

Abstract
Microsatellite DNA loci or short tandem repeats (STRs) are abundant in eukaryotic genomes and are often used for constructing phylogenetic trees of closely related populations or species. These phylogenetic trees are usually constructed by using some genetic distance measure based on allele frequency data, and there are many distance measures that have been proposed for this purpose. In the past the efficiencies of these distance measures in constructing phylogenetic trees have been studied mathematically or by computer simulations. Recently, however, allele frequencies of 783 STR loci have been compiled from various human populations. We have therefore used these empirical data to investigate the relative efficiencies of different distance measures in constructing phylogenetic trees. The results showed that (1) the probability of obtaining the correct branching pattern of a tree (PC) is generally highest for DA distance; (2) FST*, standard genetic distance (DS), and 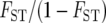 give similar PC-values, FST* being slightly better than the other two; and (3) (δμ)2 shows PC-values much lower than the other distance measures. To have reasonably high PC-values for trees similar to ours, at least 30 loci with a minimum of 15 individuals are required when DA distance is used.
give similar PC-values, FST* being slightly better than the other two; and (3) (δμ)2 shows PC-values much lower than the other distance measures. To have reasonably high PC-values for trees similar to ours, at least 30 loci with a minimum of 15 individuals are required when DA distance is used.
BECAUSE microsatellite DNA loci or short tandem repeat (STR) loci are highly polymorphic, they are very useful for studying the evolutionary relationships of closely related populations or species. However, a number of statistical problems should be solved before this approach can be used effectively. Some of the major problems are: (1) What kind of distance measures should be used for constructing phylogenetic trees?, (2) How many loci should be used?, and (3) How many individuals per locus should be used? Some of these problems have been studied theoretically or by computer simulation with specific mathematical models (e.g., Zhivotovsky and Feldman 1995; Takezaki and Nei 1996; Zhivotovsky et al. 2001; Kalinowski 2002). However, the assumptions for constructing mathematical models are not always realistic, and for this reason these studies may give wrong conclusions. Fortunately, a large number of data about STR loci have recently been accumulated from various human populations (Ramachandran et al. 2005; Rosenberg et al. 2005). We have therefore decided to study the above problems using these empirical data.
MATERIALS AND METHODS
Microsatellite data:
Genotypic data of 783 STR loci of 1027 individuals from 53 populations that were used by Ramachandran et al. (2005) for studying isolation by distance were downloaded from http://rosenberglab.bioinformatics.med.umich.edu/data/ramachandranEtAl2005/combinedmicrosats-1027.stru. Allele frequencies were computed by excluding missing data. Information regarding the STR loci such as the unit of nucleotide repeats and genomic locations of STR loci was obtained from http://rosenberglab.bioinformatics.med.umich.edu/data/rosenbergEtAl2002/diversityloci.txt and by internet search including databases of Marshfield (http://marshfieldclinic.org/research/pages/index.aspx) and UniSTS at NCBI (http://www.ncbi.nlm.nih.gov/). These data were essentially the same as the microsatellite data used by Rosenberg et al. (2005), although Rosenberg et al.'s data included insertion/deletion polymorphism data and data from the Surui population in Brazil. In our data set of 783 STR loci, there were 45 dinucleotide, 175 trinucleotide, 555 tetranucleotide, and 8 pentanucleotide repeat loci. Examining 1056 individuals from 52 populations, Zhivotovsky et al. (2003) studied the extent of variation in 377 STR loci, of which 45 were dinucleotide, 58 were trinucleotide, and 274 were tetranucleotide repeat loci. The same data set was also used by Rosenberg et al. (2002). The dinucleotide repeat loci used by Zhivotovsky et al. (2003) were the same as those in this study, and their trinucleotide and tetranucleotide repeat loci were a subset of the loci used in this study.
Genomic locations were available for ∼300 loci, but only a few pairs of loci were within a genomic distance of 1 Mbp. Therefore, the extent of linkage disequilibrium between the loci seems to be negligible.
The heterozygosity for a locus was calculated by 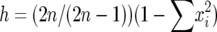 , where
, where 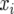 is the frequency of the ith allele in the sample, and n is the number of diploid individuals examined at the locus. In this study null alleles were excluded when allele frequencies were computed. Therefore, the actual 2n value could be smaller than two times the number of individuals examined. The average heterozygosity (H) for r loci was estimated by
is the frequency of the ith allele in the sample, and n is the number of diploid individuals examined at the locus. In this study null alleles were excluded when allele frequencies were computed. Therefore, the actual 2n value could be smaller than two times the number of individuals examined. The average heterozygosity (H) for r loci was estimated by 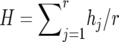 , whereas the sampling variance of H is computed by
, whereas the sampling variance of H is computed by 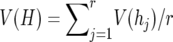 , where
, where 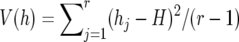 (Nei 1987). The expected value of heterozygosity [E(H)] in an equilibrium population is
(Nei 1987). The expected value of heterozygosity [E(H)] in an equilibrium population is 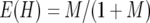 for the infinite-allele model (IAM) (Kimura and Crow 1964) and
for the infinite-allele model (IAM) (Kimura and Crow 1964) and 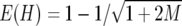 for the stepwise mutation model (SMM) (Ohta and Kimura 1973), where M = 4Nv, and N and v are the effective population size and mutation rate, respectively. We also examined the distribution of the average of the number (na) of alleles per locus within populations.
for the stepwise mutation model (SMM) (Ohta and Kimura 1973), where M = 4Nv, and N and v are the effective population size and mutation rate, respectively. We also examined the distribution of the average of the number (na) of alleles per locus within populations.
Genetic distance measures:
We examined the statistical properties of several genetic distance measures that are often used in actual data analysis. Most of them were originally developed for classical genetic markers such as blood group and isozyme loci that approximately follow the IAM, in which a new allele is always created by mutation. The DA distance (Nei et al. 1983) is defined as
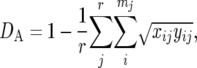 |
where xij and yij are the frequencies of the ith allele at the jth locus in populations X and Y, respectively, and mj is the number of alleles at the jth locus. Cavalli-Sforza and Edwards (1967) proposed the chord distance (DC) defined by
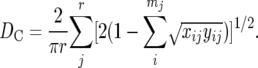 |
In the computer simulation by Takezaki and Nei (1996), the DA and DC distances showed the highest probabilities of obtaining the correct tree. However, since DA performed slightly better than DC in constructing phylogenetic trees, we used only DA in this study.
Nei's (1972) standard genetic distance (DS) is given by
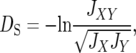 |
where 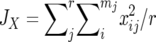 and
and 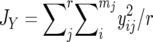 are the average homozygosities over the loci for populations X and Y, respectively, and
are the average homozygosities over the loci for populations X and Y, respectively, and 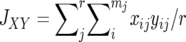 . Under the IAM with mutation–drift balance, E[DS], the expectation of DS, is given by 2vt, where v is a mutation rate per locus per generation and t is the time measured in generations after the two populations diverged. Therefore, DS is expected to increase linearly with time under the IAM.
. Under the IAM with mutation–drift balance, E[DS], the expectation of DS, is given by 2vt, where v is a mutation rate per locus per generation and t is the time measured in generations after the two populations diverged. Therefore, DS is expected to increase linearly with time under the IAM.
The FST* distance proposed by Latter (1972) is as follows:
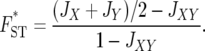 |
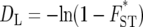 (Latter 1972; Reynolds et al. 1983) was also proposed as a distance measure. However, our previous studies showed that the probabilities of obtaining correct tree topologies were very similar for DL and FST* or slightly lower for DL than for FST*. Therefore, DL is not considered in this study. In this study we examine the following distance measure related to FST*, which was not included in our previous computer simulation:
(Latter 1972; Reynolds et al. 1983) was also proposed as a distance measure. However, our previous studies showed that the probabilities of obtaining correct tree topologies were very similar for DL and FST* or slightly lower for DL than for FST*. Therefore, DL is not considered in this study. In this study we examine the following distance measure related to FST*, which was not included in our previous computer simulation:
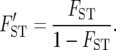 |
FST in the above formula is equivalent to Nei's (1973) GST (Slatkin 1995), and GST can be estimated by FST* in the case of two populations. Therefore, FST′ was computed by FST*/(1 − FST*) in this study. FST′ was originally proposed as a measure of isolation by distance (Slatkin 1993; Rousset 1997). It was shown to increase approximately linearly with time after the two populations separated for the case of low mutation rate (Slatkin 1995).
Some distance measures were developed specifically for STR loci in which the number of short nucleotide repeats was assumed to change following the SMM. Distance measure (δμ)2 was developed for this model in which the number of nucleotide repeats (i) changes to i − 1 or i + 1 with an equal probability (Goldstein et al. 1995). It is defined by
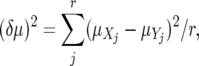 |
where 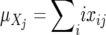 and
and 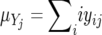 are the average repeat numbers of alleles at the jth locus, and xij and yij are the frequencies of the allele with repeat number (allele size) i at the jth locus in populations X and Y, respectively. Under the SMM with mutation–drift balance, the expected value of (δμ)2 is 2vt, where v is a mutation rate per locus per generation and t is the time measured in generations after the two populations diverged. Therefore, (δμ)2 is expected to increase linearly with time under the SMM.
are the average repeat numbers of alleles at the jth locus, and xij and yij are the frequencies of the allele with repeat number (allele size) i at the jth locus in populations X and Y, respectively. Under the SMM with mutation–drift balance, the expected value of (δμ)2 is 2vt, where v is a mutation rate per locus per generation and t is the time measured in generations after the two populations diverged. Therefore, (δμ)2 is expected to increase linearly with time under the SMM.
Measures of the efficiency of phylogenetic construction:
In this study, phylogenetic trees were constructed by the neighbor-joining method (Saitou and Nei 1987) with the genetic distance measures considered above. We examined the efficiencies of phylogenetic construction of different genetic distance measures by the nonparametric bootstrap approach. In this approach a certain number of loci (10, 20, 30, 50, 100, 300, 500, and 700) were chosen at random with replacement from the actual STR data set and neighbor-joining trees were constructed with the different distance measures mentioned above. In the examination of the effect of sample size on the efficiency of phylogenic construction, 5, 10, 15, 20, or 25 individuals were chosen at each locus, but in the other cases all the individuals were used. To examine the accuracy of a constructed tree, we need a true tree or a model tree with which the topology of the tree constructed can be compared. In the case of computer simulation, this model tree can be predetermined as in the case of Takezaki and Nei's (1996) study. In a study with empirical data there is no way to know the true tree that would reflect the historical relationship of the populations used. We have therefore decided to use a crude model tree based on the biogeographical distribution of populations and the consensus tree constructed by using all 783 STR loci with all distance measures used. Such a convergent topology is presented in Figure 1, and we used this tree topology as a model tree. We are aware of many arguments that can be raised against this approach. Interestingly, however, the same tree topology as this one was obtained by all distance measures when all STR loci were used. Therefore, this tree is not an arbitrary one but is supported by both biogeographical and genetic data. As far as the STR data are concerned, this is the tree to which all tree estimates are supposed to converge as the number of loci increases.
Figure 1.—

The phylogenetic trees of 12 representative populations. The phylogenetic trees were constructed with different distance measures using 783 STR loci. The phylogenetic trees with all the distance measures have the same topology. This tree topology is used as a model tree for testing the relative efficiencies of the different distance measures. (A) The DA tree. The numbers above and below the branches of the tree are bootstrap values and numbers assigned to the branches, respectively. (B) The DS tree. (C) The FST* tree. (D) The FST′ tree. (E) The (δμ)2 tree. (B–E) The numbers on the branches of the trees are bootstrap values. The countries where the populations are located are as follows: Yoruba, Nigeria; Mandenka, Senegal; Biaka Pygmy, Central African Republic; French, France; Basque, France; Russian, Russia; Japanese, Japan; Han, China; Cambodian, Cambodia; Pima, Mexico; Colombian, Columbia; and Karitiana, Brazil. There are two Han population samples in the data of Ramachandran et al. (2005) (see supplemental Tables 1–4 at http://www.genetics.org/supplemental/), Han from China and Han from northern China. Here we exclude the Han samples from northern China.
To measure the accuracy of a reconstructed tree, we computed the average of the topological distances (dT) (Robinson and Foulds 1981; Nei and Kumar 2000) of constructed trees from the model tree, the probability (PC) of obtaining the correct topology, and the probabilities (P1) of obtaining the groupings (partitions) of the populations separated by each interior branch of the model tree with 10,000 replications. It should be noted that dT is twice the number of partitions in a constructed tree different from those in the model tree. In this study we did not consider branch length errors, because it was difficult to determine the true or model branch lengths in the present case.
We computed the means and coefficients of variation (CVs) (standard deviation/mean) for the distance measures by the nonparametric bootstrap approach. The averages and the standard deviations of the distance values were computed for the distance values for 30 loci chosen at random in each replication by carrying out 10,000 replications.
RESULTS
Extent of variation in di-, tri-, and tetranucleotide repeat loci:
Because the extent of genetic polymorphism affects the efficiency of phylogenetic construction, we first examined the levels of heterozygosity for three different groups of STR loci with respect to the size of the unit of nucleotide repeats (Table 1). Average heterozygosities (H) were similar for three kinds of loci across all populations (0.715 for dinucleotide, 0.696 for trinucleotide, and 0.718 for tetranucletide repeat loci).
TABLE 1.
Average heterozygosities and numbers of alleles per locus for populations in different geographic regions
| Geographic region | No. of populations | Heterozygosity (H)
|
No. of alleles per locus per population (na)
|
||||||
|---|---|---|---|---|---|---|---|---|---|
| All | Dinucleotide | Trinucleotide | Tetranucleotide | All | Dinucleotide | Trinucleotide | Tetranucleotide | ||
| Africa | 8 | 0.757 | 0.793 | 0.763 | 0.753 | 5.79 | 6.84 | 5.84 | 5.69 |
| Europe | 8 | 0.729 | 0.745 | 0.716 | 0.732 | 5.88 | 6.54 | 5.76 | 5.88 |
| Middle East | 4 | 0.735 | 0.759 | 0.726 | 0.736 | 6.99 | 8.28 | 6.97 | 6.91 |
| Central/South Asia | 9 | 0.729 | 0.744 | 0.716 | 0.733 | 6.14 | 6.98 | 6.10 | 6.10 |
| Oceania | 2 | 0.667 | 0.657 | 0.627 | 0.681 | 5.07 | 5.31 | 4.77 | 5.16 |
| America | 4 | 0.606 | 0.564 | 0.561 | 0.625 | 4.41 | 4.60 | 4.10 | 4.51 |
| East Asia | 18 | 0.701 | 0.683 | 0.680 | 0.710 | 5.11 | 5.47 | 4.95 | 5.14 |
| All populations | 53 | 0.713 | 0.715 | 0.696 | 0.718 | 5.59 | 6.23 | 5.48 | 5.59 |
| SDa | 0.078 | 0.086 | 0.081 | 0.076 | 1.25 | 1.33 | 0.95 | 1.32 | |
| SEb | 0.004 | 0.020 | 0.010 | 0.005 | |||||
Standard deviation of heterozygosity (h) and the average of the number of alleles per locus per population (na).
Standard errors for average heterozygosities (H) (see materials and methods).
Table 1 also shows the average number (na) of alleles per locus for populations in different geographical regions. The average na for the entire set of populations is somewhat higher for dinucleotide repeat loci (6.23) than those for trinucleotide (5.48) and tetranucleotide repeat loci (5.58). The na's for trinucleotide and tetranucleotide repeat loci are similar, but the peak of the frequency distribution of na for tetranucleotide repeat loci is slightly shifted to the left and wider in comparison to that of trinucleotide repeat loci (Figure 2). Zhivotovsky et al. (2003) showed the frequency distributions of the number of alleles per locus for the entire population, not the average of the number of alleles per locus per population (na) that we examined in this study. We also computed the number of alleles per locus for the entire population (see supplemental Figure 1 at http://www.genetics.org/supplemental/). The shapes of the distributions of this quantity for the different repeat sizes are similar to those in Zhivotovsky et al. (2003), who used the subset of the loci used in this study, and to those of the averages of na in Figure 2.
Figure 2.—

Frequency distribution of average number of alleles per locus per population (na). (A) Dinucleotide repeat loci. (B) Trinucleotide repeat loci. (C) Tetranucleotide repeat loci.
Although the distributions of the average of na seem to be somewhat different among different types of STR loci of different repeat sizes, H values are similar for all three types of loci. Considering the large standard deviation associated with the na and relatively small numbers of dinucleotide (45) and trinucleotide (175) repeat loci, we decided to use all 783 loci together in the analysis of this study.
The values of H are the highest for the African populations and the smallest for the Oceanian and American populations (Table 1; for the heterozygosity values for all 53 populations see supplemental Tables 1–4 at http://www.genetics.org/supplemental/). The values of H are likely to reflect the large population size of Africans and the small population size of Oceanians and Americans. na is also smaller in these populations. But the na values of the Middle Eastern and Central/South Asian populations are higher than those of the African population. This could be due to the recent gene admixture that occurred in these regions.
Efficiency of constructing phylogenetic trees using different distance measures:
Table 2 shows the average topological distance (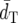 ) (measure of the extent of difference in topologies of constructed trees and the model tree) and the probabilities (PC) of obtaining the topology of the tree of 12 populations constructed from the 783 loci (model tree) (Figure 1) for the cases of 10, 20, 30, 50, 100, 300, 500, and 700 loci computed by the nonparametric bootstrap approach (see materials and methods).
) (measure of the extent of difference in topologies of constructed trees and the model tree) and the probabilities (PC) of obtaining the topology of the tree of 12 populations constructed from the 783 loci (model tree) (Figure 1) for the cases of 10, 20, 30, 50, 100, 300, 500, and 700 loci computed by the nonparametric bootstrap approach (see materials and methods).
TABLE 2.
Average topological distance and the probabilities of obtaining the correct tree topology for different distance measures
Average topological distance (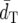 ) )
|
Probability of obtaining the correct topology (PC) (%)
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| No. loci | DA | DS | FST* | FST′ | (δμ)2 | DA | DS | FST* | FST′ | (δμ)2 |
| 10 | 4.8 ± 2.5 | 6.5 ± 2.8 | 5.9 ± 2.8 | 6.6 ± 2.8 | 10.7 ± 3.0 | 5.1 | 1.6 | 2.9 | 1.6 | 0.1 |
| 20 | 3.1 ± 2.1 | 4.6 ± 2.4 | 3.7 ± 2.9 | 4.5 ± 2.4 | 8.4 ± 2.8 | 16.1 | 6.2 | 10.8 | 6.8 | 0.4 |
| 30 | 2.2 ± 1.8 | 3.6 ± 2.2 | 2.8 ± 2.1 | 3.5 ± 2.2 | 7.3 ± 2.7 | 27.3 | 11.4 | 19.8 | 12.9 | 0.9 |
| 50 | 1.4 ± 1.5 | 2.6 ± 1.9 | 1.8 ± 1.7 | 2.4 ± 1.9 | 6.1 ± 2.5 | 46.9 | 22.2 | 35.7 | 24.9 | 1.7 |
| 100 | 0.5 ± 1.0 | 1.5 ± 1.5 | 0.9 ± 1.2 | 1.3 ± 1.4 | 4.8 ± 2.3 | 74.8 | 41.0 | 59.9 | 47.4 | 4.3 |
| 300 | 0.1 ± 0.4 | 0.8 ± 1.0 | 0.4 ± 0.8 | 0.6 ± 0.9 | 3.1 ± 2.0 | 95.1 | 62.5 | 80.7 | 72.9 | 14.8 |
| 500 | 0.0 ± 0.3 | 0.6 ± 0.9 | 0.3 ± 0.7 | 0.4 ± 0.8 | 2.4 ± 1.8 | 98.4 | 68.9 | 86.9 | 79.9 | 22.9 |
| 700 | 0.0 ± 0.1 | 0.5 ± 0.9 | 0.2 ± 0.6 | 0.3 ± 0.7 | 2.0 ± 1.6 | 99.5 | 72.8 | 90.4 | 84.2 | 28.8 |
Among the distance measures compared, DA generally shows the smallest 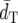 and the highest PC-values. Following DA, FST* has the second smallest
and the highest PC-values. Following DA, FST* has the second smallest 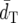 and the second highest PC.
and the second highest PC. 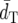 and PC-values of DS and FST′ are similar to each other, but FST′ has slightly lower
and PC-values of DS and FST′ are similar to each other, but FST′ has slightly lower 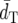 and higher PC-values than DS. (δμ)2 shows much higher
and higher PC-values than DS. (δμ)2 shows much higher 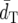 and lower PC-values than the other distance measures. This is because (δμ)2 has low efficiency in phylogeny construction because of its large sampling error (Zhivotovsky and Feldman 1995; Takezaki and Nei 1996; Goldstein and Pollock 1997; Zhivotovsky et al. 2001). With 700 loci, while the PC-value of DA becomes almost 100% and PC-values of FST*, DS, and FST′ are >70%, the PC-value of (δμ)2 is only <30%.
and lower PC-values than the other distance measures. This is because (δμ)2 has low efficiency in phylogeny construction because of its large sampling error (Zhivotovsky and Feldman 1995; Takezaki and Nei 1996; Goldstein and Pollock 1997; Zhivotovsky et al. 2001). With 700 loci, while the PC-value of DA becomes almost 100% and PC-values of FST*, DS, and FST′ are >70%, the PC-value of (δμ)2 is only <30%.
Table 3 shows the probabilities (P1) of obtaining the grouping of the populations (partitions) separated by each interior branch of the model tree (Figure 1). For example, at branch 1 of the model tree the populations are separated into Yoruba and Mandenka and the rest. The P1-value at branch 1 is a frequency that a branch separating populations into Yoruba and Mandenka and the rest appeared in a constructed tree in the nonparametric bootstrap replications. The general tendency of relative efficiencies of the different distance measures at each interior branch is similar to that observed in 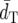 and PC as described above. DA has the highest efficiency in phylogeny construction among all the distance measures examined, FST* has the second highest, DS and FST′ have similar values, and (δμ)2 has much lower efficiency than the other distance measures. However, FST* has the highest P1-value and the P1-value of DA becomes the second highest at branch 6.
and PC as described above. DA has the highest efficiency in phylogeny construction among all the distance measures examined, FST* has the second highest, DS and FST′ have similar values, and (δμ)2 has much lower efficiency than the other distance measures. However, FST* has the highest P1-value and the P1-value of DA becomes the second highest at branch 6.
TABLE 3.
Probabilities (P1) of obtaining the correct partitions for different distance measures
| Probability of obtaining the correct partition (P1) (%)
|
|||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Branch 1
|
Branch 2
|
Branch 3
|
|||||||||||||
| No. loci | DA | DS | FST* | FST′ | (δμ)2 | DA | DS | FST* | FST′ | (δμ)2 | DA | DS | FST* | FST′ | (δμ)2 |
| 10 | 63 | 54 | 61 | 56 | 42 | 100 | 98 | 98 | 98 | 75 | 62 | 54 | 59 | 55 | 31 |
| 20 | 74 | 62 | 70 | 63 | 49 | 100 | 100 | 100 | 100 | 93 | 77 | 70 | 74 | 71 | 41 |
| 30 | 80 | 66 | 77 | 69 | 52 | 100 | 100 | 100 | 100 | 98 | 84 | 79 | 82 | 80 | 46 |
| 50 | 89 | 74 | 85 | 77 | 56 | 100 | 100 | 100 | 100 | 100 | 92 | 89 | 91 | 90 | 53 |
| 100 | 97 | 84 | 94 | 88 | 62 | 100 | 100 | 100 | 100 | 100 | 99 | 97 | 98 | 98 | 63 |
| 300 | 100 | 97 | 100 | 98 | 76 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 82 |
| 500 | 100 | 99 | 100 | 100 | 84 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 91 |
| 700 | 100 | 100 | 100 | 100 | 89 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 95 |
| Probability of obtaining the correct partition (P1) (%)
|
|||||||||||||||
| Branch 4
|
Branch 5
|
Branch 6
|
|||||||||||||
| No. loci | DA | DS | FST* | FST′ | (δμ)2 | DA | DS | FST* | FST′ | (δμ)2 | DA | DS | FST* | FST′ | (δμ)2 |
| 10 | 84 | 67 | 74 | 65 | 45 | 47 | 35 | 42 | 36 | 23 | 59 | 53 | 63 | 52 | 28 |
| 20 | 95 | 82 | 90 | 81 | 63 | 58 | 43 | 52 | 45 | 28 | 73 | 67 | 80 | 66 | 40 |
| 30 | 98 | 89 | 95 | 88 | 72 | 64 | 48 | 57 | 50 | 31 | 80 | 74 | 87 | 73 | 47 |
| 50 | 100 | 95 | 99 | 94 | 83 | 72 | 52 | 64 | 57 | 34 | 89 | 83 | 95 | 81 | 54 |
| 100 | 100 | 99 | 100 | 99 | 94 | 82 | 58 | 71 | 65 | 38 | 97 | 93 | 99 | 91 | 64 |
| 300 | 100 | 100 | 100 | 100 | 100 | 95 | 65 | 81 | 74 | 43 | 100 | 100 | 100 | 100 | 81 |
| 500 | 100 | 100 | 100 | 100 | 100 | 98 | 69 | 87 | 80 | 46 | 100 | 100 | 100 | 100 | 88 |
| 700 | 100 | 100 | 100 | 100 | 100 | 100 | 73 | 90 | 84 | 46 | 100 | 100 | 100 | 100 | 91 |
| Probability of obtaining the correct partition (P1) (%)
|
|||||||||||||||
| Branch 7
|
Branch 8
|
Branch 9
|
|||||||||||||
| No. loci | DA | DS | FST* | FST′ | (δμ)2 | DA | DS | FST* | FST′ | (δμ)2 | DA | DS | FST* | FST′ | (δμ)2 |
| 10 | 81 | 66 | 67 | 67 | 34 | 99 | 88 | 87 | 84 | 55 | 65 | 57 | 57 | 57 | 36 |
| 20 | 93 | 83 | 83 | 85 | 49 | 100 | 99 | 98 | 97 | 77 | 76 | 67 | 67 | 67 | 42 |
| 30 | 97 | 90 | 90 | 92 | 58 | 100 | 100 | 100 | 99 | 89 | 83 | 74 | 74 | 75 | 45 |
| 50 | 99 | 96 | 96 | 97 | 69 | 100 | 100 | 100 | 100 | 97 | 91 | 83 | 83 | 83 | 49 |
| 100 | 100 | 100 | 100 | 100 | 83 | 100 | 100 | 100 | 100 | 100 | 98 | 93 | 93 | 93 | 54 |
| 300 | 100 | 100 | 100 | 100 | 98 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 67 |
| 500 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 73 |
| 700 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 79 |
P1-values at the branches that separate the clusters of the four geographic regions require smaller numbers of loci to reach high values (e.g., 90%) than those at the branches within the major geographic clusters. P1-values for DA, DS, FST*, and FST′ are already ≥98% with 10 loci at branch 2 that separates Africans and non-Africans and ≥97% with 20 loci at branch 8 for the cluster of Americans. However, P1-values of the four distance measures for the clusters of Europeans and East Asians require 50 and 100 loci to reach 90%. P1-values of (δμ)2 are lower than those for the other distances, but they reach 90% with 20 loci at the branch for the African cluster and with 50 loci for the American cluster. P1-values of (δμ)2 for the cluster of East Asians and for the clusters of East Asians and Americans need 300–500 loci to reach 90%. P1-values of DA, DS, FST*, and FST′ at the branches within the clusters of the four geographic regions become ≥90% with 100 loci at branches 3 and 9 and with 300 loci at branch 1. P1-values at branch 5 become 95% for DA with 300 loci, but 70–80% for DS, FST*, and FST′. The P1-value of (δμ)2 at branch 5 is <50% even with 700 loci and needs ≥500 loci to reach 90% at the other branches within the clusters of the major geographic regions.
Means and sampling errors of distance measures:
The efficiency of constructing phylogenetic trees by distance measures depends on their linearity with time and sampling errors (Goldstein and Pollock 1994; Tajima and Takezaki 1994; Takezaki and Nei 1996). Therefore, we examined the means and CVs of the distance measures. Figure 3 shows the means and CVs of the distance measures of each pair of the 12 populations (Figure 1) in relation to the mean (δμ)2 computed for the same pair of populations in the abscissa by choosing 30 loci in the nonparametric bootstrap approach. In the data set used in this study, the assumptions for the linear increase of the expected (δμ)2 with time apparently do not hold. The different heterozygosity values for the populations of the different geographic origins (Table 1) indicate that population sizes have changed some times. Further, the mutational pattern of microsatellite loci does not strictly follow the SMM (Di Rienzo et al. 1994; Ellegren 2000; Xu et al. 2000; Huang et al. 2002; Ellegren 2004). The other distance measures increase approximately linearly in short-term evolution, but lose the linearity with time quickly under the SMM (Takezaki and Nei 1996). Therefore, the means and CVs of the distance measures are shown in relation to the mean (δμ)2 in the abscissa.
Figure 3.—

Relationships of the means and coefficients of variation of different distance measures in relation to the mean (δμ)2. Average distance values and the coefficients of variation were computed for the 12 populations in Figure 1 by choosing 30 loci in each replication of the nonparametric bootstrap approach. (A) Means. (B) Coefficients of variation.
The slope of the mean DS is the steepest among those of DA, DS, FST*, and FST′. When the mean (δμ)2 in the abscissa is close to zero, the mean DS is smaller than that of DA, but slightly larger than those of FST* and FST′ (Figure 3A). However, as the average (δμ)2 increases, the mean DS becomes larger than the mean DA and becomes the largest of the means of the four distance measures. The slopes of mean DA, FST*, and FST′ are similar to one another, although the mean DA is much larger than those of FST* and FST′ for all the (δμ)2 values. Means of FST* and FST′ are similar to each other, but the mean FST′ is larger than that of FST* particularly for the larger abscissa values.
CVs of (δμ)2 are much higher than those of the other distance measures (Figure 3B) as expected from theoretical and computer simulation studies (Zhivotovsky and Feldman 1995; Takezaki and Nei 1996; Zhivotovsky et al. 2001). By contrast, DA has the smallest CVs for all the abscissa values. CVs of DS, FST*, and FST′ are intermediate between those of (δμ)2 and DA, although CVs of these three distance measures are extremely high for small abscissa values (<0.2). CVs of DS, FST*, and FST′ are very similar to one another, but DS generally has the largest CVs of the three, FST′ has the second largest CVs, and CVs of FST* are the smallest.
In general, CV values are inversely related to the PC-values of the distance measures. DA with the highest PC-values has the smallest CVs, and FST* with the second-highest PC-values has the second-smallest CVs. (δμ)2 that has the largest CVs shows the lowest PC-values. Thus, as far as the data set used in this study is concerned, the relative values of CVs of the distance measures are a main factor that affects their performance in constructing phylogenies, and the effect of the linear increase of the distance measure with time seems minor.
Sample size:
To investigate the effect of sample size on the efficiency of phylogeny construction, we constructed phylogenetic trees for the 12 populations by sampling a different number of individuals (n) per locus. Average n values of the 783 loci are 20–45 for most of the populations (see supplemental Tables 1–4 at http://www.genetics.org/supplemental/). Average n values of the 12 populations in the model tree (Figure 1) are ∼25 except for Cambodian and Colombian populations whose average n values are 11.5 and 12.5, respectively.
We chose 5–25 individuals per locus for each population at random without replacement and recalculated the allele frequencies and computed PC for a different number of loci (10–700). If the number of individuals at a locus was <5–25, we used all the available samples. Table 4 shows PC-values for DA distance. Note that the result of the other distance measures was essentially the same as that for DA (data not shown). As expected for the data of high heterozygosities (0.6–0.8) (Table 1 and supplemental Tables 1–4 at http://www.genetics.org/supplemental/) (Takezaki and Nei 1996), the effect of sample size on PC-values is quite large. By increasing n from 5 to 15, PC-values increase to a great extent. In the case of 300–700 loci PC-values are >80–90% for n = 15–25, but PC-values are 40–60% for n = 5. It should be noted, however, that the number of loci generally has a larger effect on PC-values than the sample size when n ≥ 15.
TABLE 4.
The probabilities of obtaining the correct tree topology (PC) (%) for different sample sizes
| Diploid sample size (n)
|
|||||
|---|---|---|---|---|---|
| No. loci | 5 | 10 | 15 | 20 | 25 |
| 10 | 0 | 2 | 3 | 4 | 5 |
| 20 | 2 | 6 | 10 | 14 | 15 |
| 30 | 4 | 12 | 19 | 23 | 26 |
| 40 | 6 | 18 | 27 | 33 | 37 |
| 50 | 8 | 23 | 32 | 41 | 46 |
| 100 | 21 | 44 | 58 | 67 | 71 |
| 200 | 34 | 64 | 79 | 84 | 87 |
| 300 | 44 | 74 | 86 | 90 | 92 |
| 500 | 56 | 83 | 89 | 95 | 97 |
| 700 | 61 | 88 | 93 | 96 | 98 |
DA distance was used. n is the number of individuals sampled from the data. If the n values in the data were smaller than the values in the table, all of the samples were used.
DISCUSSION
Using microsatellite data of human populations, this study showed that DA distance is the most efficient in obtaining a correct branching pattern, followed by FST*, FST′, and DS, and that (δμ)2 has much lower efficiency than the other distance measures. This result is consistent with the previous computer simulation study, although efficiency of FST′ was not examined (Takezaki and Nei 1996). DA, DS, FST*, and FST′ were originally developed for classical genetic markers that the IAM can apply. Mean values for these distance measures approximately increase linearly with time after separation of the two populations for a small 2vt value (expected number of mutations) even under the SMM, but the rate of the increase slows down for large 2vt values. Theoretically, (δμ)2 is expected to be proportional to mutation rate under the SMM. However, (δμ)2 has a much larger sampling error than the other distance measures. As far as the data examined in this study are concerned, the extent of sampling error is a major factor that affects the efficiencies of phylogenetic construction of the distance measures.
The PC-values for the actual STR loci obtained in this study are comparable to those in the case of H = 0.8 and the largest 2vt between populations = 0.56 in the computer simulation with the SMM (Takezaki and Nei 1996). They tend to be lower than those in the simulation for all the distance measures. But PC-values of (δμ)2 are particularly lower in this data analysis than in the simulation in comparison to the other distance measures. The mutational pattern of STR loci is believed to roughly follow the SMM. However, there are irregular changes such as a large number of nucleotide repeat changes or disruption of nucleotide repeats, the constraints for the repeat number, and the asymmetric mutation rate for contraction and expansion of the allele (Di Rienzo et al. 1994; Goldstein and Pollock 1997; Kruglyak et al. 1998) at STR loci. Furthermore, the mutation rates vary among the STR loci (Ellegren 2004). Such complication of the mutational pattern in these loci may have affected the efficiency of (δμ)2 in phylogeny construction. Cooper et al. (1999) found that the standard deviation of (δμ)2 is much larger than the expected value (Zhivotovsky and Feldman 1995; Goldstein and Pollock 1997) by studying 213 dinucleotide STR loci of four human populations. At any rate, the present study showed that (δμ)2 is not efficient in reconstructing phylogenetic trees of populations even if a large number of loci are used.
We already have a DOS version of the computer program (POPTREE) for constructing phylogenetic trees for STR data with bootstrapping. It is available free of charge from http://www.bio.psu.edu/People/Faculty/Nei/Lab/software.htm. We are currently in the process of converting the program to a Windows version.
References
- Cavalli-Sforza, L. L., and A. W. Edwards, 1967. Phylogenetic analysis. Models and estimation procedures. Am. J. Hum. Genet. 19: 233–257. [PMC free article] [PubMed] [Google Scholar]
- Cooper, G., W. Amos, R. Bellamy, M. R. Siddiqui, A. Frodsham et al., 1999. An empirical exploration of the (δμ)2 genetic distance for 213 human microsatellite markers. Am. J. Hum. Genet. 65: 1125–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Rienzo, A., A. C. Peterson, J. C. Garza, A. M. Valdes, M. Slatkin et al., 1994. Mutational processes of simple-sequence repeat loci in human populations. Proc. Natl. Acad. Sci. USA 91: 3166–3170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellegren, H., 2000. Heterogeneous mutation processes in human microsatellite DNA sequences. Nat. Genet. 24: 400–402. [DOI] [PubMed] [Google Scholar]
- Ellegren, H., 2004. Microsatellites: simple sequences with complex evolution. Nat. Rev. Genet. 5: 435–445. [DOI] [PubMed] [Google Scholar]
- Goldstein, D. B., and D. D. Pollock, 1994. Least squares estimation of molecular distance–noise abatement in phylogenetic reconstruction. Theor. Popul. Biol. 44: 219–226. [DOI] [PubMed] [Google Scholar]
- Goldstein, D. B., and D. D. Pollock, 1997. Launching microsatellites: a review of mutation processes and methods of phylogenetic inference. J. Hered. 88: 335–342. [DOI] [PubMed] [Google Scholar]
- Goldstein, D. B., A. Ruiz Linares, L. L. Cavalli-Sforza and M. W. Feldman, 1995. Genetic absolute dating based on microsatellites and the origin of modern humans. Proc. Natl. Acad. Sci. USA 92: 6723–6727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, Q. Y., F. H. Xu, H. Shen, H. Y. Deng, Y. J. Liu et al., 2002. Mutation patterns at dinucleotide microsatellite loci in humans. Am. J. Hum. Genet. 70: 625–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalinowski, S. T., 2002. Evolutionary and statistical properties of three genetic distances. Mol. Ecol. 11: 1263–1273. [DOI] [PubMed] [Google Scholar]
- Kimura, M., and J. F. Crow, 1964. The number of alleles that can be maintained in a finite population. Genetics 49: 523–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruglyak, S., R. T. Durrett, M. D. Schug and C. F. Aquadro, 1998. Equilibrium distributions of microsatellite repeat length resulting from a balance between slippage events and point mutations. Proc. Natl. Acad. Sci. USA 95: 10774–10778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latter, B. D., 1972. Selection in finite populations with multiple alleles. III. Genetic divergence with centripetal selection and mutation. Genetics 70: 475–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nei, M., 1972. Genetic distance between populations. Am. Nat. 106: 283–291. [Google Scholar]
- Nei, M., 1973. Analysis of gene diversity in subdivided populations. Proc. Natl. Acad. Sci. USA 70: 3321–3323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nei, M., 1987. Molecular Evolutionary Genetics. Columbia University Press, New York.
- Nei, M., and S. Kumar, 2000. Molecular Phylogenetics and Evolution. Oxford University Press, New York.
- Nei, M., F. Tajima and Y. Tateno, 1983. Accuracy of estimated phylogenetic trees from molecular data. II. Gene frequency data. J. Mol. Evol. 19: 153–170. [DOI] [PubMed] [Google Scholar]
- Ohta, T., and M. Kimura, 1973. A model of mutation appropriate to estimate the number of electrophoretically detectable alleles in a finite population. Genet. Res. 22: 201–204. [DOI] [PubMed] [Google Scholar]
- Ramachandran, S., O. Deshpande, C. C. Roseman, N. A. Rosenberg, M. W. Feldman et al., 2005. Support from the relationship of genetic and geographic distance in human populations for a serial founder effect originating in Africa. Proc. Natl. Acad. Sci. USA 102: 15942–15947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds, J., B. D. Weir and C. C. Cockerham, 1983. Estimation of the coancestry coefficient: basis for a short-term genetic distance. Genetics 105: 765–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson, D. F., and L. R. Foulds, 1981. Comparison of phylogenetic trees. Math. Biosci. 53: 131–147. [Google Scholar]
- Rosenberg, N. A., J. K. Pritchard, J. L. Weber, H. M. Cann, K. K. Kidd et al., 2002. Genetic structure of human populations. Science 298: 2381–2385. [DOI] [PubMed] [Google Scholar]
- Rosenberg, N. A., S. Mahajan, S. Ramachandran, C. Zhao, J. K. Pritchard et al., 2005. Clines, clusters, and the effect of study design on the inference of human population structure. PLoS Genet. 1: e70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousset, F., 1997. Genetic differentiation and estimation of gene flow from F-statistics under isolation by distance. Genetics 145: 1219–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitou, N., and M. Nei, 1987. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 4: 406–425. [DOI] [PubMed] [Google Scholar]
- Slatkin, M., 1993. Isolation by distance in equilibrium and non-equilibrium populations. Evolution 47: 264–279. [DOI] [PubMed] [Google Scholar]
- Slatkin, M., 1995. A measure of population subdivision based on microsatellite allele frequencies. Genetics 139: 457–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajima, F., and N. Takezaki, 1994. Estimation of evolutionary distance for reconstructing molecular phylogenetic trees. Mol. Biol. Evol. 11: 278–286. [DOI] [PubMed] [Google Scholar]
- Takezaki, N., and M. Nei, 1996. Genetic distances and reconstruction of phylogenetic trees from microsatellite DNA. Genetics 144: 389–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, X., M. Peng, Z. Fang and X. Xu, 2000. The direction of microsatellite mutations is dependent upon allele length. Nat. Genet. 24: 396–399. [DOI] [PubMed] [Google Scholar]
- Zhivotovsky, L. A., and M. W. Feldman, 1995. Microsatellite variability and genetic distances. Proc. Natl. Acad. Sci. USA 92: 11549–11552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhivotovsky, L. A., D. B. Goldstein and M. W. Feldman, 2001. Genetic sampling error of distance (δμ)2 and variation in mutation rate among microsatellite loci. Mol. Biol. Evol. 18: 2141–2145. [DOI] [PubMed] [Google Scholar]
- Zhivotovsky, L. A., N. A. Rosenberg and M. W. Feldman, 2003. Features of evolution and expansion of modern humans, inferred from genomewide microsatellite markers. Am. J. Hum. Genet. 72: 1171–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]