeast two-hybrid assays with HPAAs of ∼30 residues in length. Among the novel interactions identified was the self-interaction of polyalanine tracts. We report a drastic structural alteration of the expanded polyalanine protein and a self-association likely to contribute to the formation of dimers or multimers. This is the first report comparing HPAA–HPAA interactions and demonstrating the length dependency of the self-interaction of polyalanine-containing proteins.
Results
Hydrophobic homopolymeric amino acid tracts interact with each other
To test for interactions between HPAAs, all 20 possible HPAA proteins, we performed yeast two-hybrid assays. Protein–protein interactions were detected by the activity of reporter gene HIS3, histidine sythase 3. The strength of the interaction was estimated using increasing concentrations of 3-AT (0–40 mM), a competitive inhibitor of histidine synthetic enzymes, in the SD/−Leu/−Trp/−His medium for the assay plates. Although this method was not quantitative, the survival of the yeast on plates with higher 3-AT concentration implied a stronger interaction. We first conducted control experiments in which HPAA–AD (activation domain) fusion genes were cotransformed with the nonfusion BD (binding domain) gene or HPAA–BD fusions were cotransformed with the nonfusion AD. This was necessary to establish the level of 3-AT resistance mediated by individual HPAAs in a protein–protein interaction-independent manner. We next examined combinations of HPAA–AD fusions and HPAA–BD fusions. For several combinations, the yeast transformants survived at higher 3-AT concentration than in corresponding controls, representing the interaction between the HPAAs (Fig. 1A). Many hydrophobic HPAAs interacted with themselves and/or with other hydrophobic HPAAs, except for the highly hydrophobic Val28 and Val29 proteins, which showed no apparent interaction. Other types of interactions were also detected, including polycysteine with polyhistidine and polyasparagine with polyasparagine. However, some interactions could not be reproduced when the prey and bait were exchanged. For example, BD–Leu28 and AD–Pro30 interacted, but BD–Pro27 and AD–Leu30 did not. These interactions may represent either weak or false positive interactions. Figure 1B summarizes the different HPAA interactions. The interactions that were detected even when the bait and prey were exchanged are shown in color.
Figure 1.
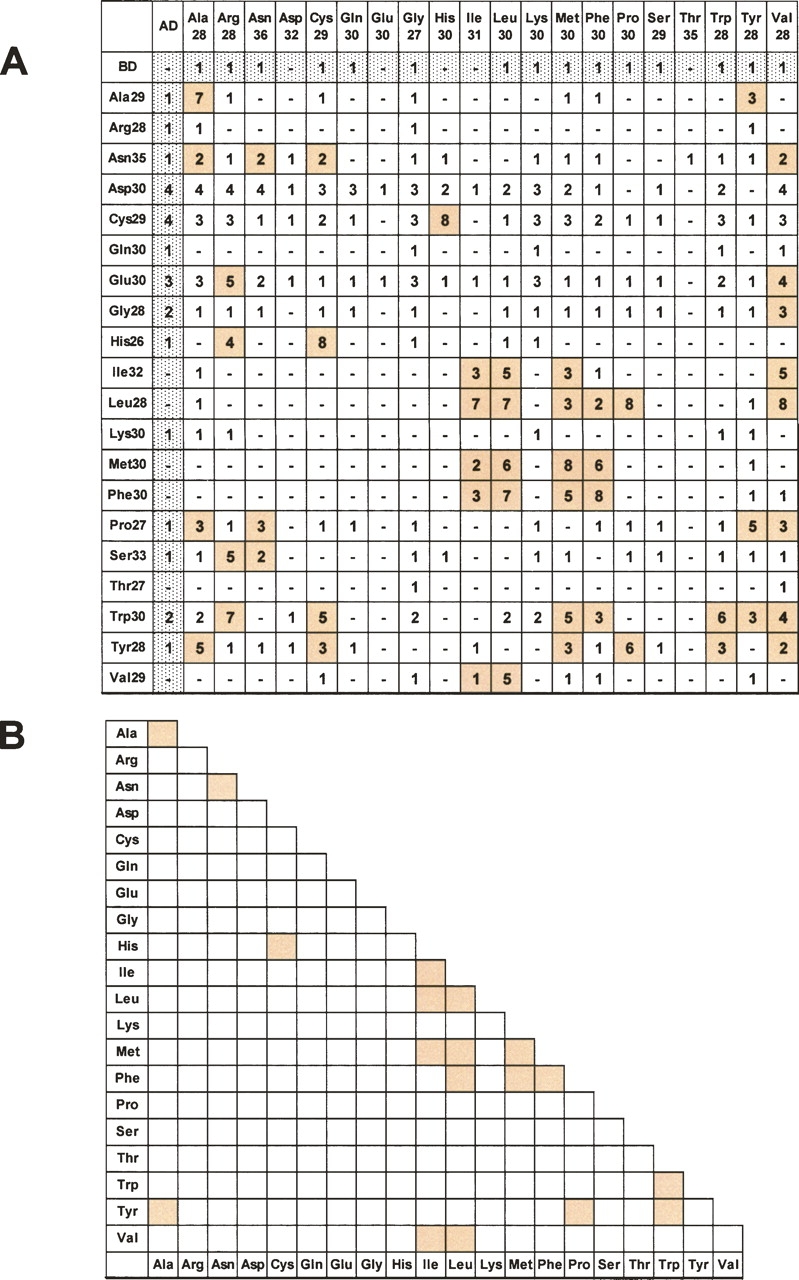
HPAA–HPAA interactions examined by yeast two-hybrid assay. Yeast two-hybrid assays were performed with all 20 possible HPAA types (each ∼30 residues in length), with each HPAA fused to a DNA-binding domain (BD) and to an activation domain (AD). The binding strength was assigned according to the 3-AT concentration, ranging from 1 (without 3-AT) to 8 (with 40 mM 3-AT). (A) Interactions stronger than the control value (shown in gray) are indicated by colored columns. (B) Summary of HPAA interactions. Clearly detected interactions are colored.
We confirmed the yeast two-hybrid positive interactions by using GST pulldown assays. GST-fused HPAAs ∼30 residues in length were expressed, purified from Escherichia coli, and mixed with a cell lysate from COS-7 cells expressing YFP-fused HPAAs ∼30 residues in length. Interacting proteins were detected on Western blots using an anti-YFP antibody. The following interactions were confirmed by GST pulldown assays: Ala35 with Ala30, Asn35 with Asn35, Ala35 with Tyr28, and Cys29 with His26 (Fig. 2A–C). We were unable to examine the hydrophobic HPAA interactions using GST–Leu30 versus each hydrophobic YFP–HPAA, because the expression and purification of GST–Leu30 were hampered by a low-expression level and insolubility of the recombinant protein. These properties of GST–Leu30 are consistent with our previous finding that hydrophobic HPAAs such as polyleucine form aggregates and are highly cytotoxic for mammalian cultured cells (Oma et al. 2004, 2005).
Figure 2.
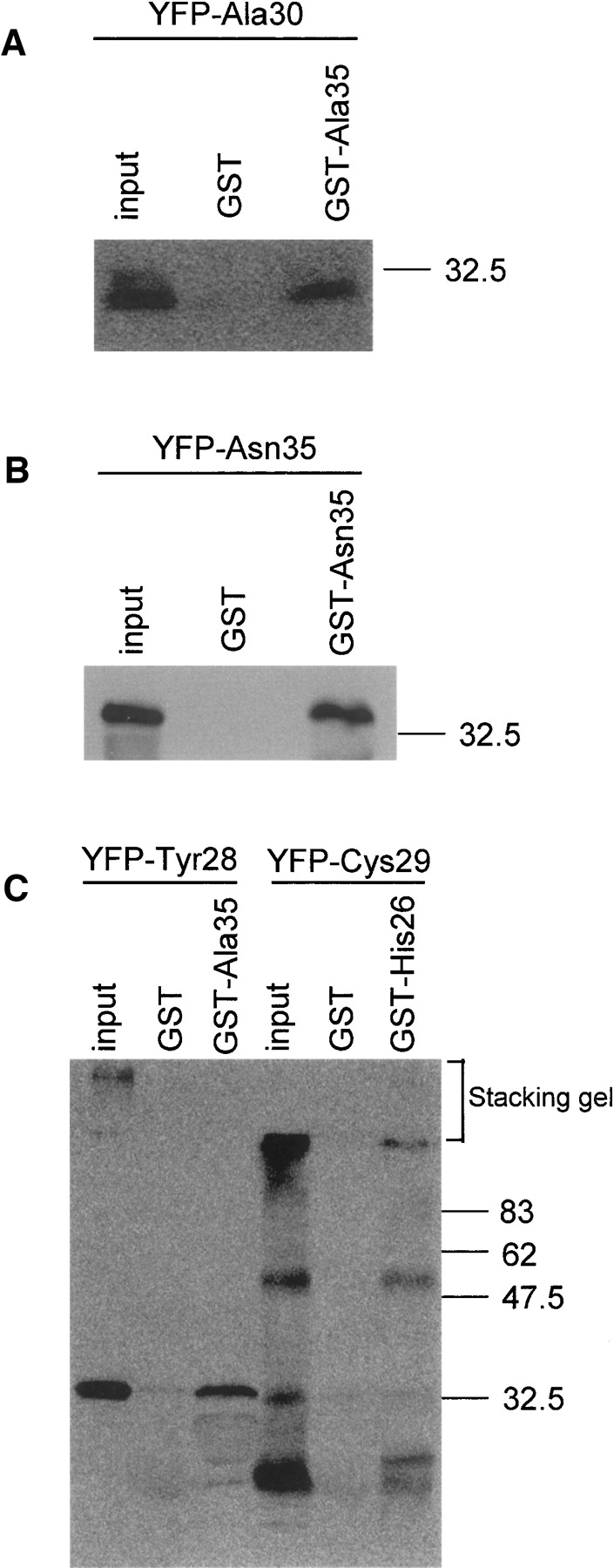
HPAA–HPAA interactions confirmed by GST pulldown assays. GST pulldown assays were performed using GST-fused HPAAs, ∼30 residues in length, expressed in E. coli, and YFP-fused HPAAs, ∼30 residues in length, expressed in COS-7 cells. Interactions between the following proteins were detected on Western blots using anti-YFP antibody: (A) Ala35 with Ala30, (B) Asn35 with Asn35, and (C) Ala35 with Tyr28, and Cys29 with His26.
Self-interaction of polyalanine is length dependent
Among the observed HPAA–HPAA interactions, the Ala28–Ala29 interaction was notable because several human diseases are caused by expanded polyalanine tracts of the length used in this study, 28–29 residues. To assess the effect of tract length on the interaction, we created polyalanine expression vectors with six and 18 repeated residues and performed a yeast two-hybrid assay. The results showed that the polyalanine interaction was length dependent. Long polyalanine (Ala29, Ala28) interacted with Ala18 and Ala28–Ala29, whereas Ala18 did not interact with Ala18 itself (Fig. 3A). Similar results were obtained by performing GST pulldown assay, in which interactions between GST–Ala35 and polyalanine tracts of variable lengths were analyzed. YFP–Ala29, YFP–Ala30, YFP–Ala35, and YFP–Ala70 showed positive interactions with GST–Ala35, whereas YFP–Ala7, YFP–Ala10, YFP–Ala15, and YFP–Ala23 did not (Fig. 3B,C).
Figure 3.

Length dependency of the polyalanine–polyalanine interaction. A length-dependent polyalanine–polyalanine interaction was detected by both yeast two-hybrid assay (A) and GST pulldown assay (B,C).
Polyalanine changes its properties in a length-dependent manner
To investigate the length-dependent differences between polyalanine-containing proteins, SDS-PAGE and native PAGE were conducted using purified GST-fusion proteins of increasing size (GST–Ala7 to GST–Ala35). Native PAGE is performed without SDS or any reducing agents; therefore, proteins retain their “native” conformation during electrophoresis, while in SDS-PAGE the proteins' conformation is essentially unfolded and linearized. After elution from Sepharose beads, 2.5 μg of each protein was loaded onto a gel, electrophoresed, and stained with Coomassie Brilliant Blue. On SDS-PAGE, all proteins migrated to their expected sizes, indicating that polyalanine tracts containing up to 35 residues did not form SDS-resistant structures in vitro (Fig. 4A). However, in the native gels, a drastic difference was observed between the mobility of GST–Ala23 and GST–Ala29. The GST–Ala29 to GST–Ala35 products were all retained in the upper part of the running gel, whereas the GST–Ala7 to GST–Ala23 products migrated into the running gel (Fig. 4B).
Figure 4.
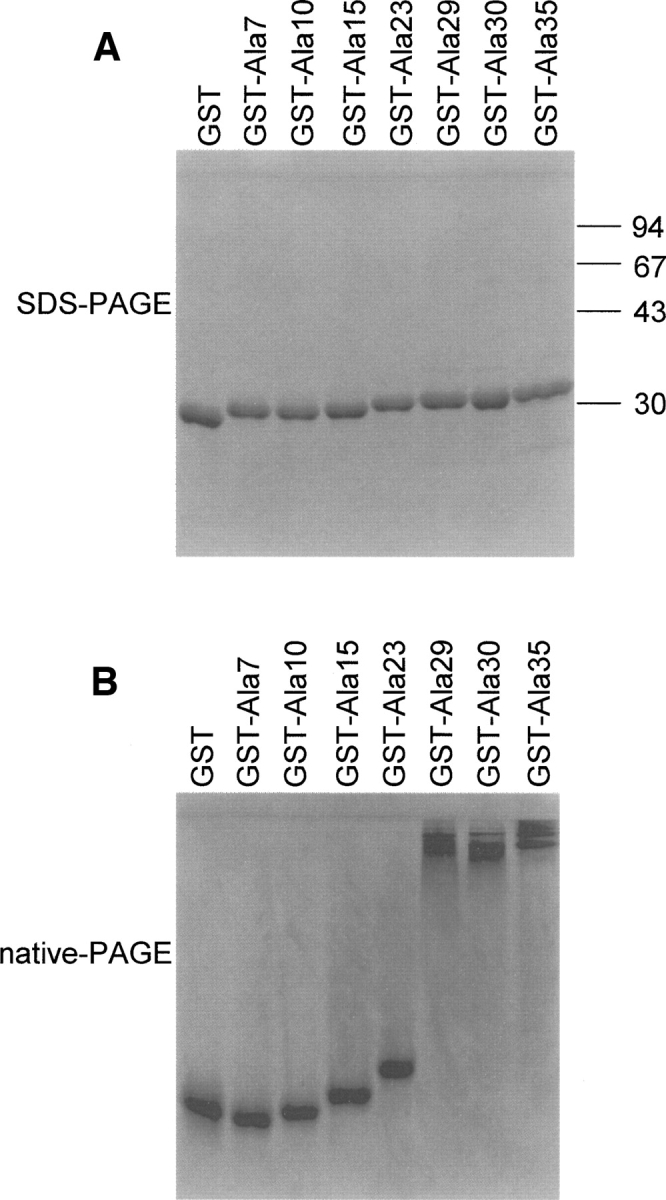
GST–polyalanine alteration. SDS-PAGE (A) and native PAGE (B) were performed using 2.5 μg each of the purified GST–Ala7 to GST–Ala35 fusion proteins. The proteins are stained with Coomassie Brilliant Blue.
Next, we performed SDS-PAGE and native PAGE followed by Western blotting using COS-7 cell lysates transfected with the same series of YFP–polyalanine constructs and an anti-YFP antibody. On SDS-PAGE, YFP–Ala70 was retained in the stacking gel, whereas YFP–Ala7 to YFP–Ala35 migrated to the expected sizes of monomer proteins (Fig. 5A), indicating that the self-interactions of YFP–Ala29 to YFP–Ala35 are not SDS resistant. On native gels, unlike the drastic change in mobility observed for GST–polyalanine (Fig. 4B), gradually increasing amounts of protein were observed in the stacking gel as the tract length increased from YFP–Ala23 to YFP–Ala35 (Fig. 5B). Thus, moderately long polyalanine tracts (Ala23 to Ala35) might form large SDS-sensitive complexes in the cell, and extremely long polyalanine tracts such as Ala70 might form SDS-resistant complexes.
Figure 5.
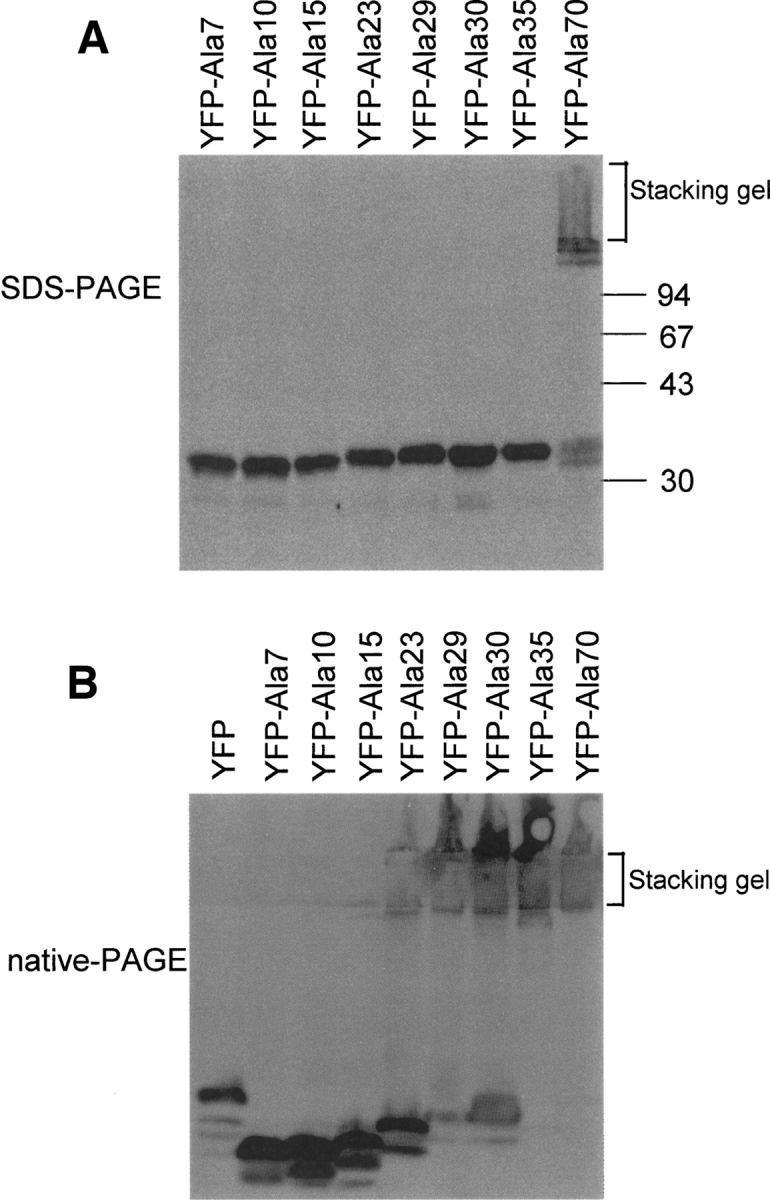
SDS-PAGE and native PAGE of YFP–polyalanine constructs. SDS-PAGE (A) and native PAGE (B) were performed using a series of YFP constructs (YFP–Ala7 to YFP–Ala70) expressed in COS-7 cells. Western blots using anti-YFP antibody reveal that the amount of protein remaining in the stacking gel in native PAGE gradually increased with increasing tract length, from YFP–Ala23 to YFP–Ala35 protein (B).
Cellular localization of polyalanine-containing proteins changes with tract length
To examine how this length-dependent alteration of polyalanine affects its protein localization, the series of YFP–polyalanine constructs were expressed and observed in cultured cells. We previously reported that YFP–Ala29 localizes exclusively in the cytoplasm in COS-7 cells (Oma et al. 2004). In the present study, we examined the localization of polyalanine-containing YFP-fusion proteins of increasing tract lengths (Ala7 to Ala35). The cellular localization of the proteins was observed under a fluorescent microscope 48 h after transfection. Polyalanine repeats of up to 15 residues in length showed the same cytosolic and nuclear pattern of fluorescence as that observed in the control (YFP only), as did 98% of the cells expressing YFP–Ala23. However, 2% of the cells expressing YFP–Ala23 and 100% of the cells expressing YFP–Ala29 to YFP–Ala35 showed exclusively cytoplasmic localization (Fig. 6), suggesting that polyalanine longer than 23 prevents the YFP protein from being transported inside the nucleus.
Figure 6.
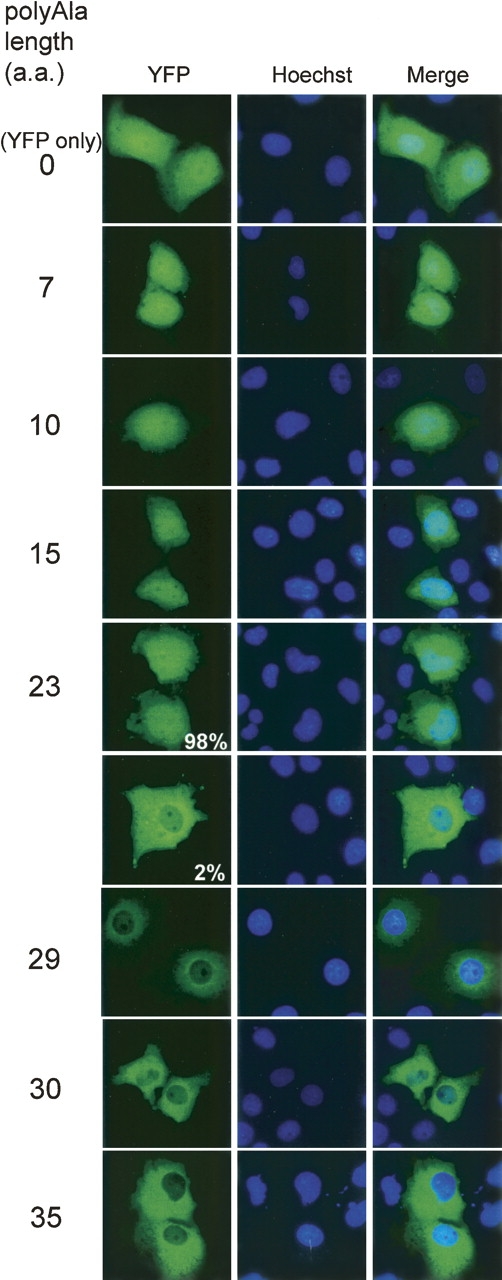
Difference in cellular localization between YFP–polyalanine with 23 and 29 repeats. Cellular localization was observed 48 h after transfection in COS-7 cells. Polyalanine repeats of up to 15 residues in length exhibit the same pattern of fluorescence as that observed in the control (YFP only), and 98% of the cells expressing YFP–Ala23 also show the same control pattern. However, 2% of the cells expressing YFP–Ala23 and 100% of the cells expressing YFP–Ala29 to YFP–Ala35 show exclusively cytoplasmic fluorescence.
Discussion
Using a yeast two-hybrid system, we demonstrated that hydrophobic HPAAs interact with themselves and with other hydrophobic HPAAs. Previously, we reported that highly hydrophobic HPAAs form large aggregates in cultured cells (Oma et al. 2004). Here, these same HPAAs showed significant interactions with themselves, suggesting that self-associations play a role in aggregate formation. In addition, different hydrophobic HPAA species interacted with each other, suggesting that hydrophobicity itself might be the mechanism of protein aggregation.
Other types of HPAA interactions were also found in this study. Polyasparagine interacted with itself in the yeast two-hybrid and pulldown assays (Figs. 1, 2). YFP–Asn35 did not form visible aggregates in mammalian cells (Oma et al. 2004), and long polyAsn stretches are rare, with none exceeding eight residues in human proteins. Nevertheless, the yeast prion proteins Sup35p and Ure2p, which contain Asn/Gln-rich sequences, are known to form aggregates (Tuite 2000), and thus, the self-interaction of polyasparagine might contribute to the process.
Polyglutamine repeats are known to form intracellular aggregates when it is longer than around 38–40 residues (Michalik and Van Broeckhoven 2003). It has been predicted that long stretches of polyglutamine form antiparallel β-strands termed “polar zipper” (Perutz et al. 1994). Expanded polyglutamine tract is also reported to interact with normal length polyglutamine-containing transcription factors (Kazantsev et al. 1999) and with polyglutamine-rich protein (Dunah et al. 2002). However, no interaction has been reported between normal-length polyglutamine tracts, and this is consistent with our two-hybrid results in which no interaction was observed between ∼30 residues of polyglutamine (Fig. 1).
Positively (polyarginine and polylysine) and negatively charged HPAAs (polyaspartic acid and polyglutamic acid) did not interact with each other in the two-hybrid assay. Moreover, no self-interactions of polyarginine and polylysine were detected, even though these HPAAs can form aggregate-like structures in the nucleus of the transfected cells (Oma et al. 2004). There are two possible explanations for this apparent discrepancy. First, the ionic interactions might be too weak to be detected in a two-hybrid assay. Second, the long stretch of positively charged arginine and lysine might have interacted aberrantly with negatively charged nucleic acids in yeast nucleus.
Among the HPAA–HPAA interactions observed in this study, the polyalanine self-interaction is intriguing because the tract lengths used in this study, 28–29 residues, correspond to the pathogenic tract lengths reported for several polyalanine-related diseases. The interaction was length dependent in both the yeast two-hybrid and GST pulldown assays (Fig. 3). Moreover, dimerization or multimerization of the expanded polyalanine were predicted from native PAGE using in vitro-purified GST–polyalanine and cellular expressed YFP–polyalanine (Figs. 4, 5). GST–polyalanine showed a drastic band shift between GST–Ala23 and GST–Ala29, whereas YFP–polyalanine showed a gradual band shift that starts from YFP–Ala23. The observed differences between the purified proteins in vitro and the proteins expressed in the cultured cells may simply be attributable to the difference in the property of YFP and GST. Another possible explanation is that ubiquitination and/or sequestration of some endogenous factors (molecular chaperones, etc.), which occurs in mammalian cells, as reported in several cases of polyglutamine and polyalanine complexes (Albrecht and Mundlos 2005; Gatchel and Zoghbi 2005), contributes to the gradual shift and sequent larger complex formation.
Concomitantly with producing self-association, the expansion of a polyalanine tract affected its intracellular localization. After reaching a threshold of ∼23 repeated residues, YFP–Ala shifted its intracellular localization pattern from diffuse to exclusively cytoplasmic (Fig. 6). Protein transport between the cytoplasm and nucleus occurs via nuclear pore complexes, and small proteins under ∼60 kDa can generally diffuse passively in and out of the nucleus (Gorlich and Kutay 1999). Monomers of YFP–Ala7 to YFP–Ala35 are about 31–33 kDa as seen under denaturing conditions (Fig. 5A). However, under nondenaturing conditions YFP–Ala29, YFP–Ala30, and YFP–Ala35 were shown to form dimers, oligomers, or multimers resulting from their self-association properties, suggesting that these proteins form over 60 kDa complexes in cells, thereby preventing their move into the nucleus. It is also possible that interaction with other cytoplasmic proteins or ubiquitin tagging contributes to the cytoplasmic localization of these proteins. Changes in intracellular localization attributable to expanded polyalanine tracts have been reported in polyalanine-related diseases. HOXD13, RUNX2, HOXA13, SOX3, FOXL2, and PHOX2B, causative proteins of polyalanine-related diseases, are reported to shift its localization from nucleus to perinucleus in the cytoplasm with polyalanine expansion (Albrecht et al. 2004; Caburet et al. 2004; Bachetti et al. 2005). Abnormal trapping of these transcription factors to cytoplasm might be one of the common pathogenesis of these polyalanine-related diseases. This study clearly shows that the aggregation of expanded polyalanine protein is initiated by the dimerization/multimerization processes that occur via the expanded polyalanine tracts.
We previously reported that 20% of the cells expressing YFP–Ala70 exhibited visible cytoplasmic aggregates and that YFP–Ala70 showed an SDS-resistant pattern on Western blots (Oma et al. 2004). The present study confirms that observation and additionally reveals that, among the various polyalanine constructs tested, only Ala70 formed SDS-resistant aggregates, which are retained in the stacking gel in SDS-PAGE. This is similar to the behavior of polyglutamine tracts beyond the pathogenic length, which form SDS-resistant aggregates (Scherzinger et al. 1997; Huang et al. 1998). However, in the case of polyalanine, Ala35 exhibited self-aggregation but did not form SDS-resistant insoluble aggregates, even though the tract exceeds pathogenic length. Maximum size of a nonpathogenic polyalanine tract in human proteome is 20 residues in PHOX2B protein. The SDS-sensitive multimerization of polyalanine tracts with more than 23 residues may contribute to the molecular pathogenesis of polyalanine-related diseases.
Altered intracellular localization of expanded GFP–polyalanine has been reported previously (Rankin et al. 2000; Ravikumar et al. 2002; Webb et al. 2004), which is consistent with our results that GFP–Ala25 localized exclusively in the cytoplasm, whereas the localization pattern of GFP–Ala7 was the same as that of the control in COS-7 cells. However, we did not observe visible aggregates in any cells expressing YFP–polyalanine constructs, with the exception of YFP–Ala70 (Fig. 4; Oma et al. 2004). These results are not consistent with the results in which GFP–Ala19, GFP–Ala25, and GFP–Ala35 were used (Rankin et al. 2000; Ravikumar et al. 2002; Webb et al. 2004). This inconsistency might be attributable to differences in the amino acid sequences flanking the polyalanine stretch. We have constructed additional polyalanine peptides with different flanking sequences; however, any peptides with a polyalanine tract of fewer than 36 residues that we tested did not form visible intracellular aggregates (data not shown).
Importantly, the threshold length, i.e., the length at which the properties of the polyalanine-containing protein change, might be influenced by the amino acid sequences flanking the polyalanine tract, as in the case of polyglutamine tracts of disease-related proteins (Nozaki et al. 2001). Among the polyalanine-related diseases, only OPMD is related to a shorter expansion length (10–12–17 residues); in eight other diseases, the pathogenic expansion is 18–33 residues in length. This may be partly related to the protein context of each protein. Indeed, several studies have reported that normal-length PABPN1 is capable of aggregation (Berciano et al. 2004; Tavanez et al. 2005; Marie-Josee Sasseville et al. 2006) and that the polyalanine tract is not responsible for its oligomerization and/or aggregation (Wang et al. 2005).
Interestingly, all of the polyalanine disease causative proteins contain multiple HPAA tracts as well as the polyalanine tract, which is expanded in disease. For example, ZIC2 has Ala9, Ala9, Ala5, His9, Gly6, and Gly6 in addition to Ala15, which expands to Ala25 in the disease condition. Proteins with multiple HPAA tracts generally include charge clusters, high counts of multiples, some histidine-rich motifs, and other unusual properties (Karlin et al. 2002a,b). It is possible that these multiple HPAA tracts in polyalanine disease causative proteins are involved in molecular pathogenesis of these diseases. This remains to be examined in future studies.
Beyond those proteins causing the nine known polyalanine-related diseases, there are many more polyalanine-containing proteins (Lavoie et al. 2003), some of which have been reported to interact with each other (Charron et al. 1999; Firulli et al. 2000); however, none have polyalanine tracts of disease-related lengths, and none have been reported to interact with each other via their polyalanine tracts. The present study is the first to directly demonstrate the self-interaction of expanded polyalanine tracts. This self-interaction may be a prerequisite for the abnormal folding of long polyalanine tracts and may alter the functions of other host proteins, which is thought to be a common aspect of the molecular pathogenesis of polyalanine-related diseases.
Materials and Methods
Plasmid construction
We annealed two complementary trinucleotide repeat oligonucleotides (90 or 21–105 bases; Proligo) by incubation at 100°C for 5 min, followed by cooling to 20°C. The double-stranded oligonucleotides were ligated into each vector (see details below). Restriction enzymes and T4 polymerase were used to make necessary frame adjustments. The integrity of each sequence was confirmed by sequencing.
pAS2–1 vector (TaKaRa Bio, Inc.): (CAG/CTG)30 was used for Gln, Ala, and Cys; (ATG/CAT)30 for Met, Asp, Ser, and Ile; (TGG/CCA)30 for Trp, Val, Thr, His, and Pro; (GAA/TTC)30 for Glu, Lys, and Phe; (TAC/GTA)30 for Tyr and Leu; (AAC/TTG)30 for Asn; (AGG/CCT)30 for Gly and Arg; and (CAG/CTG)n for Ala7 and Ala18. pACT2 vector (TaKaRa Bio, Inc.): (CAG/CTG)30 was used for Gln, Ser, Leu, and Cys; (ATG/CAT)30 for Met, Asp, Ile, and His; (TGG/CCA)30 for Trp, Pro, and Gly; (GGC/CCC)30 for Ala and Arg; (GAA/TTC)30 for Glu, Lys and Phe; (TAC/GTA)30 for Tyr and Val; (AAC/TTG)30 for Asn and Thr; and (CAG/CTG)n for Ala6 and Ala18. pGEX4T-2 vector (GE Healthcare Bioscience): (AAC/TTG)30 was used for Asn; (CAG/CTG)30 for Leu; (CCA/TGG)30 for His; and (GCA/GCT)7–35 for Ala7 to Ala35. pEYFP-C1 vector (Clontech): (CAG/CTG)30 was used for Cys; (TAC/GTA)30 for Tyr; (AAC/TTG)30 for Asn; and (GCA/GCT)7–35 for Ala7 to Ala35.
Yeast two-hybrid assay
Twenty types of HPAAs (each ∼30 residues in length) and two different polyalanines (six or 18 residues) were fused individually to both the GAL4 DNA-binding domain of vector pAS2-1 and the DNA-activation domain of vector pACT2. Yeast strain AH109 was cotransformed with the pACT2 and pAS2-1 vectors and selected on plates lacking leucine and tryptophan. Yeast transformants were transferred to selection plates lacking leucine, tryptophan, and histidine with or without 0.1, 0.25, 0.5, 1, 10, 20, or 40 mM 3-amino-1,2,4-triazole (3-AT). The plates were incubated at 30°C for about 2 wk, and the viability of the yeast transformants was analyzed. We classified the binding activity according to the 3-AT concentration, from 1 (without 3-AT) to 8 (with 40 mM 3-AT).
GST pulldown assay
A pGEX vector containing each HPAA was transformed into the BL21 (DE3) strain. An overnight culture of each transformant in LB medium was diluted and agitated at 37°C for 1 h (or until the optical density reached 0.4–0.6), and then 0.1 mM IPTG was added. During induction by IPTG, the culture was agitated at 25°C for 3 h. The bacterial cells were collected by centrifugation, suspended in 0.05× culture volume of sonication buffer containing 50 mM sodium phosphate (pH 7.5), 50 mM NaCl, 1 mM EDTA, 1 mM DTT, 0.2 mM PMSF, and 1/1000 vol of protease inhibitor cocktail (Sigma), and sonicated on ice 10 times for 10 sec each. Triton X-100 was added to a final concentration of 1%, and the lysate was subjected to affinity purification using glutathione Sepharose 4B (Amersham Biosciences) according to the manufacturer's protocol. The protein quantity and purity were checked by SDS-PAGE with Coomassie Brilliant Blue staining.
COS-7 cells were transiently transfected with the YFP–HPAA plasmids. After incubation for 48 h, the cells were harvested in TBS (20 mM Tris at pH 7.6, 137 mM NaCl, 0.2% Triton X-100, 1 mM DTT, 0.2 mM PMSF, and 1/1000 vol of protease inhibitor cocktail) and sonicated on ice for 15 sec. The lysate was centrifuged.
The supernatant fraction containing the cleared lysate was mixed with GST-coupled beads, rotated at 4°C overnight, and washed five times with TBS (containing 548 mM NaCl for the polyalanine–polyalanine pulldown assay and 137 mM NaCl for all other pulldown assays). Lysate samples were resolved by SDS-PAGE, and the proteins were transferred to Immobilon-P membranes (Millipore). The membranes were blocked with 5% nonfat dry milk in PBS containing 0.05% Tween-20 (TPBS) for 1 h at room temperature. The blocked membranes were incubated with anti-GFP/anti-YFP antibody (1:1000 dilution) for 1 h at 37°C, washed, and then incubated with horseradish peroxidase-conjugated anti-rabbit IgG antibody (1:5000 dilution) for 30 min at 37°C. Immunoreactive complexes on the membranes were visualized by enhanced chemiluminescence detection.
PAGE assay
GST fusion proteins were eluted from Sepharose beads with elution buffer containing 50 mM Tris-HCl (pH 8.0), 15 mM glutathione, 50 mM NaCl, and 1 mM DTT. Native PAGE was performed without SDS or any reducing agents. Electrophoresis was performed at 20 mA and was continued for 70 min after the dye exited the bottom of the gel.
Fluorescence microscopic analysis
COS-7 cells were grown in Dulbecco's Modified Eagle's Medium with 10% fetal bovine serum (Sigma-Aldrich). Transient transfection was performed using FuGENE 6 transfection reagent (Roche Diagnostics) following the manufacturer's instructions. At 48 h after transfection, the cells were treated with Hoechst33342 (Sigma-Aldrich) at 37°C for 45 min, and the medium was removed and replaced with PBS. YFP fluorescence was visualized using an IX70 fluorescence microscope (Olympus). About 300 transfected cells were counted for each sample.
Acknowledgments
Y.O. is supported by a JSPS Research Fellowship for Young Scientists. This work was supported by HFSP (RGP0024/2006-C). We thank Dr. Patrick Dion (Université de Montréal) for his comments on this article.
Footnotes
Reprint requests to: Shoichi Ishiura, Department of Life Sciences, Graduate School of Arts and Sciences, The University of Tokyo, 3-8-1 Komaba, Meguro-ku, Tokyo 153-8902, Japan; e-mail: cishiura@mail.ecc.u-tokyo.ac.jp; fax: 81-3-5454-6739.
Abbreviations: HPAA, homopolymeric amino acid; GST, glutathione S-transferase; PABPN1, poly(A) binding protein nuclear 1; OPMD, oculopharyngeal muscular dystrophy; YFP, yellow florescent protein; 3-AT, 3-amino-1,2,4-triazole; AD, activation domain; BD, binding domain; DTT, dithiothrol.
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.072955307.
References
- Abu-Baker A., Messaed, C., Laganiere, J., Gaspar, C., Brais, B., and Rouleau, G.A. 2003. Involvement of the ubiquitin-proteasome pathway and molecular chaperones in oculopharyngeal muscular dystrophy. Hum. Mol. Genet. 12: 2609–2623. [DOI] [PubMed] [Google Scholar]
- Alba M.M. and Guigo, R. 2004. Comparative analysis of amino acid repeats in rodents and humans. Genome Res. 14: 549–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albrecht A. and Mundlos, S. 2005. The other trinucleotide repeat: Polyalanine expansion disorders. Curr. Opin. Genet. Dev. 15: 285–293. [DOI] [PubMed] [Google Scholar]
- Albrecht A.N., Kornak, U., Boddrich, A., Suring, K., Robinson, P.N., Stiege, A.C., Lurz, R., Stricker, S., Wanker, E.E., and Mundlos, S. 2004. A molecular pathogenesis for transcription factor associated polyalanine tract expansions. Hum. Mol. Genet. 13: 2351–2359. [DOI] [PubMed] [Google Scholar]
- Amiel J., Trochet, D., Clement-Ziza, M., Munnich, A., and Lyonnet, S. 2004. Polyalanine expansions in human. Hum. Mol. Genet. 13: 235–243. [DOI] [PubMed] [Google Scholar]
- Bachetti T., Matera, I., Borghini, S., Di Duca, M., Ravazzolo, R., and Ceccherini, I. 2005. Distinct pathogenetic mechanisms for PHOX2B associated polyalanine expansions and frameshift mutations in congenital central hypoventilation syndrome. Hum. Mol. Genet. 14: 1815–1824. [DOI] [PubMed] [Google Scholar]
- Bao Y.P., Cook, L.J., O'Donovan, D., Uyama, E., and Rubinsztein, D.C. 2002. Mammalian, yeast, bacterial, and chemical chaperones reduce aggregate formation and death in a cell model of oculopharyngeal muscular dystrophy. J. Biol. Chem. 277: 12263–12269. [DOI] [PubMed] [Google Scholar]
- Becher M.W., Kotzuk, J.A., Davis, L.E., and Bear, D.G. 2000. Intranuclear inclusions in oculopharyngeal muscular dystrophy contain poly(A) binding protein 2. Ann. Neurol. 48: 812–815. [PubMed] [Google Scholar]
- Berciano M.T., Villagra, N.T., Ojeda, J.L., Navascues, J., Gomes, A., Lafarga, M., and Carmo-Fonseca, M. 2004. Oculopharyngeal muscular dystrophy-like nuclear inclusions are present in normal magnocellular neurosecretory neurons of the hypothalamus. Hum. Mol. Genet. 13: 829–838. [DOI] [PubMed] [Google Scholar]
- Brais B. 2003. Oculopharyngeal muscular dystrophy: A late-onset polyalanine disease. Cytogenet. Genome Res. 100: 252–260. [DOI] [PubMed] [Google Scholar]
- Brown L.Y. and Brown, S.A. 2004. Alanine tracts: The expanding story of human illness and trinucleotide repeats. Trends Genet. 20: 51–58. [DOI] [PubMed] [Google Scholar]
- Brown L., Paraso, M., Arkell, R., and Brown, S. 2005. In vitro analysis of partial loss-of-function ZIC2 mutations in holoprosencephaly: Alanine tract expansion modulates DNA binding and transactivation. Hum. Mol. Genet. 14: 411–420. [DOI] [PubMed] [Google Scholar]
- Caburet S., Demarez, A., Moumne, L., Fellous, M., De Baere, E., and Veitia, R.A. 2004. A recurrent polyalanine expansion in the transcription factor FOXL2 induces extensive nuclear and cytoplasmic protein aggregation. J. Med. Genet. 41: 932–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calado A., Tome, F.M., Brais, B., Rouleau, G.A., Kuhn, U., Wahle, E., and Carmo-Fonseca, M. 2000. Nuclear inclusions in oculopharyngeal muscular dystrophy consist of poly(A) binding protein 2 aggregates which sequester poly(A) RNA. Hum. Mol. Genet. 9: 2321–2328. [DOI] [PubMed] [Google Scholar]
- Charron F., Paradis, P., Bronchain, O., Nemer, G., and Nemer, M. 1999. Cooperative interaction between GATA-4 and GATA-6 regulates myocardial gene expression. Mol. Cell. Biol. 19: 4355–4365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chartier A., Benoit, B., and Simonelig, M. 2006. A Drosophila model of oculopharyngeal muscular dystrophy reveals intrinsic toxicity of PABPN1. EMBO J. 25: 2253–2262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cocquet J., De Baere, E., Caburet, S., and Veitia, R.A. 2003. Compositional biases and polyalanine runs in humans. Genetics 165: 1613–1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies S.W., Turmaine, M., Cozens, B.A., DiFiglia, M., Sharp, A.H., Ross, C.A., Scherzinger, E., Wanker, E.E., Mangiarini, L., and Bates, G.P. 1997. Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell 90: 537–548. [DOI] [PubMed] [Google Scholar]
- Davies J.E., Wang, L., Garcia-Oroz, L., Cook, L.J., Vacher, C., O'Donovan, D.G., and Rubinsztein, D.C. 2005. Doxycycline attenuates and delays toxicity of the oculopharyngeal muscular dystrophy mutation in transgenic mice. Nat. Med. 11: 672–677. [DOI] [PubMed] [Google Scholar]
- Dion P., Shanmugam, V., Gaspar, C., Messaed, C., Meijer, I., Toulouse, A., Laganiere, J., Roussel, J., Rochefort, D., Laganiere, S., et al. 2005. Transgenic expression of an expanded (GCG)13 repeat PABPN1 leads to weakness and coordination defects in mice. Neurobiol. Dis. 18: 528–536. [DOI] [PubMed] [Google Scholar]
- Dorsman J.C., Pepers, B., Langenberg, D., Kerkdijk, H., Ijszenga, M., den Dunnen, J.T., Roos, R.A., and van Ommen, G.J. 2002. Strong aggregation and increased toxicity of polyleucine over polyglutamine stretches in mammalian cells. Hum. Mol. Genet. 11: 1487–1496. [DOI] [PubMed] [Google Scholar]
- Dunah A.W., Jeong, H., Griffin, A., Kim, Y.M., Standaert, D.G., Hersch, S.M., Mouradian, M.M., Young, A.B., Tanese, N., and Krainc, D. 2002. Sp1 and TAFII130 transcriptional activity disrupted in early Huntington's disease. Science 296: 2238–2243. [DOI] [PubMed] [Google Scholar]
- Fan X., Dion, P., Laganiere, J., Brais, B., and Rouleau, G.A. 2001. Oligomerization of polyalanine expanded PABPN1 facilitates nuclear protein aggregation that is associated with cell death. Hum. Mol. Genet. 10: 2341–2351. [DOI] [PubMed] [Google Scholar]
- Faux N.G., Bottomley, S.P., Lesk, A.M., Irving, J.A., Morrison, J.R., de la Banda, M.G., and Whisstock, J.C. 2005. Functional insights from the distribution and role of homopeptide repeat-containing proteins. Genome Res. 15: 537–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firulli B.A., Hadzic, D.B., McDaid, J.R., and Firulli, A.B. 2000. The basic helix-loop-helix transcription factors dHAND and eHAND exhibit dimerization characteristics that suggest complex regulation of function. J. Biol. Chem. 275: 33567–33573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatchel J.R. and Zoghbi, H.Y. 2005. Diseases of unstable repeat expansion: Mechanisms and common principles. Nat. Rev. Genet. 6: 743–755. [DOI] [PubMed] [Google Scholar]
- Gorlich D. and Kutay, U. 1999. Transport between the cell nucleus and the cytoplasm. Annu. Rev. Cell Dev. Biol. 15: 607–660. [DOI] [PubMed] [Google Scholar]
- Hino H., Araki, K., Uyama, E., Takeya, M., Araki, M., Yoshinobu, K., Miike, K., Kawazoe, Y., Maeda, Y., Uchino, M., et al. 2004. Myopathy phenotype in transgenic mice expressing mutated PABPN1 as a model of oculopharyngeal muscular dystrophy. Hum. Mol. Genet. 13: 181–190. [DOI] [PubMed] [Google Scholar]
- Huang C.C., Faber, P.W., Persichetti, F., Mittal, V., Vonsattel, J.P., MacDonald, M.E., and Gusella, J.F. 1998. Amyloid formation by mutant huntingtin: Threshold, progressivity and recruitment of normal polyglutamine proteins. Somat. Cell Mol. Genet. 24: 217–233. [DOI] [PubMed] [Google Scholar]
- Karlin S., Brocchieri, L., Bergman, A., Mrazek, J., and Gentles, A.J. 2002a. Amino acid runs in eukaryotic proteomes and disease associations. Proc. Natl. Acad. Sci. 99: 333–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlin S., Chen, C., Gentles, A.J., and Cleary, M. 2002b. Associations between human disease genes and overlapping gene groups and multiple amino acid runs. Proc. Natl. Acad. Sci. 99: 17008–17013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazantsev A., Preisinger, E., Dranovsky, A., Goldgaber, D., and Housman, D. 1999. Insoluble detergent-resistant aggregates form between pathological and nonpathological lengths of polyglutamine in mammalian cells. Proc. Natl. Acad. Sci. 96: 11404–11409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavoie H., Debeane, F., Trinh, Q.D., Turcotte, J.F., Corbeil-Girard, L.P., Dicaire, M.J., Saint-Denis, A., Page, M., Rouleau, G.A., and Brais, B. 2003. Polymorphism, shared functions and convergent evolution of genes with sequences coding for polyalanine domains. Hum. Mol. Genet. 12: 2967–2979. [DOI] [PubMed] [Google Scholar]
- Marie-Josee Sasseville A., Caron, A.W., Bourget, L., Klein, A.F., Dicaire, M.J., Rouleau, G.A., Massie, B., Langelier, Y., and Brais, B. 2006. The dynamism of PABPN1 nuclear inclusions during the cell cycle. Neurobiol. Dis. 23: 621–629. [DOI] [PubMed] [Google Scholar]
- Michalik A. and Van Broeckhoven, C. 2003. Pathogenesis of polyglutamine disorders: Aggregation revisited. Hum. Mol. Genet. 12: 173–186. [DOI] [PubMed] [Google Scholar]
- Nasrallah I.M., Minarcik, J.C., and Golden, J.A. 2004. A polyalanine tract expansion in Arx forms intranuclear inclusions and results in increased cell death. J. Cell Biol. 167: 411–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nozaki K., Onodera, O., Takano, H., and Tsuji, S. 2001. Amino acid sequences flanking polyglutamine stretches influence their potential for aggregate formation. Neuroreport 12: 3357–3364. [DOI] [PubMed] [Google Scholar]
- Oma Y., Kino, Y., Sasagawa, N., and Ishiura, S. 2004. Intracellular localization of homopolymeric amino acid-containing proteins expressed in mammalian cells. J. Biol. Chem. 279: 21217–21222. [DOI] [PubMed] [Google Scholar]
- Oma Y., Kino, Y., Sasagawa, N., and Ishiura, S. 2005. Comparative analysis of the cytotoxicity of homopolymeric amino acids. Biochim. Biophys. Acta 1748: 174–179. [DOI] [PubMed] [Google Scholar]
- Perutz M.F., Johnson, T., Suzuki, M., and Finch, J.T. 1994. Glutamine repeats as polar zippers: Their possible role in inherited neurodegenerative diseases. Proc. Natl. Acad. Sci. 91: 5355–5358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rankin J., Wyttenbach, A., and Rubinsztein, D.C. 2000. Intracellular green fluorescent protein-polyalanine aggregates are associated with cell death. Biochem. J. 348: 15–19. [PMC free article] [PubMed] [Google Scholar]
- Ravikumar B., Duden, R., and Rubinsztein, D.C. 2002. Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum. Mol. Genet. 11: 1107–1117. [DOI] [PubMed] [Google Scholar]
- Scherzinger E., Lurz, R., Turmaine, M., Mangiarini, L., Hollenbach, B., Hasenbank, R., Bates, G.P., Davies, S.W., Lehrach, H., and Wanker, E.E. 1997. Huntingtin-encoded polyglutamine expansions form amyloid-like protein aggregates in vitro and in vivo . Cell 90: 549–558. [DOI] [PubMed] [Google Scholar]
- Shanmugam V., Dion, P., Rochefort, D., Laganiere, J., Brais, B., and Rouleau, G.A. 2000. PABP2 polyalanine tract expansion causes intranuclear inclusions in oculopharyngeal muscular dystrophy. Ann. Neurol. 48: 798–802. [PubMed] [Google Scholar]
- Shinchuk L.M., Sharma, D., Blondelle, S.E., Reixach, N., Inouye, H., and Kirschner, D.A. 2005. Poly-(L-alanine) expansions form core β-sheets that nucleate amyloid assembly. Proteins 61: 579–589. [DOI] [PubMed] [Google Scholar]
- Tavanez J.P., Calado, P., Braga, J., Lafarga, M., and Carmo-Fonseca, M. 2005. In vivo aggregation properties of the nuclear poly(A)-binding protein PABPN1. RNA 11: 752–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuite M.F. 2000. Yeast prions and their prion-forming domain. Cell 100: 289–292. [DOI] [PubMed] [Google Scholar]
- Tung C.H. and Weissleder, R. 2003. Arginine containing peptides as delivery vectors. Adv. Drug Deliv. Rev. 55: 281–294. [DOI] [PubMed] [Google Scholar]
- Uyama E., Tsukahara, T., Goto, K., Kurano, Y., Ogawa, M., Kim, Y.J., Uchino, M., and Arahata, K. 2000. Nuclear accumulation of expanded PABP2 gene product in oculopharyngeal muscular dystrophy. Muscle Nerve 23: 1549–1554. [DOI] [PubMed] [Google Scholar]
- Verheesen P., de Kluijver, A., van Koningsbruggen, S., de Brij, M., de Haard, H.J., van Ommen, G.J., van der Maarel, S.M., and Verrips, C.T. 2006. Prevention of oculopharyngeal muscular dystrophy-associated aggregation of nuclear polyA-binding protein with a single-domain intracellular antibody. Hum. Mol. Genet. 15: 105–111. [DOI] [PubMed] [Google Scholar]
- Wang Q., Mosser, D.D., and Bag, J. 2005. Induction of HSP70 expression and recruitment of HSC70 and HSP70 in the nucleus reduce aggregation of a polyalanine expansion mutant of PABPN1 in HeLa cells. Hum. Mol. Genet. 14: 3673–3684. [DOI] [PubMed] [Google Scholar]
- Webb J.L., Ravikumar, B., and Rubinsztein, D.C. 2004. Microtubule disruption inhibits autophagosome-lysosome fusion: Implications for studying the roles of aggresomes in polyglutamine diseases. Int. J. Biochem. Cell Biol. 36: 2541–2550. [DOI] [PubMed] [Google Scholar]