

Abstract
TNF receptor-associated periodic syndrome (TRAPS) is a dominantly inherited disease caused by missense mutations in the TNF receptor 1 (TNFR1) gene. Patients suffer from periodic bouts of severe abdominal pain, localised inflammation, migratory rashes, and fever. More than 40 individual mutations have been identified, all of which occur in the extracellular domain of TNFR1. In the present review we discuss new findings describing aberrant trafficking and function of TNFR1 harbouring TRAPS mutations, challenging the hypothesis that TRAPS pathology is driven by defective receptor shedding, and we suggest that TNFR1 might acquire novel functions in the endoplasmic reticulum, distinct from its role as a cell surface receptor. We also describe the clinical manifestations of TRAPS, current treatment regimens, and the widening array of patient mutations.
Introduction
The TNF-receptor associated periodic syndrome (TRAPS) is a dominantly inherited periodic fever syndrome characterised by prolonged episodic fevers, multiorgan involvement, and resistance to colchicine. Over 40 missense mutations in TNF receptor 1 (TNFR1), the prototypical proinflammatory receptor, have been associated with TRAPS. In the present review we discuss recent evidence that the unifying molecular defect in TRAPS-associated mutant TNFR1 is receptor misfolding and retention in the endoplasmic reticulum (ER). This finding constitutes a novel mechanism of receptor misbehaviour in genetic disease and suggests hitherto unexpected functions for intracellularly retained receptors in promoting inflammation.
Periodic fevers and TRAPS
The periodic fevers are a group of disorders characterised by unprovoked attacks of fever and localised inflammation that can affect multiple organ systems [1]. There are several periodic fever syndromes that are inherited in a recessive manner, such as familial Mediterranean fever (FMF) caused by mutations in the gene encoding the neutrophil-specific protein Pyrin [2]. Most evidence currently suggests that pyrin negatively regulates the production of IL-1β – a major proinflammatory cytokine of the innate immune response [3]. IL-1β is generated from its proform via cleavage by caspase-1, which occurs in a specialised IL-1 activating protein complex termed the inflammasome [4-6]. FMF-associated mutations in pyrin are thought to exert less inhibition of IL-1β processing [7]. Another such recessive disease is hyperimmunoglobulinemia-D with periodic fever syndrome, which arises due to mutations in mevalonate kinase – a key enzyme in cholesterol biosynthesis and the synthesis of nonsterol isoprenoid molecules [8,9]. The exact molecular basis for the disease resulting from these mutations, however, remains unclear.
The molecular basis of two groups of dominant periodic fever syndrome has been identified. Dominant mutations in the CIAS1 gene are associated with a heterogeneous group of inflammatory conditions encompassing Muckle–Wells syndrome, familial cold autoinflammatory syndrome, and the neonatal onset multisystem inflammatory disease [10]. CIAS1 is one of the intracellular sensors that activate the inflammasome, and it is thought that CIAS1 mutations associated with these diseases might lead to its constitutive activation. Through studies of cohorts of patients with dominantly inherited familial periodic fevers, formerly classified as dominant periodic syndrome and familial Hibernian fever, mutations in the TNFR1 gene on chromosome 12p13 were identified. These patients were therefore classified as having TRAPS [11-13]. TNFR1 is the archetypal proinflammatory receptor and a member of the TNF receptor superfamily – a large group of proteins with homology in their extracellular domains, which are involved in a variety of processes including inflammation, T-cell activation, B-cell homeostasis, and osteoclast function.
The mean age of onset for TRAPS is 3 years, but diagnoses have been made at ages ranging from 2 weeks to 53 years. Unlike FMF, in which attacks are predictably less than 5 days in duration, febrile attacks in TRAPS can last from a few days to months, with an average duration of 21 days. There is no fixed periodicity between attacks; they can occur as frequently as every 5–6 weeks, or patients can be symptom-free for months to years. Attacks are usually unprovoked, but they have been reported to be triggered or aggravated by several factors, such as local injury, minor infection, stress, exercise, and hormonal changes – attacks are often relieved during pregnancy [14,15]. The most common symptoms associated with an attack are fever, a migrating radial rash on the limbs, and extreme abdominal and muscle pain, but symptoms can also include conjunctivitis, headaches, chest pain, myalgia, arthralgia, and less commonly arthritis and other inflammatory manifestations including fasciitis, myocarditis, sacroiliitis, pharyngitis, and stomatitis [16-19]. The most serious complication associated with TRAPS is systemic amyloidosis – extracellular deposits of an insoluble cleavage product of the acute-phase reactant protein serum amyloid A. Many patients with renal amyloidosis develop nephrotic syndrome and progress to renal failure.
Laboratory findings in TRAPS are in accordance with a typical inflammatory episode: high levels of C-reactive protein, neutrophilia, moderate complement activation, raised mean erythrocyte sedimentation rate, and, in some cases, marginally raised IgA and IgD levels [16,17,20-22]. Importantly, acute phase proteins can also be elevated between attacks, suggesting that subclinical inflammation may be more chronic [23]. Cytokine profiling of a small group of TRAPS patients revealed that IL-6 was relatively more elevated than TNFα, when compared with a group of rheumatoid arthritis patients [24].
Corticosteroids, colchicine, and high doses of nonsteroidal anti-inflammatory drugs have all been used to treat TRAPS with varied success. Unlike FMF, TRAPS patients generally respond poorly to colchicine [15,25]. TRAPS symptoms can be relieved with high doses of prednisolone (> 20 mg), although efficacy fades with time [13]. The greatest success in the treatment of TRAPS has been observed with etanercept (soluble TNFR2 fused to the Fc region of human IgG) in a number of small uncontrolled clinical trials [26-32]. Treatment with infliximab (anti-TNFα) in some cases, however, led to exacerbation of the disease [33,34]. Interestingly, anakinra (an IL-1 receptor antagonist) was remarkably successful in the treatment of a TRAPS patient resistant to anti-TNF therapy [35]. Since no randomised controlled trials of therapy have been carried out for this rare syndrome, there is currently no single recommended treatment.
Mutations in TNFR1 associated with TRAPS
The TRAPS mutations were discovered through screening programmes of patients and families who had previously presented with unexplained periodic fever. Although initial mutations were identified in patients of Irish descent (which gave rise to its original name of familial Hibernian fever), it is increasingly evident that the disease is ethnically diverse – TRAPS mutations are being discovered in Japanese, Turkish, and Arabic populations, hinting at underdiagnosis in these populations rather than prior absence [36-38].
Discovery of the mutations in TNFR1 associated with TRAPS was a seminal finding that connected this poorly understood disease with one of the canonical receptors that mediates inflammation [13]. TNFR1 is a prototypic member of the TNF receptor superfamily, characterised by a distinct pattern of disulphide bonding in the extracellular cysteine-rich domains (CRDs). TNFR1 has four CRDs in its extracellular region and an intracellular death domain that can initiate signalling leading to two opposing outcomes – inflammation and cell survival, versus cell death via apoptosis – depending on the cell type and intracellular milieu [39].
The crystal structure of TNFR1 in complex with one of its ligands, lymphotoxin-α (TNFβ), demonstrates that the majority of contact residues between ligand and receptor are located in CRD2 [40]. Recent work has demonstrated that members of the TNF receptor superfamily, including TNFR1, can self-associate in the membrane in the absence of ligand. Self-association is mediated by the so-called preligand assembly domain, which is located in the first CRD [41]. Interestingly, a structure of TNFR1 in the absence of its ligand TNF revealed a dimer of two receptors with extensive contacts between receptors in CRD1 [42].
Figure 1 illustrates the location of the TRAPS mutations, which occur predominantly in CRD1 and CRD2; there are no known TRAPS mutations in the transmembrane or intracellular domain of TNFR1. A definitive list of all the TRAPS mutations can be found on the INFEVERS website [43]. Many of the mutations involve cysteine residues involved in intramolecular disulphide bonds, and others occur at residues predicted to have a pronounced effect on the overall secondary structure, such as the introduction or removal of a proline (P46L, L67P, S86P, R92P), or residues involved in hydrogen bonding between loops of the receptor (T50M, I170N) [44]. Other than single base pair changes, there is a splicing mutation that introduces an extra four amino acids [45], a single amino acid deletion [20], and an inframe interstitial deletion of nine amino acids, all in the extracellular domain [16]. Remarkably, there are no mutations encoding large truncations or deletions within the receptor, which suggests that synthesis of the mutant protein is important for disease pathogenesis.
Figure 1.

TNF receptor-associated periodic syndrome mutations in TNF receptor 1. Schematic of TNF receptor-associated periodic syndrome (TRAPS) mutations in TNF receptor 1 (TNFR1) illustrating the extreme clustering of mutations towards cysteine-rich domains CRD1 and CRD2. (a) The preligand assembly domain (PLAD), ligand binding domain, and other structural features of TNFR1 are highlighted. (b) A line model of CRD1 and CRD2 of TNFR1, with the sites of TRAPS mutations and amino acid (aa) changes indicated (red boxes).
In addition to the TNFR1 mutations identified in TRAPS that do not occur in the normal population, two rare coding sequence variants in TNFR1 that alter single amino acids have been identified as low-penetrance risk factors for TRAPS. The R92Q amino acid substitution is carried at a low frequency (~2%) in North American and Irish populations [23,45], and the P46L mutation was found in 9% of African populations [46]. TRAPS patients with these polymorphisms have a milder syndrome, with less severe flares and almost no incidence of amyloidosis. The R92Q mutation has also been linked with other diseases associated with inflammation, such as rheumatoid arthritis and atherosclerosis [47-49]. Although the penetrance of TRAPS is high among patients with the rare TNFR1 mutations (>80%), several asymptomatic family members have been reported, suggesting that other genetic and/or environmental factors also play a role in triggering the symptoms of TRAPS.
Molecular basis of TRAPS: new answers and new questions
In light of our current understanding of TNF receptor signalling, this disease presents a paradox. TNFα is a prototypic proinflammatory cytokine, and exerts most of its biologic effects through TNFR1. The question therefore beckons of how these mutations, which would be predicted to lead to a nonfunctional receptor and therefore less TNFR1 signalling, can drive a proinflammatory condition. Following the earliest research, several hypotheses were proposed [13,31].
Firstly, it was suggested that the mutations might render the receptor constitutively active. Since cysteines are often affected, it seemed possible that receptors could be cross-linked at the surface through the formation of aberrant disulphide bonds between receptor pairs, resulting in constitutive TNFR1 signalling.
A second theory was that TRAPS mutations increase the affinity of the receptor for TNFα – a concept difficult to rationalise as mutations would be predicted to have a profound effect on the overall structure of TNFRI, and which is not supported by binding measurements of radiolabelled TNFα to patients' leukocytes [31].
A third hypothesis is that TNFR1 mutations in TRAPS cause impaired cleavage of membrane-bound TNFR1, leading to reduced serum levels of soluble TNFR1. This hypothesis was supported by initial observations that some TRAPS patient cells are resistant to phorbol-myristate acetate-induced 'shedding' of TNFR1 from the surface, and that serum from TRAPS patients contained reduced levels of circulating soluble TNFR1, relative to controls [13].
Functional buffering of TNF by soluble TNFR1, generated through metalloprotease-dependent cleavage of cell surface TNFR1, is a well-defined phenomenon that appears to occur constitutively, but can also occur in response to several cytokines and TNFα itself (reviewed in [50]). The resultant reservoir of free soluble TNFR1 has a homeostatic effect on signalling that is twofold: decreasing the number of sites available on the membrane for binding, and providing a pool of receptors to sequester TNFα [51]. Shed TNFR1 can function as a soluble inhibitor of TNF and forms the basis for using etanercept to treat TRAPS. In addition, it has been shown that full-length TNFR1 is released in exosome-like microvesicles, which would also be expected to contribute to functional buffering of TNFα signalling [52].
In an attempt to mimic the accepted TRAPS 'phenotype' of impaired receptor shedding, Xanthoulea and colleagues created a knock-in mouse that expressed a nonsheddable form of TNFR1 [53]. Mice with either heterozygous or homozygous expression of the otherwise functional receptor showed enhanced TNF signalling, with spontaneous hepatitis and increased susceptibility and severity of induced arthritis. These data indicate that either a failure to cleave TNFR1 from the cell surface or a failure to form soluble TNFR1 can predispose spontaneous inflammation – supporting an anti-inflammatory role for soluble TNFR1.
Several metalloproteases have been implicated in the cleavage of membrane-bound TNFR1 [54]. Since these enzymes target a sequence in the membrane proximal region of the receptor, it seems unlikely that mutations in domains away from this region could cause complete inhibition of cleavage. Despite this, much of the early functional work on TRAPS concentrated on this concept, which has been termed the 'shedding hypothesis' (Figure 2). The shedding defect in TRAPS patients' cells, however, was subsequently found to be variable, especially between the type of cells studied, and did not occur in all cases [55]. Since shedding can also result from the secretion of exosomal vesicles, the observed defect could equally be due to a defect in this pathway, which can be triggered from within the cell [52]. Alternative explanations for the pathogenesis of inflammation in TRAPS have therefore been sought.
Figure 2.
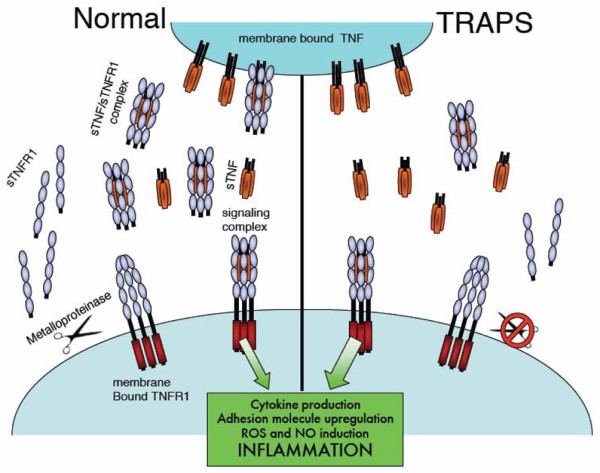
The shedding hypothesis. Mutations in TNF receptor 1 (TNFR1) were thought to impair cleavage of TNFR1 from the membrane by membrane metalloproteases, leading to depletion in the pool of soluble TNF (sTNF) receptor (sTNFR1). In this model for TNF receptor-associated periodic syndrome mutations (TRAPS), both soluble and membrane-bound TNFα are no longer sequestered by the soluble receptor, resulting in overstimulation of membrane-bound TNFR1. NO, nitric oxide; ROS, reactive oxygen species.
New concepts in the understanding of TRAPS have arisen from studies by our laboratories, and others, showing that receptor misfolding and mislocalisation is a universal feature of TNFR1 mutations in TRAPS. TNF binding studies in cells and in vitro have demonstrated that the TRAPS-associated mutant TNFR1 is unable to bind to TNF. Furthermore, we and other workers have now demonstrated that the mutant receptors fail to localise to the cell surface [56]. This defect was also observed in cells from TNFR1 'knock-in' mice expressing the T50M mutation [56]. Significantly, the TRAPS-associated mutant TNFR1 was specifically retained in the ER – the retention site for misfolded proteins in the secretory pathway.
The molecular basis of intracellular retention of TRAPS-associated mutants appears to be misfolding – based on the findings that the mutant receptors fail to bind TNF, they do not make normal preligand assembly domain-dependent interactions with wild-type TNFR1, and, due to aberrant intermolecular disulphide bonds, can form high-order oligomers [56]. At higher levels of expression, it is probable that TRAPS-associated mutant TNFR1 exits the ER through the process of ER-associated degradation and accumulates in cytoplasmic punctate structures that colocalise with chaperone proteins termed 'aggresomes', destined for proteasome-mediated disposal [56,57]. Most of this work, however, has been carried out using cDNA transfected cells, which some may argue are a poor model for ER studies. Hence, it will be interesting to see data to support the ER retention model from future studies using cells from TRAPS patients.
Unlike all the TRAPS mutations, the rare TNFR1 variant R92Q behaves similarly to wild-type TNFR1 in terms of ligand binding and trafficking, indicating that the mechanism of disease associated with this variant receptor is probably different to that of the mutations strictly associated with TRAPS. Since TRAPS is an autosomal dominant disease, with patients generally carrying one wild-type allele and one mutant allele, it was important to test the effects of coexpression of mutant and wild-type TNFR1. Despite the dramatic defects in TRAPS-associated mutant TNFR1, transfection studies found that the mutant TNFR1 did not significantly interfere with the trafficking or function of the wild-type allele [56].
These findings suggest that inflammation in TRAPS results from either haploinsufficiency of wild-type receptors or some critical function gained by the mutant TNFR1. In our opinion, haploinsufficiency of surface or soluble TNFR1 seems unlikely given the fact that no known TRAPS mutations lead to complete lack of protein expression. New studies will probably focus on the consequences of intracellularly retained mutant TNFR1 in TRAPS. It is known that TRAPS-associated mutants have functional death domains and retain the ability to activate NF-κB [56-59], but, as mentioned previously, mutant receptors do not seem to induce increased NF-κB transactivation in transfected cells.
Accumulation of mutant TNFR1 in the ER may trigger the ER stress response, which could directly or indirectly lead to inflammation. One such mechanism is the so-called ER overload response that can activate NF-κB activation [60,61]. Interestingly, CREBH, a recently identified ER sensor protein expressed in hepatocytes, can directly induce expression of acute phase proteins in response to ER stress [62]. In addition, TNFR1 itself has been implicated in the pathway leading from ER stress to activation of the JNK family of mitogen-activated protein kinases [63]. In a transfection system mutant, however, TNFR1 did not appear to directly activate ER stress responses, so this seems less likely to be at the crux of the disease mechanism [56].
A final possibility is that the retained receptors in TRAPS may alter the balance between the TNFR1-initiated signalling pathways. TNFR1 can signal via two opposing pathways that lead to either apoptosis via caspase activation or to cellular survival and inflammation via the activation of NF-κB (Figure 3). Much work has been carried out to decipher how these two opposing pathways are regulated, and it is thought that a crucial difference in the threshold of TNF activation is required for cytokine activation via NF-κB, or apoptosis induction. The accepted model for TNFR1 signalling is one in which the survival genes are the first to be activated, through the formation of a surface-bound complex. The apoptosis pathway occurs later, upon formation of a second intracellular complex in receptosomes [64,65]. Interestingly, neutrophils and dermal fibroblasts from TRAPS patients with several different mutations undergo reduced apoptosis when exposed to TNF [16,66]. Dermal fibroblasts derived from a patient with the C43S mutation showed a reduced level of apoptosis compared with normal controls, but intact production of the proinflammatory cytokines IL-6 and IL-8 when exposed to TNF. Failure of activated immune cells to undergo apoptosis in TRAPS could lead to a build up of proinflammatory cytokines. Figure 4 illustrates the possible differences in the trafficking and signalling of TNFR1 in TRAPS patient cells.
Figure 3.
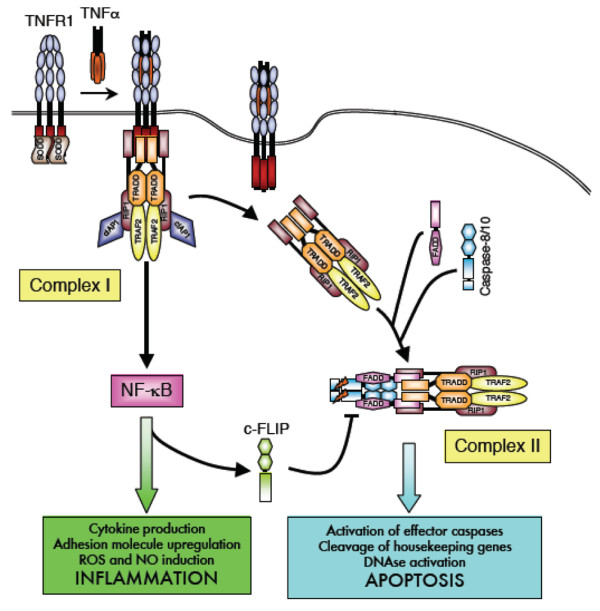
TNF receptor 1 signalling. TNF receptor 1 (TNFR1) ligation induces death domain-mediated recruitment of TNFR1/TRADD/TRAF2/RIP1 (complex I) at the cell surface, and this complex initiates NF-κB activation, which in turn drives production of cFLIP, an apoptotic inhibitor. Activated TNFR1 is then endocytosed and the TRADD/TRAF2/RIP1 complex dissociates from the receptor in a temporal manner. This complex then recruits FADD and caspase-8 (complex II), which activates the apoptotic machinery, provided that the levels of cFLIP are low enough to remove inhibition. Adapted from [64]. In a second model, proposed by Schneider-Brachart and colleagues [65], the apoptotic complex is internalised upon assembly in receptosomes, which fuse with golgi vesicles and signal for apoptosis from within the cell. NO, nitric oxide; ROS, reactive oxygen species.
Figure 4.
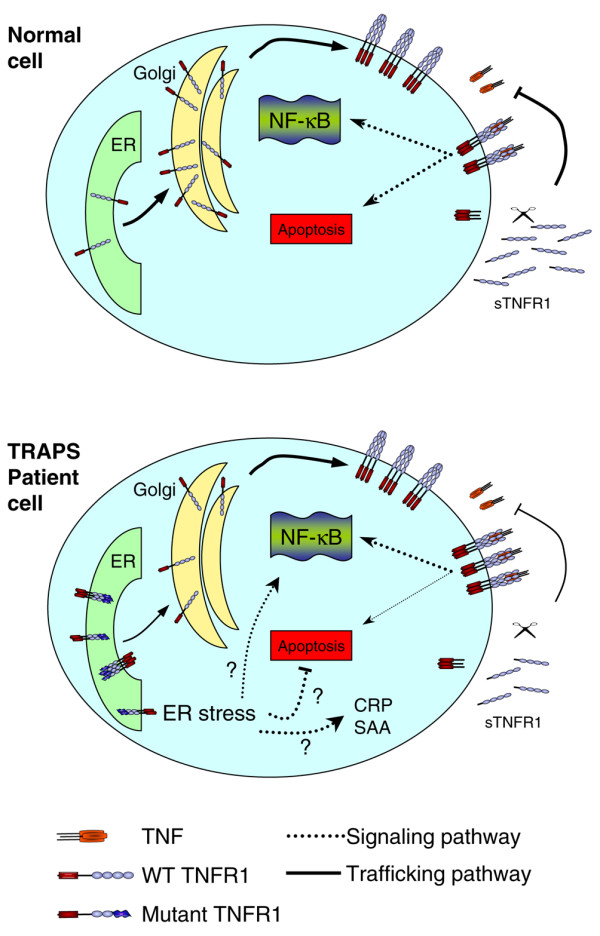
Signalling and trafficking pathways for wild-type and mutant TNF receptor 1. In TNF receptor-associated periodic syndrome mutations (TRAPS) patient cells, mutant TNF receptor 1 (TNFR1) are retained in the endoplasmic reticulum (ER) as disulphide-linked oligomers while the wild-type (WT) receptors traffic to the cell surface, leaving TNFα-induced NF-κB activation intact. ER-retained oligomers may independently activate novel signalling pathways leading to inflammation, may modify ER stress-induced responses, or may block TNFα-induced apoptosis. Mutant TNFR1 does not contribute to the antagonistic pool of soluble TNFR1 because it does not bind ligand. CRP, C-reactive protein; SAA, serum amyloid A; sTNFR1, soluble TNFR1.
Conclusion
Contrary to initial findings, the systemic inflammation that is characteristic of TRAPS may not be solely due to a defect in receptor shedding, but instead could stem from the consequences of misfolded and intracellularly retained TNFR1. It is, however, possible that there is not one unifying mechanism for all TRAPS mutations – the variability in symptoms among patients may be a clue towards this, and there is also a strong possibility of linked susceptibility genes.
At present, some success with anti-TNF agents is likely to steer therapeutics in this direction – but while these therapeutics may alleviate the inflammatory symptoms in some cases, extracellular TNF blockade may not address the underlying molecular mechanism of this disease. For the new findings to bear fruit, it will be crucial to determine the exact mechanism by which intracellular retention of the TRAPS mutant TNFR1 can drive inflammation. In neurons, the accumulation of misfolded proteins is thought to cause cell death and play a major role in the pathogenesis of neurodegenerative diseases, but the consequences of misfolded proteins in immune cells has not been well studied. Conversely, the effect of TNFR1 mutations outside immune cells also needs to be examined. Understanding the signalling pathways connecting protein misfolding and inflammation will be interesting in relation to this rare genetic syndrome, and will also be potentially relevant to more common inflammatory conditions.
Abbreviations
CRD = cysteine-rich domain; ER = endoplasmic reticulum; FMF = familial Mediterranean fever; IL = interleukin; NF = nuclear factor; TNF = tumour necrosis factor; TNFR1 = tumour necrosis factor receptor 1; TRAPS = tumour necrosis factor receptor-associated periodic syndrome.
Competing interests
The authors declare that they have no competing interests.
Acknowledgments
Acknowledgements
FCK and AAL contributed equally to this work. The authors would like to thank Anna Simon for useful comments and discussion during the preparation of this manuscript.
Contributor Information
Fiona C Kimberley, Email: F.C.Kimberley@amc.uva.nl.
Adrian A Lobito, Email: lobito.adrian@gene.com.
Richard M Siegel, Email: siegelr@mail.nih.gov.
Gavin R Screaton, Email: g.screaton@imperial.ac.uk.
References
- Drenth JP, van der Meer JW. Hereditary periodic fever. N Engl J Med. 2001;345:1748–1757. doi: 10.1056/NEJMra010200. [DOI] [PubMed] [Google Scholar]
- Centola M, Wood G, Frucht DM, Galon J, Aringer M, Farrell C, Kingma DW, Horwitz ME, Mansfield E, Holland SM, et al. The gene for familial Mediterranean fever, MEFV, is expressed in early leukocyte development and is regulated in response to inflammatory mediators. Blood. 2000;95:3223–3231. [PubMed] [Google Scholar]
- Chae JJ, Komarow HD, Cheng J, Wood G, Raben N, Liu PP, Kastner DL. Targeted disruption of pyrin, the FMF protein, causes heightened sensitivity to endotoxin and a defect in macrophage apoptosis. Mol Cell. 2003;11:591–604. doi: 10.1016/S1097-2765(03)00056-X. [DOI] [PubMed] [Google Scholar]
- Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell. 2002;10:417–426. doi: 10.1016/S1097-2765(02)00599-3. [DOI] [PubMed] [Google Scholar]
- Hiller S, Kohl A, Fiorito F, Herrmann T, Wider G, Tschopp J, Grutter MG, Wuthrich K. NMR structure of the apoptosis- and inflammation-related NALP1 pyrin domain. Structure. 2003;11:1199–1205. doi: 10.1016/j.str.2003.08.009. [DOI] [PubMed] [Google Scholar]
- Drenth JP, van der Meer JW. The inflammasome – a linebacker of innate defense. N Engl J Med. 2006;355:730–732. doi: 10.1056/NEJMcibr063500. [DOI] [PubMed] [Google Scholar]
- Chae JJ, Wood G, Masters SL, Richard K, Park G, Smith BJ, Kastner DL. The B30.2 domain of pyrin, the familial Mediterranean fever protein, interacts directly with caspase-1 to modulate IL-1beta production. Proc Natl Acad Sci USA. 2006;103:9982–9987. doi: 10.1073/pnas.0602081103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drenth JP, Cuisset L, Grateau G, Vasseur C, van de Velde-Visser SD, de Jong JG, Beckmann JS, van der Meer JW, Delpech M. Mutations in the gene encoding mevalonate kinase cause hyper-IgD and periodic fever syndrome. International Hyper-IgD Study Group. Nat Genet. 1999;22:178–181. doi: 10.1038/9696. [DOI] [PubMed] [Google Scholar]
- Houten SM, Kuis W, Duran M, de Koning TJ, van Royen-Kerkhof A, Romeijn GJ, Frenkel J, Dorland L, de Barse MM, Huijbers WA, et al. Mutations in MVK, encoding mevalonate kinase, cause hyperimmunoglobulinaemia D and periodic fever syndrome. Nat Genet. 1999;22:175–177. doi: 10.1038/9691. [DOI] [PubMed] [Google Scholar]
- Agostini L, Martinon F, Burns K, McDermott MF, Hawkins PN, Tschopp J. NALP3 forms an IL-1beta-processing inflammasome with increased activity in Muckle–Wells autoinflammatory disorder. Immunity. 2004;20:319–325. doi: 10.1016/S1074-7613(04)00046-9. [DOI] [PubMed] [Google Scholar]
- Mulley J, Saar K, Hewitt G, Ruschendorf F, Phillips H, Colley A, Sillence D, Reis A, Wilson M. Gene localization for an autosomal dominant familial periodic fever to 12p13. Am J Hum Genet. 1998;62:884–889. doi: 10.1086/301793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDermott MF, Ogunkolade BW, McDermott EM, Jones LC, Wan Y, Quane KA, McCarthy J, Phelan M, Molloy MG, Powell RJ, et al. Linkage of familial Hibernian fever to chromosome 12p13. Am J Hum Genet. 1998;62:1446–1451. doi: 10.1086/301886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDermott MF, Aksentijevich I, Galon J, McDermott EM, Ogunkolade BW, Centola M, Mansfield E, Gadina M, Karenko L, Pettersson T, et al. Germline mutations in the extracellular domains of the 55 kDa TNF receptor, TNFR1, define a family of dominantly inherited autoinflammatory syndromes. Cell. 1999;97:133–144. doi: 10.1016/S0092-8674(00)80721-7. [DOI] [PubMed] [Google Scholar]
- Kriegel MA, Huffmeier U, Scherb E, Scheidig C, Geiler T, Kalden JR, Reis A, Lorenz HM. Tumor necrosis factor receptor-associated periodic syndrome characterized by a mutation affecting the cleavage site of the receptor: implications for pathogenesis. Arthritis Rheum. 2003;48:2386–2388. doi: 10.1002/art.11169. [DOI] [PubMed] [Google Scholar]
- McDermott EM, Smillie DM, Powell RJ. Clinical spectrum of familial Hibernian fever: a 14-year follow-up study of the index case and extended family. Mayo Clin Proc. 1997;72:806–817. doi: 10.4065/72.9.806. [DOI] [PubMed] [Google Scholar]
- D'Osualdo A, Ferlito F, Prigione I, Obici L, Meini A, Zulian F, Pontillo A, Corona F, Barcellona R, Di Duca M, et al. Neutrophils from patients with TNFRSF1A mutations display resistance to tumor necrosis factor-induced apoptosis: pathogenetic and clinical implications. Arthritis Rheum. 2006;54:998–1008. doi: 10.1002/art.21657. [DOI] [PubMed] [Google Scholar]
- Saulsbury FT, Wispelwey B. Tumor necrosis factor receptor-associated periodic syndrome in a young adult who had features of periodic fever, aphthous stomatitis, pharyngitis, and adenitis as a child. J Pediatr. 2005;146:283–285. doi: 10.1016/j.jpeds.2004.09.006. [DOI] [PubMed] [Google Scholar]
- Trost S, Rose CD. Myocarditis and sacroiliitis: 2 previously unrecognized manifestations of tumor necrosis factor receptor associated periodic syndrome. J Rheumatol. 2005;32:175–177. [PubMed] [Google Scholar]
- Hull KM, Wong K, Wood GM, Chu WS, Kastner DL. Monocytic fasciitis: a newly recognized clinical feature of tumor necrosis factor receptor dysfunction. Arthritis Rheum. 2002;46:2189–2194. doi: 10.1002/art.10448. [DOI] [PubMed] [Google Scholar]
- Aganna E, Hammond L, Hawkins PN, Aldea A, McKee SA, van Amstel HK, Mischung C, Kusuhara K, Saulsbury FT, Lachmann HJ, et al. Heterogeneity among patients with tumor necrosis factor receptor-associated periodic syndrome phenotypes. Arthritis Rheum. 2003;48:2632–2644. doi: 10.1002/art.11215. [DOI] [PubMed] [Google Scholar]
- Dode C, Cuisset L, Delpech M, Grateau G. TNFRSF1A-associated periodic syndrome (TRAPS), Muckle–Wells syndrome (MWS) and renal amyloidosis. J Nephrol. 2003;16:435–437. doi: 10.1159/000072570. [DOI] [PubMed] [Google Scholar]
- Rudofsky G, Jr, Hoffmann F, Muller K, Filser M, Lohse P, Beimler J, Schwenger V. A nephrotic patient with tumour necrosis factor receptor-associated periodic syndrome, IgA nephropathy and CNS involvement. Nephrol Dial Transplant. 2006;21:1109–1112. doi: 10.1093/ndt/gfk098. [DOI] [PubMed] [Google Scholar]
- Hull KM, Drewe E, Aksentijevich I, Singh HK, Wong K, McDermott EM, Dean J, Powell RJ, Kastner DL. The TNF receptor-associated periodic syndrome (TRAPS): emerging concepts of an autoinflammatory disorder. Medicine (Baltimore) 2002;81:349–368. doi: 10.1097/00005792-200209000-00002. [DOI] [PubMed] [Google Scholar]
- Nowlan ML, Drewe E, Bulsara H, Esposito N, Robins RA, Tighe PJ, Powell RJ, Todd I. Systemic cytokine levels and the effects of etanercept in TNF receptor-associated periodic syndrome (TRAPS) involving a C33Y mutation in TNFRSF1A. Rheumatology (Oxford) 2006;45:31–37. doi: 10.1093/rheumatology/kei090. [DOI] [PubMed] [Google Scholar]
- Williamson LM, Hull D, Mehta R, Reeves WG, Robinson BH, Toghill PJ. Familial Hibernian fever. Q J Med. 1982;51:469–480. [PubMed] [Google Scholar]
- Nigrovic PA, Sundel RP. Treatment of TRAPS with etanercept: use in pediatrics. Clin Exp Rheumatol. 2001;19:484–485. [PubMed] [Google Scholar]
- Kallinich T, Briese S, Roesler J, Rudolph B, Sarioglu N, Blankenstein O, Keitzer R, Querfeld U, Haffner D. Two familial cases with tumor necrosis factor receptor-associated periodic syndrome caused by a non-cysteine mutation (T50M) in the TNFRSF1A gene associated with severe multiorganic amyloidosis. J Rheumatol. 2004;31:2519–2522. [PubMed] [Google Scholar]
- Drewe E, McDermott EM, Powell PT, Isaacs JD, Powell RJ. Prospective study of anti-tumour necrosis factor receptor superfamily 1B fusion protein, and case study of anti-tumour necrosis factor receptor superfamily 1A fusion protein, in tumour necrosis factor receptor associated periodic syndrome (TRAPS): clinical and laboratory findings in a series of seven patients. Rheumatology (Oxford) 2003;42:235–239. doi: 10.1093/rheumatology/keg070. [DOI] [PubMed] [Google Scholar]
- Drewe E, McDermott EM, Powell RJ. Treatment of the nephrotic syndrome with etanercept in patients with the tumor necrosis factor receptor-associated periodic syndrome. N Engl J Med. 2000;343:1044–1045. doi: 10.1056/NEJM200010053431412. [DOI] [PubMed] [Google Scholar]
- Haas SL, Lohse P, Schmitt WH, Hildenbrand R, Karaorman M, Singer MV, Bocker U. Severe TNF receptor-associated periodic syndrome due to 2 TNFRSF1A mutations including a new F60V substitution. Gastroenterology. 2006;130:172–178. doi: 10.1053/j.gastro.2005.09.014. [DOI] [PubMed] [Google Scholar]
- Galon J, Aksentijevich I, McDermott MF, O'Shea JJ, Kastner DL. TNFRSF1A mutations and autoinflammatory syndromes. Curr Opin Immunol. 2000;12:479–486. doi: 10.1016/S0952-7915(00)00124-2. [DOI] [PubMed] [Google Scholar]
- Arostegui JI, Solis P, Aldea A, Cantero T, Rius J, Bahillo P, Plaza S, Vives J, Gomez S, Yague J. Etanercept plus colchicine treatment in a child with tumour necrosis factor receptor-associated periodic syndrome abolishes auto-inflammatory episodes without normalising the subclinical acute phase response. Eur J Pediatr. 2005;164:13–16. doi: 10.1007/s00431-004-1563-1. [DOI] [PubMed] [Google Scholar]
- Offer M, Lachmann HJ, Bybee A, Rowczenio DM, Goodman HJ, Hawkins PN. FMF and beyond. Fourth International Conference on Systemic Inflammatory Disorders; Bethesda, MD; 2005. Phenotype and response to treatment of 22 patients with autoinflammatory disease associated with mutations in TNFRSF1A. [Google Scholar]
- Bodar EJ, Simons A, Fiselier TJ, van der Hilst JC, Drenth JP, van der Meer JW. FMF and beyond. Fourth International Conference on Systemic Inflammatory Disorders; Bethesda, MD; 2005. Intravenous interleukin-1 receptor antagonist in a patient with severe TNF-receptor associated periodic syndrome. [Google Scholar]
- Simon A, Bodar EJ, van der Hilst JC, van der Meer JW, Fiselier TJ, Cuppen MP, Drenth JP. Beneficial response to interleukin 1 receptor antagonist in traps. Am J Med. 2004;117:208–210. doi: 10.1016/j.amjmed.2004.02.039. [DOI] [PubMed] [Google Scholar]
- Kusuhara K, Nomura A, Nakao F, Hara T. Tumour necrosis factor receptor-associated periodic syndrome with a novel mutation in the TNFRSF1A gene in a Japanese family. Eur J Pediatr. 2004;163:30–32. doi: 10.1007/s00431-003-1338-0. [DOI] [PubMed] [Google Scholar]
- Dinc A, Erdem H, Rowczenio D, Simsek I, Pay S, Bahce M, Lachmann H. Autosomal dominant periodic fever with AA amyloidosis: tumor necrosis factor receptor-associated periodic syndrome (TRAPS) in a Turkish family. J Nephrol. 2005;18:626–629. [PubMed] [Google Scholar]
- Aganna E, Zeharia A, Hitman GA, Basel-Vanagaite L, Allotey RA, Booth DR, Hawkins PN, Thacker C, Syndercombe-Court D, McDermott MF. An Israeli Arab patient with a de novo TNFRSF1A mutation causing tumor necrosis factor receptor-associated periodic syndrome. Arthritis Rheum. 2002;46:245–249. doi: 10.1002/1529-0131(200201)46:1<245::AID-ART10038>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- Muppidi JR, Tschopp J, Siegel RM. Life and death decisions: secondary complexes and lipid rafts in TNF receptor family signal transduction. Immunity. 2004;21:461–465. doi: 10.1016/j.immuni.2004.10.001. [DOI] [PubMed] [Google Scholar]
- Banner DW, D'Arcy A, Janes W, Gentz R, Schoenfeld HJ, Broger C, Loetscher H, Lesslauer W. Crystal structure of the soluble human 55 kd TNF receptor-human TNF beta complex: implications for TNF receptor activation. Cell. 1993;73:431–445. doi: 10.1016/0092-8674(93)90132-A. [DOI] [PubMed] [Google Scholar]
- Chan FK, Chun HJ, Zheng L, Siegel RM, Bui KL, Lenardo MJ. A domain in TNF receptors that mediates ligand-independent receptor assembly and signaling. Science. 2000;288:2351–2354. doi: 10.1126/science.288.5475.2351. [DOI] [PubMed] [Google Scholar]
- Naismith JH, Devine TQ, Brandhuber BJ, Sprang SR. Crystallographic evidence for dimerization of unliganded tumor necrosis factor receptor. J Biol Chem. 1995;270:13303–13307. doi: 10.1074/jbc.270.22.13303. [DOI] [PubMed] [Google Scholar]
- INFEVERS website http://fmf.igh.cnrs.fr/infevers/
- Rebelo SL, Bainbridge SE, Amel-Kashipaz MR, Radford PM, Powell RJ, Todd I, Tighe PJ. Modeling of tumor necrosis factor receptor superfamily 1A mutants associated with tumor necrosis factor receptor-associated periodic syndrome indicates misfolding consistent with abnormal function. Arthritis Rheum. 2006;54:2674–2687. doi: 10.1002/art.21964. [DOI] [PubMed] [Google Scholar]
- Aksentijevich I, Galon J, Soares M, Mansfield E, Hull K, Oh HH, Goldbach-Mansky R, Dean J, Athreya B, Reginato AJ, et al. The tumor-necrosis-factor receptor-associated periodic syndrome: new mutations in TNFRSF1A, ancestral origins, genotype–phenotype studies, and evidence for further genetic heterogeneity of periodic fevers. Am J Hum Genet. 2001;69:301–314. doi: 10.1086/321976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tchernitchko D, Chiminqgi M, Galacteros F, Prehu C, Segbena Y, Coulibaly H, Rebaya N, Loric S. Unexpected high frequency of P46L TNFRSF1A allele in sub-Saharan West African populations. Eur J Hum Genet. 2005;13:513–515. doi: 10.1038/sj.ejhg.5201344. [DOI] [PubMed] [Google Scholar]
- Aksentijevich I, Galon J, Soares M, Mansfield E, Hull K, Oh HH, Goldbach-Mansky R, Dean J, Athreya B, Reginato AJ, et al. The tumor-necrosis-factor receptor-associated periodic syndrome: new mutations in TNFRSF1A, ancestral origins, genotype–phenotype studies, and evidence for further genetic heterogeneity of periodic fevers. Am J Hum Genet. 2001;69:301–314. doi: 10.1086/321976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poirier O, Nicaud V, Gariepy J, Courbon D, Elbaz A, Morrison C, Kee F, Evans A, Arveiler D, Ducimetiere P, et al. Polymorphism R92Q of the tumour necrosis factor receptor 1 gene is associated with myocardial infarction and carotid intima-media thickness – the ECTIM, AXA, EVA and GENIC studies. Eur J Hum Genet. 2004;12:213–219. doi: 10.1038/sj.ejhg.5201143. [DOI] [PubMed] [Google Scholar]
- Amoura Z, Dode C, Hue S, Caillat-Zucman S, Bahram S, Delpech M, Grateau G, Wechsler B, Piette JC. Association of the R92Q TNFRSF1A mutation and extracranial deep vein thrombosis in patients with Behcet's disease. Arthritis Rheum. 2005;52:608–611. doi: 10.1002/art.20873. [DOI] [PubMed] [Google Scholar]
- Aderka D. The potential biological and clinical significance of the soluble tumor necrosis factor receptors. Cytokine Growth Factor Rev. 1996;7:231–240. doi: 10.1016/S1359-6101(96)00026-3. [DOI] [PubMed] [Google Scholar]
- Van Zee KJ, Kohno T, Fischer E, Rock CS, Moldawer LL, Lowry SF. Tumor necrosis factor soluble receptors circulate during experimental and clinical inflammation and can protect against excessive tumor necrosis factor alpha in vitro and in vivo. Proc Natl Acad Sci USA. 1992;89:4845–4849. doi: 10.1073/pnas.89.11.4845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawari FI, Rouhani FN, Cui X, Yu ZX, Buckley C, Kaler M, Levine SJ. Release of full-length 55-kDa TNF receptor 1 in exosome-like vesicles: a mechanism for generation of soluble cytokine receptors. Proc Natl Acad Sci USA. 2004;101:1297–1302. doi: 10.1073/pnas.0307981100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xanthoulea S, Pasparakis M, Kousteni S, Brakebusch C, Wallach D, Bauer J, Lassmann H, Kollias G. Tumor necrosis factor (TNF) receptor shedding controls thresholds of innate immune activation that balance opposing TNF functions in infectious and inflammatory diseases. J Exp Med. 2004;200:367–376. doi: 10.1084/jem.20040435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy P, Slack JL, Davis R, Cerretti DP, Kozlosky CJ, Blanton RA, Shows D, Peschon JJ, Black RA. Functional analysis of the domain structure of tumor necrosis factor-alpha converting enzyme. J Biol Chem. 2000;275:14608–14614. doi: 10.1074/jbc.275.19.14608. [DOI] [PubMed] [Google Scholar]
- Huggins ML, Radford PM, McIntosh RS, Bainbridge SE, Dickinson P, Draper-Morgan KA, Tighe PJ, Powell RJ, Todd I. Shedding of mutant tumor necrosis factor receptor superfamily 1A associated with tumor necrosis factor receptor-associated periodic syndrome: differences between cell types. Arthritis Rheum. 2004;50:2651–2659. doi: 10.1002/art.20380. [DOI] [PubMed] [Google Scholar]
- Lobito AA, Kimberley FC, Muppidi JR, Komarow H, Jackson AJ, Hull KM, Kastner DL, Screaton GR, Siegel RM. Abnormal disulfide-linked oligomerization results in ER retention and altered signaling by TNFR1 mutants in the TNFR1 associated periodic fever syndrome (TRAPS) Blood. 2006;108:1320–1327. doi: 10.1182/blood-2005-11-006783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd I, Radford PM, Draper-Morgan KA, McIntosh R, Bainbridge S, Dickinson P, Jamhawi L, Sansaridis M, Huggins ML, Tighe PJ, et al. Mutant forms of tumour necrosis factor receptor I that occur in TNF-receptor-associated periodic syndrome retain signalling functions but show abnormal behaviour. Immunology. 2004;113:65–79. doi: 10.1111/j.1365-2567.2004.01942.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siebert S, Fielding CA, Williams BD, Brennan P. Mutation of the extracellular domain of tumour necrosis factor receptor 1 causes reduced NF-kappaB activation due to decreased surface expression. FEBS Lett. 2005;579:5193–5198. doi: 10.1016/j.febslet.2005.08.037. [DOI] [PubMed] [Google Scholar]
- Yousaf N, Gould DJ, Aganna E, Hammond L, Mirakian RM, Turner MD, Hitman GA, McDermott MF, Chernajovsky Y. Tumor necrosis factor receptor I from patients with tumor necrosis factor receptor-associated periodic syndrome interacts with wild-type tumor necrosis factor receptor I and induces ligand-independent NF-kappaB activation. Arthritis Rheum. 2005;52:2906–2916. doi: 10.1002/art.21268. [DOI] [PubMed] [Google Scholar]
- Pahl HL, Baeuerle PA. The ER-overload response: activation of NF-kappa B. Trends Biochem Sci. 1997;22:63–67. doi: 10.1016/S0968-0004(96)10073-6. [DOI] [PubMed] [Google Scholar]
- Schroder M, Kaufman RJ. The mammalian unfolded protein response. Annu Rev Biochem. 2005;74:739–789. doi: 10.1146/annurev.biochem.73.011303.074134. [DOI] [PubMed] [Google Scholar]
- Zhang K, Shen X, Wu J, Sakaki K, Saunders T, Rutkowski DT, Back SH, Kaufman RJ. Endoplasmic reticulum stress activates cleavage of CREBH to induce a systemic inflammatory response. Cell. 2006;124:587–599. doi: 10.1016/j.cell.2005.11.040. [DOI] [PubMed] [Google Scholar]
- Yang Q, Kim YS, Lin Y, Lewis J, Neckers L, Liu ZG. Tumour necrosis factor receptor 1 mediates endoplasmic reticulum stress-induced activation of the MAP kinase JNK. EMBO Rep. 2006;7:622–627. doi: 10.1038/sj.embor.7400687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micheau O, Tschopp J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell. 2003;114:181–190. doi: 10.1016/S0092-8674(03)00521-X. [DOI] [PubMed] [Google Scholar]
- Schneider-Brachert W, Tchikov V, Neumeyer J, Jakob M, Winoto-Morbach S, Held-Feindt J, Heinrich M, Merkel O, Ehrenschwender M, Adam D, et al. Compartmentalization of TNF receptor 1 signaling: internalized TNF receptosomes as death signaling vesicles. Immunity. 2004;21:415–428. doi: 10.1016/j.immuni.2004.08.017. [DOI] [PubMed] [Google Scholar]
- Siebert S, Amos N, Fielding CA, Wang EC, Aksentijevich I, Williams BD, Brennan P. Reduced tumor necrosis factor signaling in primary human fibroblasts containing a tumor necrosis factor receptor superfamily 1A mutant. Arthritis Rheum. 2005;52:1287–1292. doi: 10.1002/art.20955. [DOI] [PMC free article] [PubMed] [Google Scholar]