

Abstract
Cell therapies based on focal delivery of the inhibitory neuromodulator adenosine were previously shown to provide potent seizure suppression in animal models of epilepsy. However, hitherto used therapeutic cells were derived from rodents and thus not suitable for clinical applications. Autologous patient-derived adenosine-releasing cell implants would constitute a major therapeutic advance to avoid both xenotransplantation and immunosuppression. Here we describe a novel approach based on lentiviral RNAi mediated downregulation of adenosine kinase (ADK), the major adenosine-removing enzyme, in human mesenchymal stem cells (hMSCs), which would be compatible with autologous cell grafting in patients. Following lentiviral transduction of hMSCs with anti-ADK miRNA expression cassettes we demonstrate up to 80% downregulation of ADK and a concentration of 8.5 ng adenosine per ml of medium after incubating 105 cells for 8 hours. hMSCs with a knockdown of ADK or cells expressing a scrambled control sequence were transplanted into hippocampi of mice 1 week prior to the intraamygdaloid injection of kainic acid (KA). While mice with control implants expressing a scrambled miRNA sequence or sham treated control animals were characterized by KA-induced status epilepticus and subsequent CA3 neuronal cell loss, animals with therapeutic ADK knockdown implants displayed a 35% reduction in seizure duration and 65% reduction in CA3 neuronal cell loss, when analyzed 24h after KA-injection. We conclude that lentiviral expression of anti-ADK miRNA constitutes a versatile tool to generate therapeutically effective adenosine releasing hMSCs, thus representing a model system to generate patient identical autologous adult stem cell grafts.
Keywords: Adenosine, Adenosine kinase, Epilepsy, Kainic acid, Status epilepticus, Neuroprotection, RNAi, Lentivirus, Human mesenchymal stem cells, Cell therapy
Introduction
Epilepsy affects about 1% of the population; currently available antiepileptic drugs (AEDs) fail to prevent seizures in about 35 percent of patients with partial epilepsy and new treatment strategies are urgently needed (Dichter, 2007; Kwan and Brodie, 2006; Loscher and Schmidt, 2004; Remy and Beck, 2006). In addition, pronounced side effects may compromise the most favorable use of AEDs (Sabers and Gram, 2000). Therefore, focal treatment approaches for refractory epilepsy have been evaluated as an alternative to systemic drug delivery. In several approaches it was demonstrated that focal drug delivery is generally well tolerated and devoid of major side effects (Nilsen and Cock, 2004). Recently, focal adenosine-based cell therapies have been investigated as an approach for the therapy of partial epilepsy (Boison, 2005). These approaches made novel therapeutic use of adenosine, which was long known to be an endogenous neuromodulator of the brain with potent antiepileptic and neuroprotective properties (Dragunow, 1986; Dragunow, 1988; Dragunow, 1991). In these studies the proof of principle was established that focal cell-mediated release of adenosine can effectively suppress seizures by local augmentation of the adenosine system (Boison, 2007). Adenosine levels in adult brain are largely regulated by the activity of the astrocyte based enzyme adenosine kinase (ADK) (Boison, 2006). Consequently, increases in ADK lead to increased seizure activity (Fedele et al., 2005; Gouder et al., 2004) and brain vulnerability (Pignataro et al., 2006), while reduced levels of ADK augment endogenous adenosine and reduce seizure activity (Boison, 2005).
Focal cell-based augmentation of the adenosine system as a novel approach in epilepsy therapy is supported by the following arguments: (i) Deficits in the adenosine-based neuromodulatory system have been associated with epileptogenesis (Gouder et al., 2004; Rebola et al., 2003) and these deficits promote seizures (Fedele et al., 2005). Thus, reconstitution of adenosinergic neuromodulation is a rational therapeutic approach. (ii) The anticonvulsant activity of locally released adenosine is maintained in models of epilepsy, which are resistant to major antiepileptic drugs (Gouder et al., 2003). Thus, adenosine promises to be effective in drug resistant forms of epilepsy. (iii) Focal delivery of adenosine from encapsulated fibroblasts or myoblasts transiently suppresses seizures in kindled rats without overt side effects (Güttinger et al., 2005b; Huber et al., 2001). However, the hitherto used encapsulated animal cell-based adenosine delivery systems have limitations for clinical applications because of the need for xenografting.
As an alternative to xenografting, strategies have been evaluated to transplant human cells directly into the brain: Thus, intracerebral transplantation of fetal cell preparations has been widely used in a multitude of experimental paradigms and in clinical trials, most notably for the treatment of Parkinson's disease (Bjorklund, 2000; Freed et al., 2001; Lindvall, 2000; Lindvall and Bjorklund, 2004). Fetal cell transplantation studies have led to the following important results: (i) Human embryonic dopamine-neuron transplants survive in patients with severe Parkinson's disease for years. Thus, long-term survival of implants and clinical application of cell therapy is principally feasible (Freed et al., 2001). (ii) Fetal hippocampal cell grafts exhibit robust long-term survival (> six months) and integration in animal models of epilepsy (Rao et al., 2006). Thus, the condition of epilepsy does not prevent long-term graft survival. (iii) Fetal hippocampal cell grafts display endogenous protective effects in animal models of epilepsy (Rao et al., 2006; Shetty and Turner, 1996; Shetty and Turner, 2000; Zaman et al., 2000; Zaman and Shetty, 2001). Thus, potential effects of intrahippocampal cell transplants on local circuitry may be beneficial. (iv) The heterogeneous cellular composition of fetal grafts, although not optimal, does not preclude their use in clinical trials (Lindvall and Bjorklund, 2004).
Eventually, stem cells may provide a superior source for transplantation over fetal cell grafts due to their proliferative potential and the ability to standardize protocols and procedures (Vats et al., 2005). Thus, adult stem cells have been isolated from several easily accessible tissue sources (Korbling et al., 2003), including bone marrow (Barry and Murphy, 2004; Kassem, 2004; Mezey et al., 2003), skeletal muscle (Gussoni et al., 1999; Jackson et al., 1999) and skin (Fernandes et al., 2004; Fernandes et al., 2006; Toma et al., 2001). All of these stem cells preferentially generate differentiated cells of the same lineage as the tissue of origin. However, transplant studies indicate that adult stem cells can also generate cells of a different embryonic lineage in vivo. For example, neural stem cells were shown to generate blood (Bjornson et al., 1999) and skeletal muscle cells (Galli et al., 2000). Similarly, transplanted bone marrow stem cells contributed to skeletal muscle (Ferrari et al., 1998; Gussoni et al., 1999), liver (Petersen et al., 1999), and generated cells producing neuronal markers in the brain (Brazelton et al., 2000; Kassem, 2004; Mezey et al., 2000; Zhao et al., 2002). These findings have therapeutic implications, since neural stem cells can promote functional recovery upon transplantation into the injured nervous system (Chu et al., 2003; Chu et al., 2004; Jeong et al., 2003; Pluchino et al., 2003). Thus, adult stem cells provide an easily accessible source for autologous grafting of stem cell derived (neuronal) progeny.
Here we choose to engineer hMSCs for therapeutic adenosine release for the following reasons: (i) Readily accessible hMSCs would permit autologous cell transplantation in human patients. (ii) MSCs can be induced to neuronal differentiation (Black and Woodbury, 2001; Munoz-Elias et al., 2004; Woodbury et al., 2000). (iii) Gene expression with lentiviral vectors can effectively be achieved in hMSCs (Totsugawa et al., 2002) and continuous stable gene expression was demonstrated throughout differentiation (Sugiyama et al., 2005). (iv) Functional RNA interference in hMSCs throughout differentiation is well documented (Hoelters et al., 2005).
In this communication we used a lentiviral system for the expression of anti-ADK miRNA in hMSCs and tested the therapeutic potential of adenosine releasing hMSC-derived brain implants in a mouse model of kainic acid induced brain injury. In this proof-of-principle-study we conclude that lentiviral expression of anti-ADK miRNA in human adult stem cells is a novel and efficient approach to generate therapeutically active human stem cells, thus opening new perspectives for future autologous cell transplantation in epilepsy.
Materials and Methods
Generation of lentiviral constructs for the expression of anti-ADK miRNAs
Five different pre-miRNA sequences, of which four were homologous to human, and one to human, mouse and rat Adk cDNA (Figure 1A) were designed according to published recommendations (Elbashir et al., 2001a; Elbashir et al., 2001b). In addition, a randomized scrambled control (SC) sequence was synthesized (Invitrogen, Carlsbad, CA). The selected sequences were cloned into pcDNA™ 6.2-GW/EmGFP-miR expression vectors (Invitrogen), which contain an emerald green fluorescence protein (EmGFP) reporter gene and a blasticidin resistance gene, thus allowing co-expression of the respective miRNA with EmGFP and selection of stably transduced cells with blasticidin (Figure 1B). All constructs were sequenced to confirm their structure. To produce a lentiviral expression clone, the pre-miRNA expression cassettes from the pcDNA™ 6.2-GW/EmGFP-miR expression clones were transferred into a pLenti6/V5-DEST destination vector according to the manufacturer's recommendations (Invitrogen). Briefly, an entry clone was generated by first performing a recombination reaction between the attB substrate in the pcDNA™ 6.2-GW/EmGFP-miR expression clone (Fig. 1B) and the attP substrate in a pDONR ™ 221 vector using BP Clonase-II™ (Invitrogen). The second recombination reaction was then performed between the resulting entry clone containing the attL substrate and the pLenti6/V5-DEST vector containing the attR substrate. Using this approach, we created a panel of 5 lentiviral anti-ADK miRNA expression constructs H236, H239, H240, H241, H242, and one scrambled control construct containing a randomized sequence.
Figure 1. Design of lentiviral miRNAs.

A: Core target recognition sequences of miRNAs from human (p239 – p242) or human, mouse, and rat (p236) ADK cDNA. B: Schematic gene map of the pcDNA™ 6.2-GW/EmGFP-miR expression vectors into which the miRNA sequences derived from (A) were cloned. pCMV: cytomegalovirus promotor, attB1: first attB substrate, EmGFP: emerald green fluorescence protein, 5′FR: 5′ flanking region, miRNA, 3′FR: 3′ flanking region, attB2: second attB substrate, TKpA: thymidine kinase polyadenylation signal, Bla: blasticidin resistance gene.
Production of lentiviral vectors
For virus production, 293FT helper cells (Invitrogen) were cultured in DMEM containing 10% fetal bovine serum (FBS), 0.1 mM MEM non-essential amino acids, 1% penicillin/streptomycin, 1 mM sodium pyruvate, and 50μg/ml Geneticin® (Invitrogen) for at least 3 passages. For transfection 6 × 106 cells / 100 mm dish were co-transfected with 9 μg ViraPower™ Packaging Mix and 3 μg pLenti6/V5-GW/miR expression plasmid DNA using Lipofectamine™ 2000 according to the manufacturer's instructions (Invitrogen). The lentiviral stocks were collected 2 to 3 days after transfection, filtered with a 0.45 μm filter (Millipore, Bedford, MA), and stored at −80°C for future use.
To determine the titer of the virus, HT-1080 cells (ATCC, Manassas, VA) were grown in Eagle's Minimal Essential Medium with Earle's BSS and 2 mM L-Glutamine (EMEM; ATCC) containing 1 mM sodium pyruvate, 0.1 mM MEM non-essential amino acids, 10% non heat inactivated FBS (ATCC). The cells were plated in a 6 well plate at a density of 2 × 105 cell / well / 2 ml. Medium was replaced the next day by serial dilutions of lentivirus (10-2 to 10-6) in complete growth medium at 1ml / well containing 6 μg / ml polybrene (Invitrogen). To select for stably transduced cells, from day 4 on medium was replaced with fresh medium containing blasticidin (10 μg / ml) every 3 - 4 days. The selection was complete after 10 - 12 days. The numbers of surviving colonies were counted under a microscope (Nikon, ELLIPSE, TS100, Japan) and the titer of the lentiviral stocks (transducing units, TU) was calculated by the formula: lentiviral titer (TU / ml) = numbers of colonies × lentiviral dilution. The lentiviral titers from the 6 different miRNA constructs were between 106 to 107 TU. The determination of the lentiviral titer allowed us to estimate the multiplicity of infection (MOI) and thus to deduce the infectious activity of the viral stocks.
Culture and lentiviral transduction of hMSCs
Human bone marrow derived mesenchymal stem cells (hMSCs) were obtained from Cambrex (Walkersville, MD) and cultured at 37°C, 5% CO2 and 95% humidity in DMEM containing 10% FBS optimized for hMSCs (Stem Cell Technologies, Vancouver, Canada), 2 mM L-glutamine, 0.1 mM MEM non-essential amino acids solution, 1 mM sodium pyruvate and 1% penicillin/streptomycin. Medium was exchanged every 4 - 5 days. For lentiviral transduction hMSCs were replated in 6 well plates at a density of 2 × 105 cells / well. The next day, the medium was replaced with 1 ml of fresh medium containing lentivirus at an MOI of 5 and 6 μg / ml polybrene. The medium was replaced with fresh medium on the following day. After five days of culture, fresh medium containing blasticidin (5 μg / ml) was added to select for stably transduced cells. During seven days of selection, EmGFP fluorescence of transduced hMSCs was daily monitored using a phase contrast fluorescent microscope (Nikon ELLIPSE, TS100, Japan). Before and after selection hMSCs transduced with H239 lentiviruses were harvested and plated in duplicates on poly-L-lysine coated 8 well chamber slides (Nunc, Rochester, NY) at a density of 4 × 104 cells per well. The nucleus of the cells was labeled using fluorescence mounting medium containing DAPI (Vector Laboratories, Inc. Burlingame, CA). For quantitative analysis of EmGFP expression efficiencies four areas of each well were randomly selected and the number of total cells (DAPI) and EmGFP expressing cells were determined under a fluorescence microscope. To rule out unspecific effects caused by random genomic integration of the lentiviral vector in individual clonal cell populations, for all subsequent experiments polyclonal blasticidin-selected cell populations were used.
Quantitative analysis of ADK knockdown by Western blot analysis
Transduced hMSCs were trypsinized and harvested either 72 h after transduction or 7 days after blasticidin selection. The cells were washed 3 times with PBS, homogenized in cell lysis buffer (50mMTris, pH7.8, 150mM NaCl, 1% nonidet-40) containing 10 μl / ml protease inhibitor (Sigma), incubated on ice for 30 min, and then centrifuged 30 min at 10000 rpm. The aqueous supernatants were collected and quantified using Bradford Reagent (Sigma). Individual samples, each containing 30μg protein, were separated on a pre-cast SDS / 12.5% polyacrylamide gel in a Tris / HCl buffer (pH 7.4) and blotted onto a polyvinylidene fluoride (PVDF) membrane (Bio-Rad, Hercules, CA). The membrane was incubated at room temperature for 1h in PBST buffer (PBS pH 7.4, 0.05% Triton-100), containing 1% (w/v) bovine serum albumin to block nonspecific protein binding sites. After blocking, the blots were probed with a polyclonal rabbit antiserum against ADK (Fedele et al., 2005) (1:2500) overnight at 4°C followed by 5 washes with PBST. They were then incubated with an anti-rabbit IgG, horseradish peroxidase (HRP)-linked antibody (1:8000) (Cell Signaling Inc.) for 1 h at room temperature. After washing, the immunoreactive bands were detected using a chemiluminescent substrate (NEN, Life Science). Subsequently, the blots were re-probed with a β-actin control antibody (1:2000, Abcam, ab8227). The intensities of the bands in each Western blot were quantified using the Kodak Image Analysis Software (Eastman Kodak Company, NY). Immunoreactivity to β-actin was used as an internal standard to calculate relative amounts of ADK, which were then normalized to the scrambled control (= 100%) samples. The experiment for the blasticidin-selected cells was performed in duplicate.
Assay for adenosine
For the analysis of the amount of adenosine released from transduced hMSCs, cells were grown at a density 105 cells per well and incubated for 1 h in serum free culture medium followed by 2 washes with serum-free culture medium and the addition of 1 ml of fresh pre-warmed serum free medium. After 8 h of incubation the supernatants were collected for the adenosine analysis. After collection of the samples, the cells were removed with trypsin and counted. This count was used for normalization of the amount of adenosine per number of cells. Adenosine was quantified using an enzyme coupled bioluminescence assay of adenosine, which was performed as described previously (Fedele et al., 2004): Briefly, adenosine was metabolized into ATP by using recombinant ADK (own production), recombinant AMP:GTP phosphotransferase (own production), pyruvate kinase (Roche, Mannheim, Germany), GTP, and phosphoenolpyruvate. The formed ATP was then quantified with a commercial luciferase assay (ATP Bioluminescent Assay Kit, Sigma). All assays were performed in duplicates.
ADK-Immunocytochemistry
Transduced hMSCs were plated into poly-L-lysine coated 8 well chamber slides at a density of 4×104 cells. After growth for 24 h in complete culture medium the cells were fixed with 4% paraformaldehyde in phosphate buffered saline (PBS, pH 7.4) followed by 2 PBS washes. Cells were first incubated in 1% BSA, 0.2% Triton X-100 in PBS, pH 7.4 and then incubated overnight at 4°C with primary anti-ADK antiserum diluted 1:2000 in 1% BSA, 0.2% Triton X-100 in PBS, pH 7. The cells were then washed 3 times in PBS and incubated with goat anti-rabbit IgG conjugated with a Cy3 secondary antibody at 1:800 (Jackson Immuno Research, West Grove, Pennsylvania), The cover slides were then mounted in fluorescence mounting medium containing DAPI (Vector Laboratories, Inc. Burlingame, CA). Immunofluorescence on the stained slides was analyzed with a Leica DMLB microscope (Zeiss, Jena, Germany) using a high-resolution Zeiss Axiocam camera.
Animals
All animal procedures were performed in an AAALAC-accredited facility in accordance with protocols approved by the Legacy Institutional Animal Care and Use Committee and the principles outlined in the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Animals were kept in individually ventilated isolator cages and food and water were supplied ad libitum.
Cell transplantation
Adult male C57BL/6 mice weighing 25 - 30g were used. All animals, including control animals, received daily immunosuppression with cyclosporine A (15mg / kg, i.p.) initiated 2 days prior to transplantation. Anesthesia was induced with 3% isoflurane, 67% N2O, 30% O2 and maintained with 1.5% isoflurane, 68.5% N2O, 30% O2, while mice (n = 18) were placed in a Kopf stereotactic frame. Immediately before transplantation, cells were harvested and resuspended at a concentration of 2.5 × 104 cells per μl in culture medium. Cell injections (1.25 × 105 cells / per mouse) were performed using a glass capillary (inner diameter of tip: 70-90 μm). The cells (ADK-knockdown H239, n = 12; scrambled control, n = 6) were slowly injected in a volume of 5 μl using a drill hole above the left hippocampus and a single diagonal injection tract spanning from coordinate (AP +1.6; ML −1.2, DV 0.0) to coordinate (AP −2.8; ML +1.75; DV − 4.0), thus depositing the cells within the infrahippocampal cleft of the to-be-injured brain hemisphere as described previously (Li et al., 2007b). Cells were slowly injected (1 μl / min) while withdrawing (1 mm / min) the capillary. The capillary was fully retracted 5 min after injection to avoid reflux of cells. As described previously (Li et al., 2007b) this transplantation strategy has several advantages: (i) larger transplantation volumes can be used; (ii) the diagonal injection approach avoids injury of the ipsilateral hippocampus; (iii) coverage of the dorso-ventral extent of the hippocampal formation; (iv) good implant survival rate; (v) no interference of cells within the infrahippocampal with hippocampal architecture. Using an identical procedure sham-treated control animals (n = 6) received culture medium instead of cells.
Seizure model
One week after cell transplantation, animals received unilateral stereotaxic microinjection of kainic acid (KA) into the basolateral amygdala nucleus based on stereotactic coordinates relative to bregma: AP = -0.94 mm, L = -2.85 mm, V = -3.75 mm (Franklin and Paxinos, 1997) as described (Araki et al., 2002; Shinoda et al., 2004b).
Briefly, after anesthesia and catheterization of the femoral vein the animals were placed in a stereotactic frame adapted for the use of mice (Kopf Instruments). Animals were kept under anesthesia with a facemask using a mixture of 68.5% N2O, 30% O2, and 1.5% isoflurane. Animals were kept normothermic (37 ± 1°C) with a rectal thermometer connected to a feedback-controlled heating pad (Harvard Instruments, Holliston, MA) and heat lamp. Mice were affixed with three recording electrodes (Plastics One, Inc., Roanoke, VA) and a 26-gauge steel guide cannula over the intact dura using dental cement (Plastics One, Inc.). Anesthesia was discontinued, the animals were placed into a circular Plexiglas restrainer, EEG recordings commenced, and then a 31-gauge internal cannula (Plastics One, Inc.) was inserted into the lumen of the guide cannula to inject 0.3 μg KA in a volume of 0.2 μl vehicle (PBS, pH 7.4) into the amygdala. KA was injected slowly during 1 min. After the injection the cannula was kept in place for an additional 2 min to avoid reflux of the KA. A control group of H239 graft recipients (n = 6) received 8-cyclopentyl-1,3-dipropylxanthine (DPCPX, 1mg/kg, i.p.) 30 min prior to KA-injection. Cortical EEGs indicating electrographic SE not accompanied by motor SE were monitored for 30 min until lorazepam (6 mg/kg, i.p.) was administered to terminate seizures. The EEG was further monitored for up to 30 min to ensure seizure cessation. An observer unaware of the experimental treatment performed quantification of EEG recordings, and the cumulative duration of sustained polyspike “type IV” seizure activity (Araki et al., 2002) was calculated. Animals were killed the next day and brain tissue was collected for histology and TUNEL analysis.
Histological and KA-induced cell death analysis
Brains were obtained 24 h after KA injection and immediately frozen in 2-methylbutane (-30°C), sectioned at 12 μm on a cryostat, and kept at −80°C until further use. Coronal sections −1.7 mm to −2.1 mm caudal to bregma (Franklin and Paxinos, 1997) were air dried (15 min), postfixed in 10% formalin (15 min), washed twice in PBS, and then processed for histological analysis. Every third section was stained either with cresyl violet (section 1, 4, 7,…), 4′,6-diamidino-2-phenylindole (DAPI, Vector Laboratories Inc., Burlingame, CA, USA) to assess nuclear morphology (section 2, 5, 8,…) or for detection of DNA fragmentation (section 3, 6, 9,…) using the fluorescin-based terminal deoxynucleotidyl transferase (TdT)-mediated dUTP nick end-labeling (TUNEL) technique (Roche Molecular Biochemicals, Indianapolis, IN). TUNEL cell counts were performed on n = 3 brain sections from the entire hippocampal CA1/3 subfields. In every animal the three central slices showing most TUNEL reactivity, typically slice # 12, 15, 18 were chosen for quantification of the TUNEL data. To visualize the intrahippocampal implants, graft-based EmGFP fluorescence was determined on the DAPI-stained sections. Images were visualized using a Leica microscope under Ex/Em wavelength of 500/550 nm (green), collected using an Optronics DEI-750 three-chip camera equipped with a BQ 8000 sVGA frame grabber, and analyzed using Bioquant software (Nashville, TN).
Statistics
Unless stated otherwise, statistical variability is indicated as ± SD and data were analyzed using one-way ANOVA with Student-Newman-Keuls Test. *P<0.05, **P<0.01, ***P<0.001.
Results
Lentiviral transduction of hMSCs
Lentiviral vectors can effectively transduce a wide range of cell types, including dividing and non-dividing mammalian cells. To determine the lentiviral transduction efficiency in hMSCs, all lentiviral vectors described in this study (H236, H239-H242, scrambled control) were transduced into hMSCs at an MOI of 5. EmGFP expression in these cells was monitored daily by fluorescence microscopy. Green fluorescent cells indicate bicistronic expression of EmGFP with anti-ADK miRNA. Weak EmGFP expression was first detected as early as 2 days after infection and a prominent green fluorescent signal was detected after 3 - 5 days. At 5 days the ratios of EmGFP positive cells and total cell numbers from eight randomly selected fields from H239 transduced colonies were 36/46, 26/31, 22/26, 19/28, 16/20, 17/24, 17/26, 26/46. Thus, 65-85% of hMSCs transduced with H239 lentiviruses expressed green fluorescence (Fig. 2A, B). Similar transduction efficiencies were obtained with vectors H236, H240-H242, and scrambled control (data not shown). To further enrich transduced cells, blasticidin selection was initiated 5 days after transduction. After 7 days of selection the ratios of EmGFP positive H239 transduced cells and total cell numbers were 34/35, 21/21, 45/45, 25/26, 43/45, 26/27, 22/22. Thus, 95-100% of the selected cells expressed green fluorescence (Fig. 2C,D). Blasticidin selection thus significantly increased the number of EmGFP positive cells from 76 ± 7.3% to 98 ± 2.0%, p<0.001, chi-square test (Fig. 2E). Although not further quantified here, blasticidin selection generally led to an increase in the intensities of EmGFP fluorescence indicating the selection for cells with higher expression levels of the virus. Compared to anti-ADK miRNA transduced cells, scrambled control vector -transduced cells had a slightly reduced intensity of EmGFP fluorescence (data not shown). Transduced hMSCs were maintained in culture as polyclonal cell populations for at least 8 weeks and during this time >95% of the cells maintained EmGFP expression. To avoid unspecific effects caused by random integration of the lentiviral vector in individual clonal cell populations, all subsequent experiments were performed with polyclonal cell populations of blasticidin-selected cells.
Figure 2. Lentiviral transduction efficiency in hMSCs.

EmGFP expression (green) in hMSCs was assessed 5 days after transduction with vector H239 (A) or 7 days after selecting these cells with blasticidin (5 μg/ml) (C). Cells were counterstained with DAPI to visualize cell nuclei (B, D). Arrows indicate EmGFP negative cells. E: Before blasticidin selection 76 ±7.3 % of the cells were positive for EmGFP, while after selection the percentage of EmGFP positive cells had increased to 98 ± 2.0%. *** P < 0.001, chi-square test, n = 8.
Knockdown of ADK in hMSCs by lentiviral RNAi
To quantify the reduction of ADK in hMSCs, Western blot analysis was performed using lysates of hMSCs transduced with the ADK knockdown vectors H236, H239, H240, H241, H242, a corresponding scrambled control vector, or a mouse specific vector (M237). ADK-specific immunoreactivity was quantified and normalized according to β-actin immunoreactivity. To exclude unspecific effects on ADK-expression caused by vector integration we compared ADK expression 72 hours after transduction (Fig. 3A). In this study, the scrambled control vector did not downregulate ADK, while downregulation of ADK was observed after transducing cells with the human specific anti-ADK miRNA vectors H236 to H242 leading to residual ADK levels of 43 to 68% of control. In contrast to the human vectors, the mouse-specific vector M237 (containing one non-matching nucleotide compared to the human sequence) was less efficient in reducing ADK (74% of control). Following one week of blasticidin selection in a duplicate study, the hMSCs transduced with the vectors H236, H239, H240, H241, and H242 had further reduced levels of ADK with a range of 20 to 54% of residual ADK compared to scrambled control (= 100%) (Fig. 3). hMSC clone H239 displayed the strongest reduction of ADK (20-28% residual ADK) and was therefore selected for all subsequent experiments. In this study, we did not include mock-transduced cells, because they did not survive blasticidin selection. Thus, in the following, all control experiments are based on blasticidin-selected scrambled control vector transduced hMSCs. To provide an independent qualitative demonstration of the H239-mediated knockdown of ADK an immunocytochemical analysis of H239 transduced hMSCs was performed 72 h after transduction (Fig. 4). EmGFP negative cells displayed strong immunostaining against ADK, particularly in the cell nucleus and perinuclear region, whereas, ADK immunoreactivity was strongly reduced in EmGFP positive cells. To investigate whether downregulation of ADK translates into the cellular release of adenosine, supernatants of duplicate samples of 105 cultured cells each were analyzed for adenosine. After incubating 105 H239 cells for 8 hours, adenosine concentrations of 8.55 ± 0.05 ng/ml were found, while adenosine levels in comparable supernatants from scrambled control vector transduced hMSCs were not detectable. Thus, downregulation of ADK by lentiviral expression of anti-ADK miRNA induces cellular release of adenosine.
Figure 3. ADK-knockdown in hMSCs by lentiviral RNAi.
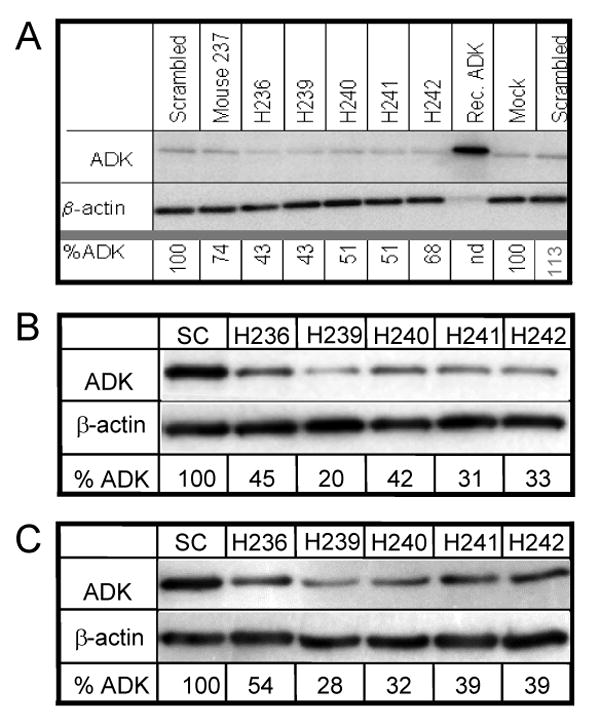
(A) In a primary assessment Western Blot analysis was performed on cell lysates (15 μg each) 72h after lentiviral transduction with MOI's of 25. ADK staining top and β actin staining bottom; Controls: Mock transduction, transduction with scrambled control virus, or with mouse specific virus 237; rec. ADK = recombinant ADK. H236 - H242: hMSCs transduced with 5 different human anti-ADK miRNA vectors. (B, C) Western blots from samples (30μg, each) derived from duplicate transductions of hMSCs with a scrambled control vector (SC) or with the anti-ADK miRNA vectors H236 - H242. Samples were prepared 7 days after blasticidin selection. The blots were probed with antibodies directed against ADK (top) or β-actin. Residual ADK (%ADK) was calculated from scanned intensities of bands in the Western Blots, and normalized to β-actin and respective control hMSCs (=100%). Note that blasticidin-selected hMSC cells H239 displayed the strongest reduction of ADK expression.
Figure 4. miRNA-mediated knockdown of ADK in cultured hMSCs.

Triple-immunofluorescence analysis of two cells from H239 transduced hMSCs stained with the nuclear stain DAPI in blue and for ADK in red. The transduced EmGFP positive cell (green) is negative for ADK, while the EmGFP negative cell is positive for ADK.
Reduction of KA-induced seizures by adenosine-releasing hMSC grafts
To assess the therapeutic potential of adenosine releasing hMSC brain implants we selected a paradigm of kainic acid (KA) induced brain injury (Araki et al., 2002; Shinoda et al., 2004b), which allowed us to assess both seizure suppression and neuroprotection by cell mediated adenosine release. Under standard immunosuppression (cyclosporine 12.5 mg/kg, i.p., daily) adult male C57BL/6 mice received intrahippocampal implants of 125,000 H239-hMSCs (n = 12 mice), scrambled control hMSCs (n = 6), or a respective sham treatment (n = 6). We used a previously established transplantation strategy to deposit the implanted cell within the infrahippocampal cleft (Li et al., 2007b) to allow distribution of the implanted cells along the dorso-ventral extend of the ipsilateral hippocampus without parenchymal integration of the cells. To determine the susceptibility to seizures, the graft recipients and corresponding controls, received intraamygdaloid injections of KA (0.3 μg in 0.2 μl) delivered one week after grafting. To demonstrate that therapeutic effects are due to graft-mediated adenosine acting via A1Rs six of the H239-treated mice received an injection of the adenosine A1R antagonist DPCPX (1mg/kg, i.p.) 30 min prior to KA-administration. Following the KA injections, electrographic seizure activity, which was not accompanied by motor seizures, was monitored in EEG recordings with a set of three cortical electrodes. 30 min after KA injection seizure activity was terminated with lorazepam (6 mg/kg, i.p.). Seizure activity during 30 min of recording was graded into type I-IV activities as described previously (Araki et al., 2002). All animals developed comparable type I-III patterns indicating the cortical response to intraamygdaloid application of KA (Fig. 5). However, the cumulative duration of all type IV injurious seizure activity was significantly reduced in H239 hMSC graft recipients (245 ± 84 sec), compared to sham treated (453 ± 130 sec, p < 0.05), scrambled control-hMSC treated (445 ± 131 sec, p < 0.05), or H239 treated control animals pretreated with DPCPX (538 ± 141.8 sec, p < 0.01). We conclude that seizure suppression is due to graft mediated adenosine release and that miRNA mediated knockdown of ADK in intrahippocampal hMSC implants is an effective strategy to reduce acute seizure duration.
Figure 5. Electrographic seizure patterns recorded after injection of KA.
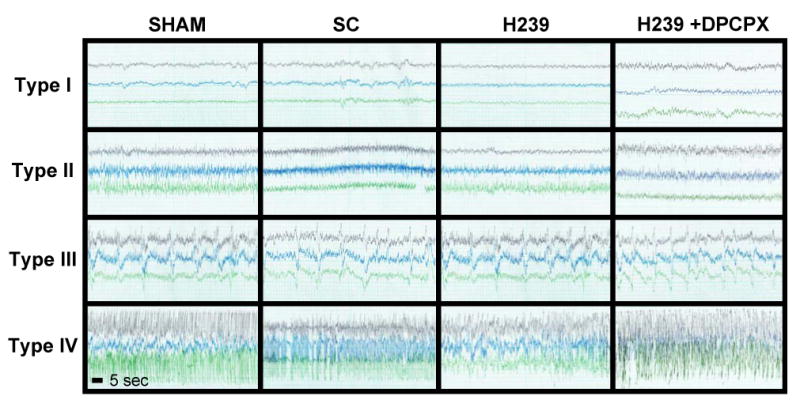
Representative traces show type I (baseline), type II (ictal fast activity), type III (high voltage spikes of < 1 Hz frequency superimposed over type II activity), and type IV (high-voltage, polyspike paroxysmal discharges of > 1 Hz frequency) electrographic activity recorded from 3 cortical electrodes after the injection of 0.3 μg KA into the basolateral amygdala of sham treated control mice (SHAM), recipients of scrambled control miRNA expressing hMSCs (SC), H239 hMSC graft recipients, or H239 graft recipients in which KA was paired with DPCPX (1mg/kg, i.p.). Typical traces representing each of the 4 seizure types were selected from a period spanning 0 to 30 min after KA-injection. Scale: each trace represents 20 sec recording time.
Reduction of KA-induced brain injury by adenosine-releasing hMSC implants
To investigate the influence of the hMSC-derived grafts on the development of acute seizure-induced hippocampal injury, all animals were analyzed histologically by Cresyl violet and TUNEL staining 24 h after KA injection (Fig. 6). The average number of TUNEL positive cells per brain slice was quantified by counting TUNEL positive cells from 3 adjacent brain sections, encompassing the entire CA1/3 fields, derived from 6 animals from each of the three different treatment groups. KA injected scrambled control-hMSC graft recipients as well as sham treated control animals and H239 graft recipients pretreated with DPCPX developed a well-defined lesion in the ipsilateral CA3 region of the hippocampal formation, which was characterized by prominent TUNEL-positive cells, indicative of ongoing apoptosis (Fig. 6A-F). In contrast, and in accordance with our previous data (Araki et al., 2002; Shinoda et al., 2004a; Shinoda et al., 2004b), in sham treated control animals neither the contralateral hippocampus, nor any other brain region, was affected by the KA-induced seizures (Fig. 6A,D). It is important to note that in contrast to one of our previous studies (Araki et al., 2002), under the conditions described here, injury in sham treated control animals and in the scrambled control and H239 graft recipients was consistently restricted to the ipsilateral CA3. The situation was completely different in H239-hMSC graft recipients (n = 6), which were characterized by a marked reduction in the CA3-selective injury (Fig. 6H). In contrast, neuroprotection by H239 cells was abolished, when KA was paired with the A1R antagonist DPCPX (Fig. 6J-L), indicating that neuroprotection depends on graft mediated adenosine release acting via A1Rs. Neuroprotection in H239 hMSC graft recipients was quantified by counting TUNEL-positive cells in the ipsilateral CA3 (Fig. 6C,F, I). In H239-hMSC graft recipients the number of TUNEL positive cells (27.3 ± 13.1) was significantly (P < 0.01) reduced compared to sham (77.5 ± 16.3) scrambled control (72.5 ± 15.2) or DPCPX pre-treated (74.7 ± 19.5) controls (Fig. 6).
Figure 6. Epilepsy associated cell loss.

Representative micrographs of the hippocampal formation from coronal brain sections taken from sham treated control mice (SHAM), recipients of scrambled control miRNA expressing hMSCs (SC), or H239 hMSC graft recipients 24 h after intraamygdaloid injection of 0.3 μg KA. Sections were stained either with cresyl violet (A,B,D,E,G,H,J,L) or with TUNEL (C,F,I,L green). (A-C) Typical apoptotic cell death in the CA3 region of the hippocampus of sham controls ipsilateral (ipsi) to the KA-injected amygdala. (D-F) CA3 selective apoptotic cell death in scrambled control-graft recipients was comparable to the sham-treated control animals. (G-I) H239 hMSC graft recipients show markedly decreased ipsilateral cell loss, which becomes evident by a hardly discernable lesion within the stratum pyramidale and by a prominent reduction in the number of TUNEL positive cells (I). (J-L) Sham- and scrambled- control like CA3 injury in H239 graft recipients in which KA was paired with DPCPX (1 mg/kg, i.p.). Scale bars: A,B,D,E,G,H,J,K: 100 μm; C,F,I,L: 25 μm.
Since H239-hMSC derived brain implants led to pronounced seizure suppression and neuroprotection at the time point of analysis (8 days after cell transplantation) we expected to find viable grafts in the treated animals. In coronal brain sections from animals sacrificed at that time-point we detected dense cellular transplants in the vicinity of the surgical injection tract, located exclusively within the infra-hippocampal cleft (Fig. 7). Cells were not found elsewhere, in particular not alongside the injection tract. Thus, we can likely exclude reflux of injected cells and conclude that all injected cells have populated the infrahippocampal cleft. In all animals (n = 6 per group) the location of the graft was confined to the ipsilateral hippocampus while the contralateral hippocampus as well as all other brain areas analyzed, were devoid of graft-derived cells.
Figure 7. Morphology of cell grafts.

Representative DAPI stained coronal brain sections from mice 8 days after transplantation of H239 transduced hMSCs cells. (A) Composite fluorescence image at lower magnification showing general graft morphology and location within the infrahippocampal cleft (arrows). (B) Fluorescence image of the boxed area in (A) showing the high density of nuclei from the graft (arrow). Note the presence of intact nuclei being indicative for graft survival. (C) Same image as (B) viewed under a GFP specific filter. Scale bars: A: 100 μm, B,C: 25 μm.
Discussion
Temporal lobe epilepsy is one of the most common forms of partial epilepsy and also one of the most difficult forms of epilepsy to treat since seizures frequently progress from focal, to secondarily generalized, and, frequently, to pharmacoresistant seizures. Thus, therapeutic alternatives are urgently needed and several focal treatment approaches for refractory epilepsy have been evaluated as potential strategies to circumvent side-effects frequently associated with classical antiepileptic drugs (Nilsen and Cock, 2004).
Cell transplantation for epilepsy can most easily be achieved by grafting of fetal tissue or cells. Thus, grafting of fetal hippocampal tissue with the aim to repair hippocampal networks in a kainic acid model of TLE led to partial reversal of hippocampal sclerosis (Rao et al., 2006; Shetty et al., 2005). Although cell transplantation approaches using fetal tissue or cells have yielded important basic results, stem cells have in recent years reached most attention and publicity as an alternative cell source for regenerative medicine. Thus, mouse embryonic stem cells have been engineered to release adenosine by bi-allelic genetic disruption of ADK induced by gene targeting (Fedele et al., 2004). Encapsulated intraventricular implants of these cells or their differentiated progeny provided transient but complete protection from kindled seizures (Güttinger et al., 2005a). More recently, ADK-deficient ES cell derived brain implants provided marked neuroprotection in a stroke model (Pignataro et al., 2006) and reduced kindling epileptogenesis when directly transplanted into the hippocampus of rats prior to the onset of kindling (Li et al., 2007b). Thus, focal augmentation of adenosine by stem cell derived brain implants has potent neuroprotective and antiepileptic properties in line with previous findings derived from encapsulated fibroblast and myoblast based cell systems (Boison, 2007). We have previously demonstrated that in contrast to pharmacological activation of A1 receptors focal cell-based adenosine release is not associated with sedative side-effects (Güttinger et al., 2005b). This is conceivable, since adenosine released from cellular brain implants will feed into the pool of endogenous (host-derived) adenosine and thus augment endogenous neuroprotective and antiepileptic functions of adenosine (Boison, 2007). In addition, adenosine is not expected to accumulate to undue levels since it is readily metabolized by host ADK (Boison, 2006). Thus, transplanted adenosine-releasing cells have the unique capability to augment the adenosinergic system in a region where this effect is needed (e.g. an epileptic focus), without having much impact on surrounding healthy brain areas. Although not observed in this study, migration of transplanted cells into other brain region is not expected to lead to unwanted adenosine-related side effects.
In the present study we developed novel methods and a proof-of-principle, which can be adapted to engineer patient identical hMSCs for therapeutic adenosine release and thus may provide the foundation for future clinical applications, in which autologous cell grafting of patient-derived adult stem cells might combine major advantages compared to encapsulated cell grafts, fetal cell grafts, or ES-cell derived grafts, such as (i) immunocompatibility; (ii) potential for long-term survival; and (iii) standardization prior to grafting. To implement focal adenosine delivery by autologous adult stem cell derived brain implants the following requirements have to be met: (i) identification of an easily accessible source of adult stem cells, which are amenable to genetic modification; (ii) development of an efficient technology to induce therapeutic adenosine delivery in these cells, and (iii) to demonstrate that these engineered cells have therapeutic potential. Based on these considerations, we developed here an efficient model system to engineer human mesenchymal stem cells for therapeutic adenosine release based on lentiviral miRNA-induced downregulation of the major adenosine-metabolic enzyme adenosine kinase (ADK). We demonstrate in a short-term proof-of-feasibility study that hMSCs engineered to release adenosine (i) have the potential to ameliorate acute seizures and (ii) to reduce the number of apoptotic neurons after brain injury. Based on these biological readouts we conclude that hMSCs with a knockdown of ADK release adenosine in therapeutically relevant amounts. Therapeutic long-term effects (over 26 days) of adenosine releasing embryonic stem cell derived brain implants were recently evaluated in a in a separate study in rats, in which we demonstrated suppression of kindling development, location of implanted cells within the infrahippocampal cleft, migration of graft derived cells into the ipsilateral CA1 region, expression of the neuronal marker NeuN, and lack of reactive gliosis in response to the graft (Li et al., 2007b). Since the major aim of the present study is the documentation that knockdown of ADK in hMSCs leads to therapeutically relevant effects, we chose a short-term model, in which we rapidly could assess the antiepileptic and neuroprotective potential of the cells. Any long-term uses of these cells including the analysis of host responses to the implants will be the subject of future studies.
Lentiviral expression of anti-ADK miRNA significantly reduces ADK expression in hMSCs
RNA interference (RNAi) has emerged as a specific and efficient method to silence gene expression in mammalian cells. Lentiviral RNAi based systems have thus been developed to generate replication incompetent lentiviral vectors that efficiently transduce both dividing and non-dividing mammalian cells and provide stable, long term expression of an miRNA of interest (Abbas-Terki et al., 2002; Stewart et al., 2003). The miRNA expression vectors used here enable bicistronic expression of an EmGFP reporter gene with the miRNA, thus allowing reliable tracking of miRNA expression in mammalian cells. It has been demonstrated that this bicistronic gene expression provides strong correlation of EmGFP fluorescence intensity with the knockdown of the target gene (Anderson et al., 2007). In our hands, we obtained transduction efficiencies ranging from 76% to 98% before, and after selection, respectively (Fig. 2). Most importantly, ADK expression in these transduced EmGFP positive cells was significantly down regulated to 20 to 54% of residual ADK levels (Fig. 3), and the expression of the transgene was stable for at least 8 weeks in vitro. We chose to work with polyclonal cell populations of blasticidin-selected cells to avoid unspecific effects caused by random genomic integration of the lentiviral vector. We have previously shown that even subtle changes in ADK activity lead to significant physiological changes. Thus, 1.8 to twofold increases in hippocampal ADK enzyme activity in vivo were associated with enhanced seizure susceptibility of the brain (Fedele et al., 2005; Gouder et al., 2004). Conversily, downregulation of ADK expression to 60% of wild-type levels in the forebrain of transgenic mice led to adenosine-mediated prevention of epileptogenesis (Li et al., 2007a). Thus, miRNA-mediated downregulation of ADK to up to 20% of baseline levels in hMSCs was expected to be of therapeutic relevance. Indeed, after 8 hours of incubation supernatants from 105 H239-hMSC contained 8.5 ng adenosine per ml medium, an amount about 25% lower than the release of adenosine from ADK-deficient C2C12-cells (11.5 ng adenosine / ml after incubating 105 cells for 8 hours), which was shown to be effective in preventing kindled seizures (Güttinger et al., 2005b).
Anti-ADK miRNA transduced hMSC implants prevent acute brain injury
To demonstrate the therapeutic potential of our novel approach to downregulate ADK in hMSCs using lentiviral expression of an miRNA directed against ADK, we chose a short-term seizure/cell death model, in which we assessed seizure duration and degree of neuronal cell death as read-outs. 8 days after cell transplantation and one day after KA injection the EmGFP-expressing hMSCs were found within the infrahippocampal cleft in a similar location reported previously using embryonic stem cell derived implants (Li et al., 2007b). The therapeutic effects of hMSC grafts with downregulated ADK were demonstrated by significantly reduced duration of type IV seizure activity and by significantly reduced cell loss compared to recipients of scrambled control cells and sham treated control animals. In contrast, recipients of scrambled control cells and sham treated control animals did neither differ in the duration of type IV seizures nor in the severity of injury. Specificity of the therapeutic effect to graft mediated adenosine was demonstrated in a control experiment in which we paired KA-injection with application of the selective A1R antagonist DPCPX. Under these conditions H239-dependent seizure suppression and neuroprotection was abolished. Since the severity of hippocampal injury is thought to depend on the severity of a preceding status epilepticus, which in turn depends on the tone of ambient adenosine, pharmacological blockade of A1 receptors increases both SE and hippocampal injury (Young and Dragunow, 1995). Conversely, the adenosine releasing hMSC implants described here reduced both the duration of type IV seizures and the associated CA3 neuronal cell death. Thus, the neuroprotective effects of the implant can be explained by both an indirect (by preventing SE) and a direct (adenosine mediated) neuroprotective effect.
Conclusions and outlook
Here we describe a novel strategy to engineer hMSCs to release therapeutically relevant amounts of adenosine. The hMSC grafts sufficiently suppress seizure activity and protect hippocampal neurons from damage. This approach provides the foundation to engineer patient derived adult stem cells for therapeutic adenosine delivery in autologous cell transplantation approaches. However, prior to clinical implementation of this novel strategy, several requirements need to be addressed in future studies such as (i) long-term survival of hMSC-derived brain implants, (ii) cellular differentiation of hMSC-derived brain implants, (iii) the potential to suppress chronic seizures, (iv) the potential to structurally integrate into hippocampal circuitry, and (v) the lack of insertional mutagenesis with the attendant risk of tumorigenicity. Apart from seizure control in epilepsy, adenosine releasing patient-derived hMSCs may become useful tools for the treatment of a variety of disorders, such as cerebral hypoxia and ischemia (Fredholm et al., 2001; Pignataro et al., 2006; Pignataro et al., 2007), chronic and neuropathic pain (Jarvis et al., 2002), or multiple sclerosis (Stevens et al., 2002).
Acknowledgments
This project was supported by grant R01 NS047622-01 from the National Institutes of Health, the Good Samaritan Hospital Foundation, and by the Epilepsy Research Foundation through the generous support of Arlene & Arnold Goldstein Family Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abbas-Terki T, Blanco-Bose W, Deglon N, Pralong W, Aebischer P. Lentiviral-mediated RNA interference. Hum Gene Ther. 2002;13:2197–2201. doi: 10.1089/104303402320987888. [DOI] [PubMed] [Google Scholar]
- Anderson GR, Semenov A, Song JH, Martemyanov KA. The membrane anchor R7BP controls the proteolytic stability of the striatal specific RGS protein, RGS9-2. J Biol Chem. 2007;282:4772–4781. doi: 10.1074/jbc.M610518200. [DOI] [PubMed] [Google Scholar]
- Araki T, Simon RP, Taki W, Lan JQ, Henshall DC. Characterization of neuronal death induced by focally evoked limbic seizures in the C57BL/6 mouse. J Neurosci Res. 2002;69:614–621. doi: 10.1002/jnr.10356. [DOI] [PubMed] [Google Scholar]
- Barry FP, Murphy JM. Mesenchymal stem cells: clinical applications and biological characterization. Int J Biochem Cell Biol. 2004;36:568–584. doi: 10.1016/j.biocel.2003.11.001. [DOI] [PubMed] [Google Scholar]
- Bjorklund A. Cell replacement strategies for neurodegenerative disorders. Novartis Found Symp. 2000;231:7–15. doi: 10.1002/0470870834.ch2. [DOI] [PubMed] [Google Scholar]
- Bjornson CRR, Rietze RL, Reynolds BA, Magli MC, Vescovi AL. Turning brain into blood: a hematopoietic fate adopted by adult neural stem cells in vivo. Science. 1999;283:534–537. doi: 10.1126/science.283.5401.534. [DOI] [PubMed] [Google Scholar]
- Black IB, Woodbury D. Adult rat and human bone marrow stromal stem cells differentiate into neurons. Blood Cells Molecules and Diseases. 2001;27:632–636. doi: 10.1006/bcmd.2001.0423. [DOI] [PubMed] [Google Scholar]
- Boison D. Adenosine and epilepsy: from therapeutic rationale to new therapeutic strategies. Neuroscientist. 2005;11:25–36. doi: 10.1177/1073858404269112. [DOI] [PubMed] [Google Scholar]
- Boison D. Adenosine kinase, epilepsy and stroke: mechanisms and therapies. Trends Pharmacol Sci. 2006;27:652–658. doi: 10.1016/j.tips.2006.10.008. [DOI] [PubMed] [Google Scholar]
- Boison D. Adenosine-based cell therapy approaches for pharmacoresistant epilepsies. Neurodegenerative Diseases. 2007;4:28–33. doi: 10.1159/000100356. [DOI] [PubMed] [Google Scholar]
- Brazelton TR, Rossi FM, Keshet GI, Blau HM. From marrow to brain: expression of neuronal phenotypes in adult mice. Science. 2000;290:1775–1779. doi: 10.1126/science.290.5497.1775. [DOI] [PubMed] [Google Scholar]
- Chu K, Kim M, Jeong SW, Kim SU, Yoon BW. Human neural stem cells can migrate, differentiate, and integrate after intravenous transplantation in adult rats with transient forebrain ischemia. Neurosci Lett. 2003;343:129–133. doi: 10.1016/s0304-3940(03)00174-5. [DOI] [PubMed] [Google Scholar]
- Chu K, Kim M, Jung KH, Jeon D, Lee ST, Kim J, Jeong SW, Kim SU, Lee SK, Shin HS, Roh JK. Human neural stem cell transplantation reduces spontaneous recurrent seizures following pilocarpine-induced status epilepticus in adult rats. Brain Res. 2004;1023:213–221. doi: 10.1016/j.brainres.2004.07.045. [DOI] [PubMed] [Google Scholar]
- Dichter MA. Innovative clinical trial designs for future antiepileptic drugs. Epilepsia. 2007;48 1:26–30. doi: 10.1111/j.1528-1167.2007.00996.x. [DOI] [PubMed] [Google Scholar]
- Dragunow M. Adenosine: the brain's natural anticonvulsant? Trends Pharmacol Sci. 1986;7:128. [Google Scholar]
- Dragunow M. Purinergic mechanisms in epilepsy. Prog Neurobiol. 1988;31:85–108. doi: 10.1016/0301-0082(88)90028-7. [DOI] [PubMed] [Google Scholar]
- Dragunow M. Adenosine and seizure termination. Ann Neurol. 1991;29:575. doi: 10.1002/ana.410290524. [DOI] [PubMed] [Google Scholar]
- Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature. 2001a;411:494–498. doi: 10.1038/35078107. [DOI] [PubMed] [Google Scholar]
- Elbashir SM, Lendeckel W, Tuschl T. RNA interference is mediated by 21- and 22-nucleotide RNAs. Genes Dev. 2001b;15:188–200. doi: 10.1101/gad.862301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedele DE, Koch P, Brüstle O, Scheurer L, Simpson EM, Mohler H, Boison D. Engineering embryonic stem cell derived glia for adenosine delivery. Neurosci Lett. 2004;370:160–165. doi: 10.1016/j.neulet.2004.08.031. [DOI] [PubMed] [Google Scholar]
- Fedele DE, Gouder N, Güttinger M, Gabernet L, Scheurer L, Rulicke T, Crestani F, Boison D. Astrogliosis in epilepsy leads to overexpression of adenosine kinase resulting in seizure aggravation. Brain. 2005;128:2383–2395. doi: 10.1093/brain/awh555. [DOI] [PubMed] [Google Scholar]
- Fernandes KJ, McKenzie IA, Mill P, Smith KM, Akhavan M, Barnabe-Heider F, Biernaskie J, Junek A, Kobayashi NR, Toma JG, Kaplan DR, Labosky PA, Rafuse V, Hui CC, Miller FD. A dermal niche for multipotent adult skin-derived precursor cells. Nat Cell Biol. 2004;6:1082–1093. doi: 10.1038/ncb1181. [DOI] [PubMed] [Google Scholar]
- Fernandes KJL, Kobayashi NR, Gallagher CJ, Barnabe-Heider F, Aumont A, Kaplan DR, Miller FD. Analysis of the neurogenic potential of multipotent skin-derived precursors. Exp Neurol. 2006;201:32–48. doi: 10.1016/j.expneurol.2006.03.018. [DOI] [PubMed] [Google Scholar]
- Ferrari G, Cusella-De Angelis G, Coletta M, Paolucci E, Stornaiuolo A, Cossu G, Mavilio F. Muscle regeneration by bone marrow-derived myogenic progenitors. Science. 1998;279:1528–1530. doi: 10.1126/science.279.5356.1528. [DOI] [PubMed] [Google Scholar]
- Franklin KBJ, Paxinos G. The mouse brain in stereotaxic coordinates. Academic Press, Inc.; San Diego: 1997. [Google Scholar]
- Fredholm BB, Ijzerman AP, Jacobson KA, Klotz KN, Linden J. International Union of Pharmacology. XXV. Nomenclature and classification of adenosine receptors. Pharmacol Rev. 2001;53:527–552. [PMC free article] [PubMed] [Google Scholar]
- Freed CR, Greene PE, Breeze RE, Tsai WY, DuMouchel W, Kao R, Dillon S, Winfield H, Culver S, Trojanowski JQ, Eidelberg D, Fahn S. Transplantation of embryonic dopamine neurons for severe Parkinson's disease. N Engl J Med. 2001;344:710–719. doi: 10.1056/NEJM200103083441002. [DOI] [PubMed] [Google Scholar]
- Galli R, Borello U, Gritti A, Minasi MG, Bjornson C, Coletta M, Mora M, De Angelis MG, Fiocco R, Cossu G, Vescovi AL. Skeletal myogenic potential of human and mouse neural stem cells. Nat Neurosci. 2000;3:986–991. doi: 10.1038/79924. [DOI] [PubMed] [Google Scholar]
- Gouder N, Fritschy JM, Boison D. Seizure suppression by adenosine A1 receptor activation in a mouse model of pharmacoresistant epilepsy. Epilepsia. 2003;44:877–885. doi: 10.1046/j.1528-1157.2003.03603.x. [DOI] [PubMed] [Google Scholar]
- Gouder N, Scheurer L, Fritschy JM, Boison D. Overexpression of adenosine kinase in epileptic hippocampus contributes to epileptogenesis. J Neurosci. 2004;24:692–701. doi: 10.1523/JNEUROSCI.4781-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gussoni E, Soneoka Y, Strickland CD, Buzney EA, Khan MK, Flint AF, Kunkel LM, Mulligan RC. Dystrophin expression in the mdx mouse restored by stem cell transplantation. Nature. 1999;401:390–394. doi: 10.1038/43919. [DOI] [PubMed] [Google Scholar]
- Güttinger M, Fedele DE, Koch P, Padrun V, Pralong W, Brüstle O, Boison D. Suppression of kindled seizures by paracrine adenosine release from stem cell derived brain implants. Epilepsia. 2005a;46:1–8. doi: 10.1111/j.1528-1167.2005.61804.x. [DOI] [PubMed] [Google Scholar]
- Güttinger M, Padrun V, Pralong W, Boison D. Seizure suppression and lack of adenosine A1 receptor desensitization after focal long-term delivery of adenosine by encapsulated myoblasts. Exp Neurol. 2005b;193:53–64. doi: 10.1016/j.expneurol.2004.12.012. [DOI] [PubMed] [Google Scholar]
- Hoelters J, Ciccarella M, Drechsel M, Geissler C, Gulkan H, Bocker W, Schieker M, Jochum M, Neth P. Nonviral genetic modification mediates effective transgene expression and functional RNA interference in human mesenchymal stem cells. Journal of Gene Medicine. 2005;7:718–728. doi: 10.1002/jgm.731. [DOI] [PubMed] [Google Scholar]
- Huber A, Padrun V, Deglon N, Aebischer P, Mohler H, Boison D. Grafts of adenosine-releasing cells suppress seizures in kindling epilepsy. Proc Natl Acad Sci USA. 2001;98:7611–7616. doi: 10.1073/pnas.131102898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson KA, Mi T, Goodell MA. Hematopoietic potential of stem cells isolated from murine skeletal muscle. Proc Natl Acad Sci U S A. 1999;96:14482–14486. doi: 10.1073/pnas.96.25.14482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarvis MF, Mikusa J, Chu KL, Wismer CT, Honore P, Kowaluk EA, McGaraughty S. Comparison of the ability of adenosine kinase inhibitors and adenosine receptor agonists to attenuate thermal hyperalgesia and reduce motor performance in rats. Pharmacol Biochem Behav. 2002;73:573–581. doi: 10.1016/s0091-3057(02)00840-7. [DOI] [PubMed] [Google Scholar]
- Jeong SW, Chu K, Jung KH, Kim SU, Kim M, Roh JK. Human neural stem cell transplantation promotes functional recovery in rats with experimental intracerebral hemorrhage. Stroke. 2003;34:2258–2263. doi: 10.1161/01.STR.0000083698.20199.1F. [DOI] [PubMed] [Google Scholar]
- Kassem M. Mesenchymal stem cells: biological characteristics and potential clinical applications. Cloning Stem Cells. 2004;6:369–374. doi: 10.1089/clo.2004.6.369. [DOI] [PubMed] [Google Scholar]
- Korbling M, Estrov Z, Champlin R. Adult stem cells and tissue repair. Bone Marrow Transplant. 2003;32 1:S23–24. doi: 10.1038/sj.bmt.1703939. [DOI] [PubMed] [Google Scholar]
- Kwan P, Brodie MJ. Refractory epilepsy: mechanisms and solutions. Expert Rev Neurother. 2006;6:397–406. doi: 10.1586/14737175.6.3.397. [DOI] [PubMed] [Google Scholar]
- Li T, Ren G, Lan JQ, Iwasato T, Itohara S, Fredholm B, Simon RP, Boison D. Adenosine kinase deficiency prevents epileptogenesis. 2007a under review. [Google Scholar]
- Li T, Steinbeck JA, Lusardi T, Koch P, Lan JQ, Wilz A, Segschneider M, Simon RP, Brustle O, Boison D. Suppression of kindling epileptogenesis by adenosine releasing stem cell-derived brain implants. Brain. 2007b;130:1276–1288. doi: 10.1093/brain/awm057. [DOI] [PubMed] [Google Scholar]
- Lindvall O. Neural transplantation in Parkinson's disease. Novartis Found Symp. 2000;231:110–123. 123–118, 145–117. discussion. [PubMed] [Google Scholar]
- Lindvall O, Bjorklund A. Cell therapy in Parkinson's disease. NeuroRx. 2004;1:382–393. doi: 10.1602/neurorx.1.4.382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loscher W, Schmidt D. New horizons in the development of antiepileptic drugs: the search for new targets. Epilepsy Res. 2004;60:77–159. doi: 10.1016/j.eplepsyres.2004.06.004. [DOI] [PubMed] [Google Scholar]
- Mezey E, Chandross KJ, Harta G, Maki RA, McKercher SR. Turning blood into brain: cells bearing neuronal antigens generated in vivo from bone marrow. Science. 2000;290:1779–1782. doi: 10.1126/science.290.5497.1779. [DOI] [PubMed] [Google Scholar]
- Mezey E, Key S, Vogelsang G, Szalayova I, Lange GD, Crain B. Transplanted bone marrow generates new neurons in human brains. Proc Natl Acad Sci U S A. 2003;100:1364–1369. doi: 10.1073/pnas.0336479100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz-Elias G, Marcus AJ, Coyne TM, Woodbury D, Black IB. Adult bone marrow stromal cells in the embryonic brain: engraftment, migration, differentiation, and long-term survival. J Neurosci. 2004;24:4585–4595. doi: 10.1523/JNEUROSCI.5060-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsen KE, Cock HR. Focal treatment for refractory epilepsy: hope for the future? Brain Res Brain Res Rev. 2004;44:141–153. doi: 10.1016/j.brainresrev.2003.11.003. [DOI] [PubMed] [Google Scholar]
- Petersen BE, Bowen WC, Patrene KD, Mars WM, Sullivan AK, Murase N, Boggs SS, Greenberger JS, Goff JP. Bone marrow as a potential source of hepatic oval cells. Science. 1999;284:1168–1170. doi: 10.1126/science.284.5417.1168. [DOI] [PubMed] [Google Scholar]
- Pignataro G, Studer FE, Wilz A, Simon RP, Boison D. Neuroprotection in ischemic mouse brain induced by stem cell derived brain implants. J Cereb Blood Flow Metab. 2006 Nov 22; doi: 10.1038/sj.jcbfm.9600422. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Pignataro G, Simon RP, Boison D. Transgenic overexpression of adenosine kinase aggravates cell death in ischemia. J Cereb Blood Flow Metab. 2007;27:1–5. doi: 10.1038/sj.jcbfm.9600334. [DOI] [PubMed] [Google Scholar]
- Pluchino S, Quattrini A, Brambilla E, Gritti A, Salani G, Dina G, Galli R, Del Carro U, Amadio S, Bergami A, Furlan R, Comi G, Vescovi AL, Martino G. Injection of adult neurospheres induces recovery in a chronic model of multiple sclerosis. Nature. 2003;422:688–694. doi: 10.1038/nature01552. [DOI] [PubMed] [Google Scholar]
- Rao MS, Hattiangady B, Shetty AK. Fetal hippocampal CA3 cell grafts enriched with FGF-2 and BDNF exhibit robust long-term survival and integration and suppress aberrant mossy fiber sprouting in the injured middle-aged hippocampus. Neurobiol Dis. 2006;21:276–290. doi: 10.1016/j.nbd.2005.07.009. [DOI] [PubMed] [Google Scholar]
- Rebola N, Coelho JE, Costenla AR, Lopes LV, Parada A, Oliveira CR, Soares-da-Silva P, de Mendonca A, Cunha RA. Decrease of adenosine A1 receptor density and of adenosine neuromodulation in the hippocampus of kindled rats. Eur J Neurosci. 2003;18:820–828. doi: 10.1046/j.1460-9568.2003.02815.x. [DOI] [PubMed] [Google Scholar]
- Remy S, Beck H. Molecular and cellular mechanisms of pharmacoresistance in epilepsy. Brain. 2006;129:18–35. doi: 10.1093/brain/awh682. [DOI] [PubMed] [Google Scholar]
- Sabers A, Gram L. Newer anticonvulsants: comparative review of drug interactions and adverse effects. Drugs. 2000;60:23–33. doi: 10.2165/00003495-200060010-00003. [DOI] [PubMed] [Google Scholar]
- Shetty AK, Turner DA. Development of fetal hippocampal grafts in intact and lesioned hippocampus. Progress in Neurobiology. 1996;50:597–653. doi: 10.1016/s0301-0082(96)00048-2. [DOI] [PubMed] [Google Scholar]
- Shetty AK, Turner DA. Fetal hippocampal grafts containing CA3 cells restore host hippocampal glutamate decarboxylase-positive interneuron numbers in a rat model of temporal lobe epilepsy. J Neurosci. 2000;20:8788–8801. doi: 10.1523/JNEUROSCI.20-23-08788.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shetty AK, Zaman V, Hattiangady B. Repair of the injured adult hippocampus through graft-mediated modulation of the plasticity of the dentate gyrus in a rat model of temporal lobe epilepsy. J Neurosci. 2005;25:8391–8401. doi: 10.1523/JNEUROSCI.1538-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinoda S, Araki T, Lan JQ, Schindler CK, Simon RP, Taki W, Henshall DC. Development of a model of seizure-induced hippocampal injury with features of programmed cell death in the BALB/c mouse. J Neurosci Res. 2004a;76:121–128. doi: 10.1002/jnr.20064. [DOI] [PubMed] [Google Scholar]
- Shinoda S, Schindler CK, Meller R, So NK, Araki T, Yamamoto A, Lan JQ, Taki W, Simon RP, Henshall DC. Bim regulation may determine hippocampal vulnerability after injurious seizures and in temporal lobe epilepsy. J Clin Invest. 2004b;113:1059–1068. doi: 10.1172/JCI19971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens B, Porta S, Haak LL, Gallo V, Fields RD. Adenosine: a neuron-glial transmitter promoting myelination in the CNS in response to action potentials. Neuron. 2002;36:855–868. doi: 10.1016/s0896-6273(02)01067-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart SA, Dykxhoorn DM, Palliser D, Mizuno H, Yu EY, An DS, Sabatini DM, Chen IS, Hahn WC, Sharp PA, Weinberg RA, Novina CD. Lentivirus-delivered stable gene silencing by RNAi in primary cells. Rna. 2003;9:493–501. doi: 10.1261/rna.2192803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiyama O, An DS, Kung SPK, Feeley BT, Gamradt S, Liu NQ, Chen ISY, Lieberman JR. Lentivirus-mediated gene transfer induces long-term transgene expression of BMP-2 in vitro and new bone formation in vivo. Molecular Therapy. 2005;11:390–398. doi: 10.1016/j.ymthe.2004.10.019. [DOI] [PubMed] [Google Scholar]
- Toma JG, Akhavan M, Fernandes KJL, Barnabe-Heider F, Sadikot A, Kaplan DR, Miller FD. Isolation of multipotent adult stem cells from the dermis of mammalian skin. Nat Cell Biol. 2001;3:778–784. doi: 10.1038/ncb0901-778. [DOI] [PubMed] [Google Scholar]
- Totsugawa T, Kobayashi N, Okitsu T, Noguchi H, Watanabe T, Matsumura T, Maruyama M, Fujiwara T, Sakaguchi M, Tanaka N. Lentiviral transfer of the LacZ gene into human endothelial cells and human bone marrow mesenchymal stem cells. Cell Transplantation. 2002;11:481–488. [PubMed] [Google Scholar]
- Vats A, Bielby RC, Tolley NS, Nerem R, Polak JM. Stem cells. Lancet. 2005;366:592–602. doi: 10.1016/S0140-6736(05)66879-1. [DOI] [PubMed] [Google Scholar]
- Woodbury D, Schwarz EJ, Prockop DJ, Black IB. Adult rat and human bone marrow stromal cells differentiate into neurons. J Neurosci Res. 2000;61:364–370. doi: 10.1002/1097-4547(20000815)61:4<364::AID-JNR2>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- Young D, Dragunow M. Neuronal injury following electrically induced status epilepticus with and without adenosine receptor antagonism. Exp Neurol. 1995;133:125–137. doi: 10.1006/exnr.1995.1015. [DOI] [PubMed] [Google Scholar]
- Zaman V, Turner DA, Shetty AK. Survival of grafted fetal neural cells in kainic acid lesioned CA3 region of adult hippocampus depends upon cell specificity. Exp Neurol. 2000;161:535–561. doi: 10.1006/exnr.1999.7304. [DOI] [PubMed] [Google Scholar]
- Zaman V, Shetty AK. Fetal hippocampal CA3 cell grafts transplanted to lesioned CA3 region of the adult hippocampus exhibit long-term survival in a rat model of temporal lobe epilepsy. Neurobiol Dis. 2001;8:942–952. doi: 10.1006/nbdi.2001.0440. [DOI] [PubMed] [Google Scholar]
- Zhao LR, Duan WM, Reyes M, Keene CD, Verfaillie CM, Low WC. Human bone marrow stem cells exhibit neural phenotypes and ameliorate neurological deficits after grafting into the ischemic brain of rats. Exp Neurol. 2002;174:11–20. doi: 10.1006/exnr.2001.7853. [DOI] [PubMed] [Google Scholar]