

Abstract
Fragile X mental retardation is caused by silencing of the gene (FMR1) that encodes the RNA-binding protein (FMRP) that influences translation in neurons. A prominent feature of the human disorder is self-injurious behavior, suggesting an abnormality in pain processing. Moreover, FMRP regulates group I metabotropic glutamate receptor (mGluR1/5)-dependent plasticity, which is known to contribute to nociceptive sensitization. We demonstrate here, using the Fmr1 knock-out (KO) mouse, that FMRP plays an important role in pain processing because Fmr1 KO mice showed (1) decreased (∼50%) responses to ongoing nociception (phase 2, formalin test), (2) a 3 week delay in the development of peripheral nerve injury-induced allodynia, and (3) a near absence of wind-up responses in ascending sensory fibers after repetitive C-fiber stimulation. We provide evidence that the behavioral deficits are related to a mGluR1/5- and mammalian target of rapamycin (mTOR)-mediated mechanism because (1) spinal mGluR5 antagonism failed to inhibit the second phase of the formalin test, and we observed a marked reduction in nociceptive response to an intrathecal injection of an mGluR1/5 agonist (RS)-3,5-dihydroxyphenylglycine (DHPG) in Fmr1 KO mice; (2) peripheral DHPG injection had no effect in KO mice yet evoked thermal hyperalgesia in wild types; and (3) the mTOR inhibitor rapamycin inhibited formalin- and DHPG-induced nociception in wild-type but not Fmr1 KO mice. These experiments show that translation regulation via FMRP and mTOR is an important feature of nociceptive plasticity. These observations also support the hypothesis that the persistence of self-injurious behavior observed in fragile X mental retardation patients could be related to deficits in nociceptive sensitization.
Keywords: pain, translation regulation, nociceptor, FMRP, mGluR, neuropathic pain, mTOR
Introduction
Fragile X mental retardation is the most common form of inherited mental retardation (Youings et al., 2000) and is caused by the expansion of a CGG repeat in the 5′-untranslated region of the fragile X mental retardation 1 (FMR1) gene, leading to hypermethylation and transcriptional silencing. The FMR1 gene encodes the fragile X mental retardation protein (FMRP), an RNA-binding protein that participates in the trafficking of mRNAs to distal sites in neurons (Bagni and Greenough, 2005; Bardoni et al., 2006). FMRP represses translation of mRNAs that it binds (Mazroui et al., 2002; Barbee et al., 2006) but also regulates the activity-dependent translation of these mRNAs, especially in response to group I metabotropic glutamate receptor (mGluR1/5) activation (Todd et al., 2003; Weiler et al., 2004; Antar et al., 2005; Hou et al., 2006). Studies in Fmr1 knock-out (KO) mice have indicated that FMRP regulates synaptic plasticity through mechanisms that are linked to its role in activity-dependent translational regulation (Antar and Bassell, 2003; Bear et al., 2004; Bardoni et al., 2006). In the hippocampus and cerebellum of Fmr1 KO mice, long-term depression (LTD) is increased (Huber et al., 2002; Koekkoek et al., 2005). In contrast, in the somatosensory and neocortical areas of the Fmr1 KO mouse, long-term potentiation (LTP) is absent (Li et al., 2002; Wilson and Cox, 2007).
A prominent behavioral feature of fragile X mental retardation is self-injurious behavior (Symons et al., 2003), suggesting alterations in pain processing. We have previously demonstrated that FMRP is expressed by nociceptors and localizes to axons of these pain-sensing neurons as well as to neurons throughout the superficial dorsal horn (Price et al., 2006). These findings suggested that FMRP could play a role in nociception; however, this possibility has not been studied experimentally. The notion that FMRP might be involved in nociception is further supported by the established role of mGluR1/5 in pain. mGluR1/5 are involved in nociceptive sensitization both in the periphery (Bhave et al., 2001) and the CNS (Fisher and Coderre, 1996; Karim et al., 2001; Adwanikar et al., 2004). mGluR1/5 are required for spinal LTP (Azkue et al., 2003) and are implicated in the increased excitability of dorsal horn neurons evoked by a persistent noxious stimulus (Fisher and Coderre, 1996; Adwanikar et al., 2004). Despite the connection between FMRP and mGluR1/5, the role of FMRP in the function and regulation of nociceptors is unknown. Likewise, the possible pronociceptive role of activity-dependent translation in neurons, which appears to be controlled, at least in part, by the mammalian target of rapamycin (mTOR) (Kelleher et al., 2004), a kinase that regulates cap-dependent translation (Richter and Sonenberg, 2005), is also unknown. We hypothesized that FMRP might be an important regulator of nociceptor sensitization, especially as it relates to mGluR1/5 and mTOR. We have used the Fmr1 KO mouse to assess this hypothesis and have found a previously unknown role of FMRP in nociception related to mGluR1/5- and mTOR-dependent enhancement of nociceptive excitability.
Materials and Methods
Experimental animals.
Male Fmr1 KO mice and female Fmr1+/− mice on a C57BL/6 background were obtained from Neuromice.org (Northwestern University, Chicago IL). All animal care and use procedures were approved by the Animal Care and Use Ethics Committee of McGill University and were in line with National Institutes of Health (NIH) and International Association for the Study of Pain guidelines. All animals were housed on a 12 h light/dark cycle and had ad libitum access to food and water. Male Fmr1 KO mice were bred at McGill University with female Fmr1+/− mice to generate experimental animals. Only male KO and wild-type animals were used. Genotyping was performed via ear punches. All experiments were conducted on KO and wild-type littermates between the ages of 8 and 12 weeks of age.
Antibodies and chemicals.
The 7G1-1 antibody was obtained from the Iowa Hybridoma Bank (University of Iowa). Isolectin B4 (IB4) conjugated to Alexa-Fluor 488 and secondary Alexa-Fluor antibodies were from Invitrogen (Carlsbad, CA), and the calcitonin gene-related peptide (CGRP) antibody was from Sigma (St. Louis, MO). The phospho-ERK (Thr202/Tyr204) antibody was from Cell Signaling Technology (Danvers, MA), and the total ERK antibody was from Millipore (Nepean, Ontario, Canada). The mGluR5 and βIII-tubulin antibodies were from Millipore. (RS)-3,5-Dihydroxyphenylglycine (DHPG) was from Tocris Bioscience (Ellisville, MO), rapamycin was from LC Laboratories (Woburn, MA), and 2-methyl-6-(phenylethynyl)pyridine hydrochloride (MPEP) was a gift from FRAXA (produced by Technically, Woburn, MA). All other chemicals were obtained from Sigma.
Immunohistochemistry.
Slide-mounted sections were fixed in ice-cold 3.7% paraformaldehyde in 1× PBS for 1 h and then washed 3 times for 5 min in PBS. Slides were transferred to a solution containing 0.1 m sodium citrate and 0.05% Tween 20 and then microwaved on high power for 3 min in a 900 W microwave oven for antigen retrieval. After a 30 min cooling period, slides were again transferred to 1× PBS, washed 3 times for 5 min, and then permeabilized in 1× PBS, containing 0.05% Triton X-100. Slides were then blocked for at least 1 h in 1× PBS, containing 10% normal goat serum, before the addition of anti-FMRP (7G1-1; 1 μg/ml) antibody overnight at 4°C. The CGRP antibody (1:1000 dilution) and IB4 (1:1000 dilution) were applied together, and the antigen retrieval step was not used. Immunoreactivity was visualized after subsequent incubation with goat anti-rabbit or goat anti-mouse Alexa-Fluor antibody for 1 h at room temperature. All immunohistochemistry (IHC) images are representative of samples taken from three animals. Confocal IHC images were taken using an Axiovert 100M confocal microscope (Zeiss, Thornwood, NY) at McGill University. Image quantification was done as described previously (Price and Flores, 2007).
Behavior.
In all experiments, animals were habituated to a Plexiglas behavior chamber under ambient light for 1 h before the beginning of the experiment. Mechanical thresholds were measured by the up–down method (Chaplan et al., 1994) with calibrated von Frey hairs. Thermal thresholds were measured by the hot plate and radiant heat (Hargreaves et al., 1988) methods. For experiments using the radiant heat method, the radiant heat source intensity was adjusted to 70% of maximum. This is a higher intensity than is normally used in mice. This procedure was needed because of mild hyperactivity in Fmr1 KO mice (The Dutch-Belgian Fragile X Consortium, 1994). In all formalin experiments, formalin (5% in saline) was injected into the hindpaw in a volume of 20 μl, and nociceptive behaviors (licking, biting, or shaking in the affected paw) were measured for 45 min. The first phase is defined as 0–10 min and the second phase as 15–45 min. In MPEP experiments, MPEP (25 nmol in 5 μl) or dilution vehicle was diluted in 2× artificial CSF (ACSF) to yield 1× ACSF. Mice were anesthetized with isoflurane, and MPEP or vehicle was injected intrathecally (by lumbar puncture) 15 min before formalin injection. Rapamycin was diluted in 100% DMSO. For intrathecal injections with rapamycin, 2 μl injections were made 15 min before formalin injection or DHPG (see below), and vehicle controls were the same volume of DMSO. For intraplantar injections, rapamycin or DMSO (vehicle) was injected 15 min before formalin in a volume of 5 μl. DHPG or vehicle was injected intrathecally (5 μl) in anesthetized mice or into the hindpaw (20 μl). For intrathecal experiments, caudally directed nociceptive behaviors were recorded for 30 min as described. For thermal hyperalgesia experiments, thermal withdrawal latencies were recorded before injection and every 30 min after injection for 3 h by the radiant heat method. Spared nerve injury was performed as described previously (Bourquin et al., 2006), and mechanical thresholds were measured before and after surgery as noted above. In all experiments, the observer was blinded to the genotype of the mice and the drug condition.
Western blotting.
Animals were injected with DHPG or vehicle intrathecally, and spinal cords were pressure ejected 15 min after injection. Lumbar spinal cord was dissected and snap frozen in liquid nitrogen. Frozen tissues were homogenized in homogenization buffer (50 mm Tris HCl, 1% Triton X-100, 150 mm NaCl, and 1 mm EDTA at pH 7.4) containing protease and phosphatase inhibitor mixtures with a motorized Dounce homogenizer, sonicated for 15 min at 4°C, and cleared of cellular debris and nuclei by centrifugation at 13,000 relative centrifugal force for 5 min at 4°C. Ten micrograms of protein per well were loaded and separated by standard 7.5% SDS-PAGE. Proteins were transferred to Immobilon-P membranes (Millipore) and then blocked with 5% dry milk for 3 h at room temperature. Phospho-ERK antiserum (1:2000) was incubated overnight at 4°C and detected the following day with donkey anti-rabbit antibody conjugated to horseradish peroxidase (Dako, Ottawa, Canada). Signal was detected by ECL-plus (GE Healthcare, Piscataway, NJ) on chemiluminescent films (Kodak, Rochester, NY). Blots were then stripped using Restore reagent (Pierce, Rockford, IL) and reblotted with a 1:1000 dilution of anti-total ERK antibody. For total mGluR5 levels, naive mice were used, and spinal proteins were prepared by the same method. The mGluR5 antibody was used at a 1:2000 dilution, and the βIII-tubulin antibody was used at a 1:10,000 dilution. Densitometry was conducted using ImageJ version 1.36b for Macintosh OS X (NIH, Bethesda, MD).
Electrophysiology.
Extracellular multiunit and single-unit recordings of spike activity in ascending axons of the contralateral ventral quadrant were performed using a modified version of a previously described preparation (Martinez-Gomez and Lopez-Garcia, 2005). Briefly, 4-week-old mice were anesthetized with urethane (2 g/kg, i.p.), and a rostrocaudal laminectomy was performed to reveal the spinal cord. The whole cord was excised with attached dorsal roots and put in cold (0–4°C), oxygenated (95% O2/5% CO2) ACSF and cleared of dura mater. The cord was then placed in a recording chamber, pinned down to a Sylgard base, and continuously superfused with oxygenated ACSF of the following composition (in mm): 117 NaCl, 3.6 KCl, 1.2 NaH2PO4, 1.2 MgCl2, 2.5 CaCl2, 25 NaHCO3, and 11 d-glucose, pH 7.4. Multiunit and single-unit activity in ascending axons of the contralateral ventral quadrant was recorded after stimulation of sacral dorsal roots, amplified, and bandpass filtered using a commercial amplifier (CyberAmp 380; Molecular Devices, Union City, CA). The data were digitized at 10 kHz using a CED analog-to-digital converter (Cambridge Electronic Design, Cambridge, UK) and analyzed with Spike 2 software (Cambridge Electronic Design). To induce wind-up, a 1 Hz train of 15 stimuli (200 μs duration, 20 times threshold) was applied to the dorsal root. This stimulus strength was sufficient to activate C fibers (Martinez-Gomez and Lopez-Garcia, 2005). Wind-up was defined as a progressive increase in the number of spikes elicited by the test stimulation over the 15 stimulations. Wind-down was defined as a progressive decrease of the number of spikes elicited by the test stimulus over the same period.
Data and statistics.
All data are presented as mean ± SEM. Single comparisons were made by two-tailed Student's t test. Multiple comparisons by genotype were made by two-way ANOVA with Bonferroni posttest. Comparisons of incidence of wind-up in ascending fibers was made by χ2 test.
Results
FMRP expression and sensory neuron markers in Fmr1 KO and wild-type mice
We first examined the expression of FMRP in the dorsal root ganglion, trigeminal ganglion, and spinal cord of wild-type mice. Using the same antibody (7G1-1) previously used to characterize FMRP expression in rat dorsal root ganglion and spinal cord neurons, we observed FMRP immunoreactivity in all dorsal root ganglion (Fig. 1A) and trigeminal ganglion neurons (data not shown) as well as neurons throughout the spinal cord (Fig. 1A). Immunoreactivity was completely absent in Fmr1 KO mice, which confirms the specificity of this antibody (Price et al., 2006). Because fragile X mental retardation is a developmental disorder and FMRP has been suggested to play a role in axonal development (Antar et al., 2006), we assessed the distribution of two common sensory/nociceptor markers in the dorsal root ganglion and dorsal horn in Fmr1 KO mice. CGRP and the IB4-binding populations of sensory neurons are segregated in the dorsal root ganglion, and their projections to the dorsal horn have a distinct laminar distribution (Molliver et al., 1997; Zwick et al., 2002). There was no difference in the distribution of CGRP-immunoreactive (wild type, 25.9 ± 1.74%, n = 3; Fmr1 KO, 24.5 ± 1.16%, n = 3) or IB4-binding (wild-type, 24.8 ± 3.72%, n = 3; Fmr1 KO, 23.3 ± 1.15%, n = 3) sensory neuronal populations in the dorsal root ganglion, and these populations showed equivalent segregation (colocalization of CGRP and IB4: wild-type, 2.82 ± 0.30%, n = 3; Fmr1 KO, 2.36 ± 0.13%, n = 3) (Fig. 1B). Moreover, the laminar distribution of CGRP-immunoreactive and IB4-binding sensory afferents appeared normal in laminas I and II in both Fmr1 KO and wild-type mice (Fig. 1B). These observations indicate that, at least for these markers, the projection of nociceptors to the dorsal horn develops normally in Fmr1 KO mice.
Figure 1.

FMRP is expressed in dorsal root ganglion and spinal cord neurons. Distribution of CGRP and IB4 in Fmr1 KO mice: A, FMRP immunoreactivity was detected in all DRG neurons and throughout the spinal cord (SC) with the anti-FMRP 7G1-1 antibody (left). Immunoreactivity was completely absent in Fmr1 KO mice (right). Scale bars: DRG panels, 200 μm. B, Immunohistochemistry was performed on wild-type (WT) and KO dorsal root ganglion and spinal dorsal horn for CGRP (red) and IB4 (green). Top, CGRP and IB4 labeling were segregated in both wild-type and KO mouse dorsal root ganglion. Scale bars, 200 μm. Bottom, In both wild-type and KO animals, IB4 (green)-positive afferent terminals localized to the superficial portion of lamina I and lamina II inner. CGRP (red) was found in lamina I and II outer. As in wild-type mice, these subpopulations did not overlap and maintained their laminar distribution in Fmr1 KO mice. Scale bars, 50 μm.
Mechanical and thermal thresholds in Fmr1 KO mice
Next we sought to measure basal nociceptive thresholds in Fmr1 KO mice for mechanical and thermal stimuli. Mechanical thresholds, as determined by von Frey hair stimulation of the hindpaw, did not differ between wild-type (1.04 ± 0.077 g, n = 13) and KO (1.22 ± 0.10 g, n = 14) mice. Noxious heat sensitivity was assessed in the hot plate and radiant heat tests. In the 50°C hot plate test, latencies to a behavioral response were not different between KO (17.1 ± 0.90 s, n = 19) and wild-type (16.4 ± 1.70 s, n = 9) mice. In the radiant heat test, withdrawal latencies also did not differ between KO (2.58 ± 0.120 s, n = 31) and wild-type (2.27 ± 0.140 s, n = 23) mice.
Decreased formalin-evoked nocifensive behavior in Fmr1 KO mice
The formalin test of chemically induced pain is a well characterized behavioral model consisting of two consecutive pain behavior phases, of which the second has been suggested to involve sensitization of the spinal nociceptive system (Coderre et al., 1990; Coderre and Melzack, 1992; Ikeda et al., 2006). We hypothesized that spinal sensitization might be blunted in Fmr1 KO mice and have used the second phase of the formalin test to assess this hypothesis because formalin injection induces LTP in a subset of dorsal horn neurons (Ikeda et al., 2006) and nociceptive behaviors in the second phase of this model are dependent on protein synthesis (Hou et al., 1997; Kim et al., 1998). In the acute (first) phase of the formalin test, the duration of nociceptive behavior was not different between genotypes (wild-type, 271 ± 29.4 s, n = 7; KO, 196 ± 10.1 s, n = 7). However, the second phase of the test was significantly reduced in Fmr1 KO mice (345 ± 63.7 s, n = 7), by ∼50% versus their wild-type littermates (608 ± 70.7 s, n = 7) (Fig. 2A,B).
Figure 2.

Decreased second-phase formalin responses in Fmr1 KO mice, role of mGluR5. Formalin (5%) was injected directly into the hindpaw of Fmr1 KO and wild-type (WT) mice, and their nociceptive behaviors were monitored for 45 min. A, Time course of nociceptive behaviors in 5 min blocks. B, Nociceptive behaviors were significantly reduced in the second phase of the formalin test (20–45 min) in Fmr1 KO mice (**p < 0.01). Intrathecal injections of MPEP or vehicle were administered 15 min before intraplantar formalin. C, Time course of nociceptive behaviors in KO and wild-type mice with MPEP or vehicle (VEH) in 5 min blocks. D, MPEP significantly reduced phase 2 responses in wild-type mice but was without effect in Fmr1 KO mice (***p < 0.001).
mGluR1/5-mediated nociceptor plasticity is decreased in Fmr1 KO mice
mGluR1/5-mediated translation is known to be regulated by FMRP (Antar et al., 2004; Aschrafi et al., 2005) and is dysregulated in Fmr1 KO mice (Hou et al., 2006). Moreover, in the dorsal horn, mGluR1/5 are known to regulate hyperalgesia and LTP (Karim et al., 2001; Azkue et al., 2003) and play an important role in mediating nociceptive behaviors in the second phase of the formalin test (Karim et al., 2001; Zhu et al., 2004). Therefore, we hypothesized that the mGluR5 antagonist MPEP would decrease the second phase of the formalin test in wild-type mice but be ineffective in KO mice. MPEP (25 μg), injected intrathecally 15 min before formalin injection, had no effect on the first phase of the formalin test in wild-type or KO mice (Fig. 2C,D). In wild-type mice, MPEP significantly decreased the second phase of the formalin test (vehicle, 439 ± 37.5 s, n = 8; MPEP, 189 ± 43.3 s, n = 8) but did not change it in Fmr1 KO mice (vehicle, 210 ± 68.5 s, n = 5; MPEP, 231 ± 51.9 s, n = 5).
The finding that intrathecal MPEP was without effect in Fmr1 KO mice suggests that mGluR5-mediated mechanisms of nociceptive behaviors in the formalin test are absent in Fmr1 KO mice. To explore this possibility further, we examined the effect of the mGluR1/5 (mGluR1/5) agonist DHPG on nociceptive behaviors in wild-type and KO mice. When injected intrathecally, DHPG causes a robust nocifensive response that is dependent on mGluR1/5 (Karim et al., 2001). Wild-type and Fmr1 KO mice received intrathecal injections of DHPG (25 nmol), and their nociceptive behaviors were monitored for 30 min. Nociceptive behaviors were decreased in KO mice versus their wild-type littermates at time blocks of 15–20, 20–25, and 25–30 min (Fig. 3A). Over the entire 30 min time period, nociceptive behaviors were decreased by 70% in KO versus wild-type mice. This finding supports the notion that mGluR1/5-mediated spinal nociception is abrogated in Fmr1 KO mice.
Figure 3.
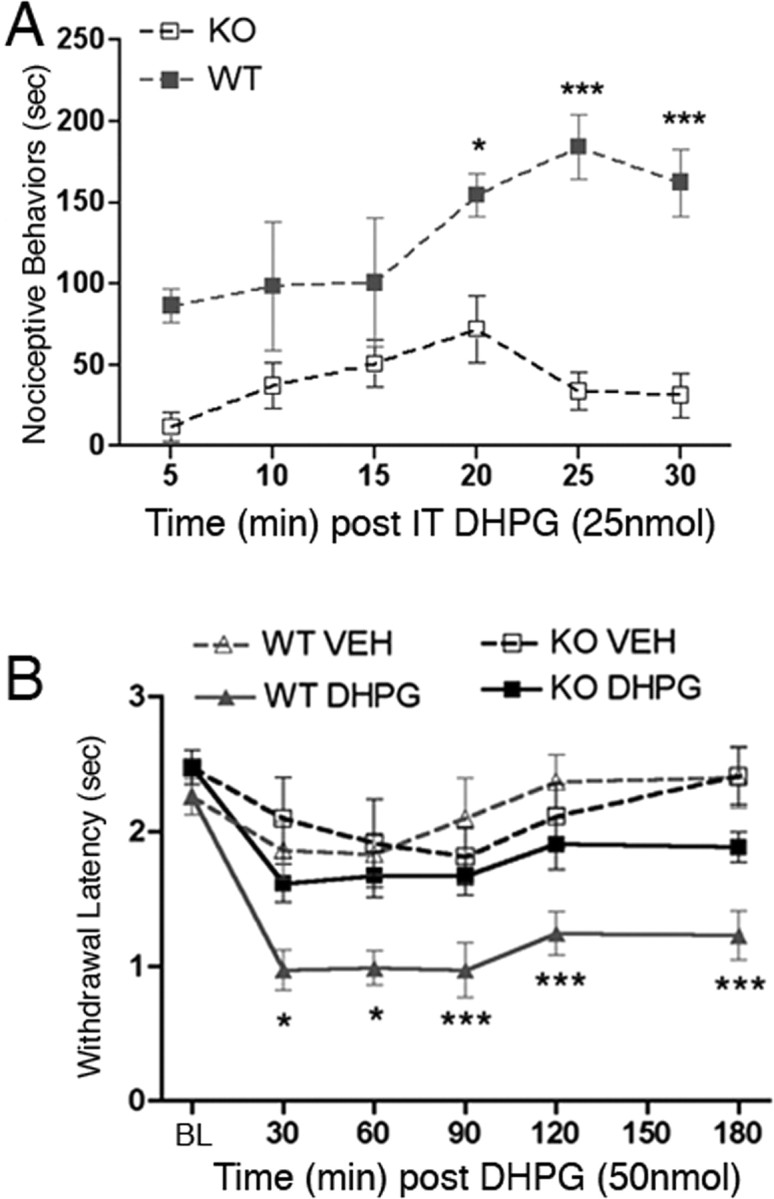
Role of central and peripheral mGluR1/5s in nociceptive sensitization in Fmr1 KO mice. DHPG was injected intrathecally (IT), and nociceptive behaviors were monitored for 30 min. A, Time course of nociceptive behaviors induced by intrathecal DHPG in 5 min blocks (*p < 0.05; ***p < 0.001). B, Baseline (BL) thermal latencies were measured and then DHPG or vehicle (VEH) was injected directly into the hindpaw. Thermal latencies were measured at the indicated time points for 180 min. Wild-type (WT) mice developed thermal hyperalgesia in response to DHPG (*p < 0.05; ***p < 0.001). Thermal hyperalgesia was completely absent in Fmr1 KO mice.
mGluR1/5 not only regulate spinal nociceptive sensitivity but are also involved in peripheral nociceptor sensitization. Peripheral injection of DHPG is known to cause thermal hyperalgesia (Bhave et al., 2001). We have previously shown that FMRP is present in the peripheral axons of nociceptors (Price et al., 2006), indicating that FMRP might play a role in regulating the effects of peripheral mGluR1/5 activation. Hence, we tested the hypothesis that peripherally mediated DHPG-induced thermal hyperalgesia would be reduced or absent in Fmr1 KO mice. We used a dose of DHPG that has been shown to be peripherally selective and mGluR1/5 dependent in mice (Bhave et al., 2001). Wild-type mice developed thermal hyperalgesia 30 min after DHPG (50 nmol) injection directly into the hindpaw. This thermal hyperalgesia was sustained over the entire 3 h time course of the experiment compared with vehicle-injected wild-type mice (DHPG, n = 7; vehicle, n = 10) (Fig. 3B). However, Fmr1 KO mice injected with DHPG did not develop thermal hyperalgesia compared with KOs that received vehicle injections (DHPG, n = 7; vehicle, n = 7) (Fig. 3B). Therefore, peripheral, thermal nociceptor sensitization to an mGluR1/5 agonist is absent in Fmr1 KO mice.
Misregulation of spinal phospho-ERK in Fmr1 KO mice
Stimulation of spinal mGluR1/5 induces ERK phosphorylation and inhibition of ERK reduces nociceptive behaviors induced by DHPG (Karim et al., 2001). In the hippocampus, DHPG also induces ERK phosphorylation, but this increase in phospho-ERK is absent in Fmr1 KO mice (Hou et al., 2006). Interestingly, hippocampal phospho-ERK levels are increased in Fmr1 KO compared with wild-type mice in the absence of mGluR1/5 activation (Hou et al., 2006). We applied DHPG or vehicle intrathecally to wild-type and KO mice and examined ERK phosphorylation status by Western blot 15 min after injection. ERK phosphorylation was standardized to total ERK levels in all conditions. In wild-type mice, DHPG induced a significant, approximately twofold increase in lumbar, spinal phospho-ERK1 and phospho-ERK2 compared with vehicle-injected wild-type mice (n = 6/group) (Fig. 4A–C). In contrast, DHPG did not induce a significant increase in phospho-ERK1 or phospho-ERK2 levels in Fmr1 KO mice compared with vehicle (n = 6/group) (Fig. 4A–C). When vehicle-injected wild-type phospho-ERK levels were compared with vehicle-injected Fmr1 KO mice, a significant, approximately twofold increase in basal phospho-ERK1 was observed (n = 6/group) (Fig. 4A,D). Therefore, DHPG fails to induce an increase in phospho-ERK in Fmr1 KO mice, even though basal phospho-ERK1 levels are increased in Fmr1 KO mice.
Figure 4.

Intrathecal DHPG-induced ERK phosphorylation is impaired in Fmr1 KO mice. DHPG or vehicle was injected intrathecally, and lumbar spinal cords were harvested 15 min later. A, A representative Western blot for phospho-ERK and total ERK in each drug and genotype condition. B, C, Phospho ERK levels are standardized to total ERK and shown as percentage of vehicle (VEH) per genotype. DHPG induced an increase in phospho-ERK1 (B) and phospho-ERK2 (C) only in wild-type (WT) mice. D, Basal phospho-ERK1 levels were elevated in Fmr1 KO mice, but a significant increase was not observed for phospho-ERK2. E, Total, spinal mGluR5 protein was also measured and standardized to β-tubulin (B-TUB), and no difference was observed between wild-type and Fmr1 KO mice. **p < 0.01.
Because differences in nociceptive behaviors and signaling between Fmr1 KO and wild-type mice could be explained by alterations in mGluR5 expression, we examined spinal mGluR5 protein levels in KO and wild-type mice. There was no change in total spinal mGluR5 protein between genotypes in naive animals (n = 6/group) (Fig. 4E).
Role of mTOR in formalin- and DHPG-induced nociceptive sensitization
Decreased nociceptive sensitization in Fmr1 KO mice suggests that activity-dependent protein synthesis plays an important role in formalin and DHPG nociception. The kinase mTOR, which phosphorylates inhibitors of elongation initiation factors (namely 4E-BP2 in neurons), allowing for the formation of the eIF–4F complex (Richter and Sonenberg, 2005), plays a key role in stimulating activity-dependent translation and is strongly implicated in synaptic plasticity (Kelleher et al., 2004). Although previous studies have demonstrated a role for protein synthesis in the second phase of the formalin test (Hou et al., 1997; Kim et al., 1998), a role for mTOR in this process has not been explored. Hence, we hypothesized that rapamycin, an inhibitor of mTOR, would inhibit nociceptive behaviors induced by formalin and DHPG in wild-type mice. Intrathecal injection of rapamycin 15 min before formalin injection had no effect on first-phase responses in wild-type or Fmr1 KO mice (Fig. 5A–C). On the other hand, rapamycin dose dependently (0.1–10 μg) decreased second-phase responses in wild-type mice with significant effects observed at 1 and 10 μg (Fig. 5A,D) (vehicle, 315.0 ± 41.0 s, n = 8; 0.1 μg of rapamycin, 252.0 ± 64.2 s, n = 5; 1 μg of rapamycin, 122.3 ± 30.8 s, n = 6; 10 μg of rapamycin, 54.4 ± 20.4 s, n = 8). In Fmr1 KO mice, rapamycin did not influence the second phase of the formalin test at any concentration tested (Fig. 5B,D) (vehicle, 186.4 ± 54.7 s, n = 7; 1 μg of rapamycin, 186. 4 ± 48.6 s, n = 7; 10 μg of rapamycin, 194.9 ± 52.0 s, n = 7).
Figure 5.

Decreased second-phase formalin responses in Fmr1 KO mice, role of spinal mTOR. Formalin (5%) was injected directly into the hindpaw of Fmr1 KO and wild-type (WT) mice, and their nociceptive behaviors were monitored for 45 min. Intrathecal injections of rapamycin (RAP) or vehicle (VEH) were administered 15 min before intraplantar formalin. A, B, Time course of nociceptive behaviors in 5 min blocks in wild-type mice with increasing intrathecal doses of rapamycin (A) and in Fmr1 KO mice (B). C, Nociceptive behaviors were not altered by rapamycin in the first phase of the formalin test in either genotype. D, Rapamycin significantly reduced phase 2 responses in wild-type mice in a dose-dependent manner but was without effect in Fmr1 KO mice (*p < 0.05, ***p < 0.001, vehicle vs [rapamycin]).
The absence of peripherally injected DHPG-induced thermal hyperalgesia in Fmr1 KO mice suggests that protein synthesis in nociceptive axons regulates nociceptor sensitization. Hence, we tested the ability of an intraplantar injection of rapamycin to inhibit a subsequent nociceptive response to formalin injection into the same hindpaw. In wild-type mice, rapamycin (10 μg) significantly inhibited the second phase of the formalin test without affecting the first phase (Fig. 6A,B) (vehicle, 427.2 ± 30.3 s, n = 9; rapamycin, 291.5 ± 35.2 s, n = 8). Rapamycin injection had no effect on formalin responses in Fmr1 KO mice (Fig. 6A,B) (vehicle, 230 ± 35.8 s, n = 6; rapamycin, 222.2 ± 31.7 s, n = 6).
Figure 6.
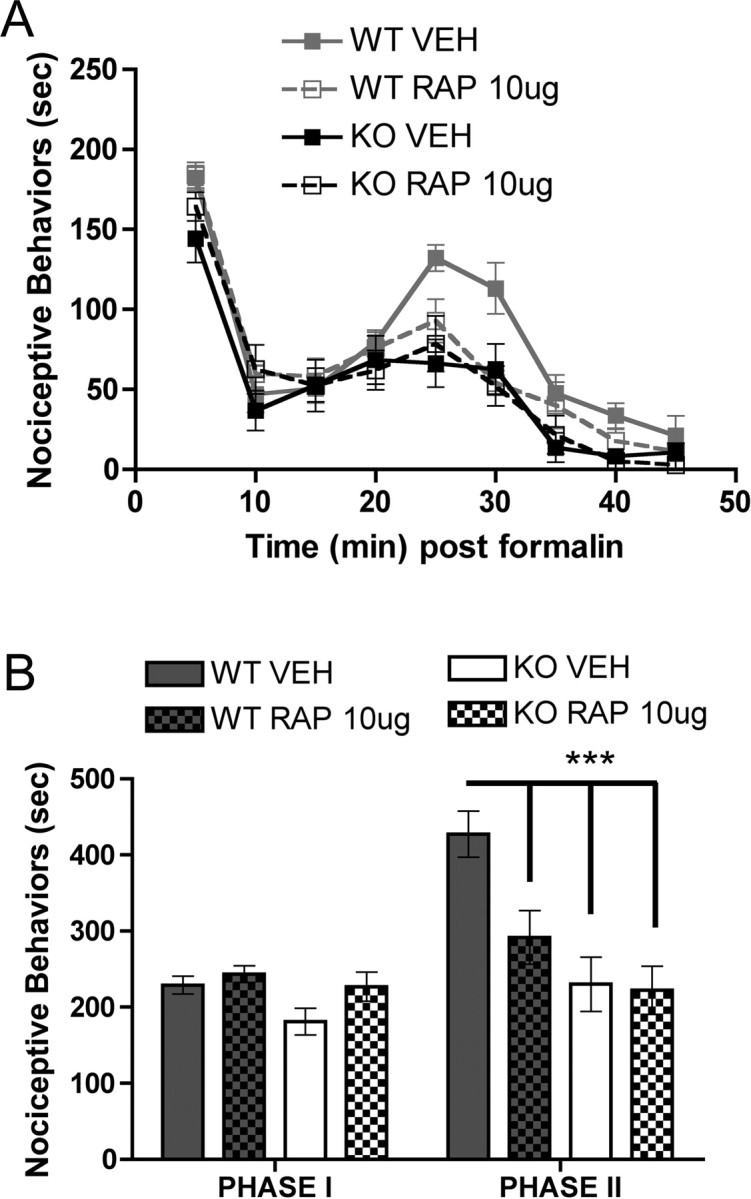
Role of peripheral mTOR in second-phase formalin responses. Rapamycin (RAP, 10 μg) or vehicle (VEH) was injected into the hindpaw 15 min before 5% formalin. A, Time course of nociceptive behaviors in 5 min blocks in wild-type (WT) and Fmr1 KO mice. B, Rapamycin had no effect on phase 1 responses in either genotype but significantly decreased phase 2 responses only in wild-type mice (***p < 0.001).
Finally, we tested the role of mTOR in intrathecally injected DHPG-stimulated nociceptive behaviors. We used a dose of rapamycin that was effective in reducing second-phase formalin responses (10 μg). Wild-type mice that received an intrathecal injection of rapamycin showed a significant decrease in nociceptive behaviors over the 30 min observation period compared with mice that received a vehicle injection (vehicle, 1294 ± 54.2 s, n = 5; rapamycin, 814 ± 135.9 s, n = 6). We did not test the effect of rapamycin in this model in Fmr1 KO mice because their DHPG-induced behaviors were dramatically decreased compared with wild-type and because rapamycin was not effective in KO mice in the formalin test. Together, these findings demonstrate that mTOR-dependent protein synthesis plays an important role at the spinal and peripheral levels in formalin-induced nociceptive behaviors in wild-type but not Fmr1 KO mice and that mTOR regulates DHPG-induced nociception at the spinal level.
Reduced wind-up in Fmr1 KO mice
To gain an independent assessment of decreased nociceptive sensitization in Fmr1 KO mice, we used an in vitro spinal cord preparation to examine the incidence of wind-up in ascending nociceptive fibers of the contralateral ventrolateral funiculus (VLF). Wind-up is a form of short-term synaptic plasticity that can be evoked in ascending VLF fibers by repetitive stimulation of the dorsal root at C-fiber strength (Martinez-Gomez and Lopez-Garcia, 2005). Of 29 units recorded from 16 wild-type mice, 13 units (44.8%) gave wind-up responses, and there was no wind-down response in any unit recorded. A representative trace of wind-up in wild-type mice is shown in Figure 8A. In eight Fmr1 KO mice, a total of 30 units were recorded. C-fiber stimulation evoked contralateral VLF unit activity in Fmr1 KO mice, further confirming the integrity of the basal nociceptive system in these mice. On the other hand, of the 30 units recorded, only five gave a wind-up response (16.7%), and four units (13.3%) displayed a wind-down response (Fig. 7B,C). The distribution of wind-up and wind-down responses in ascending, contralateral VLF nociceptive fibers was significantly different between wild-type and Fmr1 KO mice (Fig. 7C) (χ2 = 8.217, 2; p = 0.016), indicating that wind-up in response to C-fiber stimulation is severely reduced in Fmr1 KO mice, whereas wind-down (which was not observed in wild-type mice) is almost as prevalent in these mice.
Figure 8.

Delayed development of neuropathic allodynia in Fmr1 KO mice. Baseline mechanical thresholds were measured before sham or spared nerve injury (SNI) surgery. Mechanical thresholds were measured at the indicated time points after surgery. Wild-type (WT) spared nerve injury mice developed allodynia versus sham by 5 d after surgery (**p < 0.01; ***p < 0.001). Fmr1 KO spared nerve injury mice did not develop allodynia until 21 d after surgery versus sham (###p < 0.001).
Figure 7.

Reduced wind-up and increased wind-down in Fmr1 KO mice. Recordings were made in the contralateral ascending VLF fibers in an in vitro whole spinal cord preparation. A, Left, Representative trace of wind-up in a wild-type mouse preparation. B, Left, Representative trace of wind-down in an Fmr1 KO mouse preparation in response to 15 C-fiber stimuli of the dorsal root at 1 Hz. A, B, Right, The wind-up (A) or wind-down (B) response as the total number of spikes evoked by each stimulus over the stimulation period. C, The incidence of wind-up was decreased and the incidence of wind-down was increased in Fmr1 KO mice compared with wild-type (*p < 0.05).
Delayed development of nerve injury-induced allodynia in Fmr1 KO mice
Axonally synthesized proteins appear to play an important role in nerve regeneration and repair after injury (Willis and Twiss, 2006), and local, axonal translation is required for injury-induced hyperexcitability of Aplysia axons (Weragoda et al., 2004). We predicted that, if FMRP regulates the translation of proteins that mediate nerve injury-induced hyperexcitability in axons, the development of neuropathic allodynia would be delayed (because these proteins would have to be transported from the soma, rather than synthesized locally) or even absent in Fmr1 KO mice. We used the spared nerve injury model (Bourquin et al., 2006) to test this hypothesis. As previously reported (Bourquin et al., 2006), wild-type mice developed mechanical allodynia versus sham wild-type mice at 5 d after surgery, and the allodynia was maintained for the full 28 d of testing (n = 7–10/group) (Fig. 8). In contrast, Fmr1 KO mice did not develop mechanical allodynia until 3 weeks after surgery (n = 8–10/group) (Fig. 8). These findings suggest that FMRP-mediated translational regulation in injured axons is involved in the development of nerve injury-induced mechanical allodynia.
Discussion
The experiments described here demonstrate that Fmr1 KO mice have normal acute nociceptive responses (Zhao et al., 2005) and that the gross development of the dorsal root ganglion (DRG)–spinal nociceptive system is intact. On the other hand, we have observed a reduced mGluR1/5-mediated nociceptive sensitization at the level of the spinal cord and periphery in Fmr1 KO mice. Our findings also indicate that mTOR, a kinase that regulates cap-dependent translation (Richter and Sonenberg, 2005), plays an important role in nociceptive sensitization on the spinal and peripheral levels in wild-type mice and that this mechanism is lacking in Fmr1 KO mice. Moreover, Fmr1 KO mice show a drastic reduction in C-fiber stimulation-evoked wind-up in ascending nociceptive pathways and a 3-week delay in the development of allodynia in response to peripheral nerve injury. Hence, our experiments demonstrate that Fmr1 KO mice show decreased nociceptive sensitization in multiple experimental paradigms and that translation regulation via FMRP and mTOR are likely to play a critical role in sensitization of the nociceptive pathway.
New protein synthesis is required for several forms of synaptic plasticity (Steward, 2002; Schuman et al., 2006). Increased excitability in the spinal cord after a persistent noxious stimulus is associated with spinal LTP (Ji et al., 2003; Ikeda et al., 2006; Sandkuhler, 2007), and spinal late-phase LTP requires protein synthesis (Hu et al., 2003). Formalin injection induces LTP in a subset of dorsal horn neurons (Ikeda et al., 2006). Moreover, nociceptive behaviors in the second phase of the formalin test are inhibited by systemic (Hou et al., 1997) or intrathecal (Kim et al., 1998) application of protein synthesis inhibitors. Our findings suggest that FMRP and mTOR are important regulators of translation related to spinal sensitization in the formalin model. Second-phase formalin responses were strongly reduced in Fmr1 KO mice, and the mTOR inhibitor rapamycin dose dependently blocked second-phase responses in wild-type mice via intrathecal delivery. Rapamycin was without effect in Fmr1 KO mice in the formalin model, consistent with recent findings demonstrating that activity-regulated protein synthesis is dysregulated in Fmr1 KO mice (Hou et al., 2006; Nosyreva and Huber, 2006; Muddashetty et al., 2007). The lack of effect of intrathecal MPEP in the second phase of the formalin test coupled with the ∼70% decrease in DHPG-mediated nociceptive behaviors in Fmr1 KO mice indicates that these events are linked to mGluR1/5. Moreover, DHPG-induced nociceptive behaviors were attenuated by intrathecal rapamycin in wild-type mice, indicating that mTOR-dependent translation mediates part of this effect downstream of mGluR1/5. Intrathecal DHPG also failed to increase ERK phosphorylation in the spinal cord of Fmr1 KO mice, whereas basal phospho-ERK levels were increased in these mice. This finding is consistent with previous experiments in hippocampal slices from Fmr1 KO mice (Hou et al., 2006) and suggests that the temporal regulation of ERK activity in response to mGluR1/5-stimulated pathways is absent in Fmr1 KO mice. This apparent failure in mGluR1/5-mediated ERK regulation could explain the paradoxical finding of decreased nociceptive sensitization in the presence of increased ERK phosphorylation. Moreover, in the context of mGluR-dependent synaptic plasticity, ERK and mTOR signaling appear to be parallel pathways that eventually converge on eIF4E to regulate cap-dependent protein synthesis (Banko et al., 2006). Hence, understanding aberrant ERK and mTOR signaling and their effects on cap-dependent translation in Fmr1 KO mice could reveal mechanisms of disrupted activity-dependent translation that have been observed in this model of fragile X mental retardation (Hou et al., 2006; Nosyreva and Huber, 2006; Muddashetty et al., 2007) and have relevance to nociceptive sensitization.
FMRP plays a well established role in mGluR1/5-mediated translational regulation (Todd et al., 2003; Antar et al., 2004, 2005; Hou et al., 2006). In the dorsal horn, mGluR1/5 are required for the induction of LTP (Azkue et al., 2003), and mGluR5 is strongly implicated in nociceptive sensitization (Fisher and Coderre, 1996; Karim et al., 2001; Adwanikar et al., 2004). Hence, we propose that FMRP-regulated translational mechanisms play a previously unappreciated role in regulating mGluR1/5-mediated nociception. Our results in the Fmr1 KO mouse and with mTOR inhibition in wild-type mice are consistent with the hypothesis that spinal LTP is disrupted in Fmr1 KO mice or in the presence of rapamycin; however, we cannot discount the possibility that spinal LTD is enhanced in Fmr1 KO mice, as in other brain regions (Huber et al., 2002; Koekkoek et al., 2005). LTD can be induced in spinal neurons by mGluR1/5 stimulation (Heinke and Sandkuhler, 2005), and spinal LTD has been associated with antinociception (Sandkuhler et al., 1997). Hence, an enhancement of spinal LTD in Fmr1 KO mice could yield a similar behavioral phenotype to decreased LTP. The resolution of these questions will require spinal electrophysiological recordings in Fmr1 KO mice.
In addition to LTP and LTD, a shorter-term form of nociceptive sensitization, called wind-up, is normally expressed in the spinal cord and is characterized by a gradual increase in C-fiber-evoked spike activity in dorsal horn neurons and their ascending fibers (Baranauskas and Nistri, 1998; Herrero et al., 2000). Our findings demonstrate a clear reduction of wind-up in ascending fibers of the contralateral VLF in Fmr1 KO mice compared with wild-type mice, which adds additional weight to the hypothesis that spinal nociceptive sensitization is decreased in Fmr1 KO mice.
Our experiments also suggest that FMRP and mTOR contribute to the peripheral sensitization of nociceptors. Several experiments have demonstrated that dorsal root ganglion axons are translationally competent (Zheng et al., 2001; Willis et al., 2005) and that axonal translation is important for growth cone guidance and collapse (Verma et al., 2005; Wu et al., 2005) and for injury- or depolarization-induced hyperexcitability of Aplysia sensory axons (Weragoda et al., 2004). We have previously demonstrated that FMRP is transported to the peripheral axons of rat nociceptors (Price et al., 2006). It is also known that FMRP localizes to axonal growth cones, where it is involved in regulating axonal growth (Antar et al., 2006), and that FMRP is associated with RNAi machinery in sensory axons (Murashov et al., 2007). Here, we have shown that intraplantar rapamycin inhibited second-phase formalin responses in wild-type but not KO mice, demonstrating a role for peripheral mTOR-mediated translation in this model. Moreover, mGluR1/5-mediated thermal hyperalgesia is completely absent in the Fmr1 KO, suggesting that FMRP regulates mGluR1/5-mediated translation locally in peripheral nociceptors and that this process plays a role in thermal hyperalgesia, at least in response to DHPG. Finally, we have shown that the development of neuropathic allodynia is delayed in Fmr1 KO mice, which supports the notion that FMRP regulates peripheral nociceptor hyperexcitability, a role that could be linked to axonal regeneration, in the case of peripheral nerve injury (Willis and Twiss, 2006). Interestingly, in male fragile X premutation carriers, in which there is an increase in FMR1 gene transcription, peripheral neuropathy is a common clinical feature (Jacquemont et al., 2003; Berry-Kravis et al., 2007). These clinical findings have led to the hypothesis that an increase in FMR1 mRNA causes neuropathy (Tassone et al., 2007), a hypothesis supported by experiments in Drosophila models (Jin et al., 2007; Sofola et al., 2007). In Fmr1 KO animals, which lack Fmr1 mRNA and FMRP protein, we have observed the opposite: a delay in the development of neuropathic allodynia.
Finally, a major behavioral feature of fragile X mental retardation is self-injurious behavior (Symons et al., 2003). The connection between self-injurious behavior and pain sensitivity is not fully understood (Symons and Danov, 2005), but there are indications that self-injurious behaviors can be reduced by increasing pain sensitivity with opioid antagonist treatment (Osman and Loschen, 1992; Symons et al., 2004). Although deficits in nociceptive sensitization are unlikely to precipitate self-injurious behavior, this hypothesis does provide a rationale for the persistence of such behavior. We are not aware of clinical studies that address the notion that self-injurious behavior is related to decreased nociceptor sensitization in experimental pain models (and there are obvious ethical concerns in conducting such studies), but it is nonetheless interesting that self-injurious behavior is a common behavioral feature of many developmental disorders characterized by deficits in synaptic plasticity (Osman and Loschen, 1992; Percy, 2002; Symons et al., 2003), including fragile X mental retardation and Rett syndrome (Moretti et al., 2006). Here, in a mouse model of fragile X mental retardation, we have provided behavioral and electrophysiological evidence for marked decreases in plasticity of the nociceptive system. Deficits in wind-up in ascending nociceptive pathways that accompany fragile X mental retardation may be particularly significant, in that spinal wind-up is associated with temporal summation and “second pain” responses in humans and nonhuman primates (Price et al., 1977, 1978). More work is needed to establish a connection between altered nociceptive sensitization and self-injurious behavior; however, our results demonstrate that in a mouse model of a human disease in which self-injurious behavior is prominent, there are clear indications of decreased nociceptive sensitization linked to deficits in signaling pathways that regulate neuronal plasticity.
Footnotes
This work was supported by the National Institute for Neurological Disorders and Stroke Grant NS049772 (T.J.P.), the American Pain Society (T.J.P.), the Canadian Foundation for Innovation (F.C.), the Canadian Institutes of Health Research (F.C.), the Fonds de la recherche en santé du Québec (F.C.), and the Spanish Secretaria de Estado de Educacion y Universidades: Formacion de Profesorado Universitario Grant (J.M.E.). We thank Lisa Krawec and Grace Krawec for expert technical assistance, FRAXA for kindly providing MPEP, and Drs. Laura S. Stone, Christopher M. Flores, and Edward W. Khandjian for critically reading this manuscript.
References
- Adwanikar H, Karim F, Gereau RW., IV Inflammation persistently enhances nocifensive behaviors mediated by spinal group I mGluRs through sustained ERK activation. Pain. 2004;111:125–135. doi: 10.1016/j.pain.2004.06.009. [DOI] [PubMed] [Google Scholar]
- Antar LN, Bassell GJ. Sunrise at the synapse: the FMRP mRNP shaping the synaptic interface. Neuron. 2003;37:555–558. doi: 10.1016/s0896-6273(03)00090-4. [DOI] [PubMed] [Google Scholar]
- Antar LN, Afroz R, Dictenberg JB, Carroll RC, Bassell GJ. Metabotropic glutamate receptor activation regulates fragile X mental retardation protein and FMR1 mRNA localization differentially in dendrites and at synapses. J Neurosci. 2004;24:2648–2655. doi: 10.1523/JNEUROSCI.0099-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antar LN, Dictenberg JB, Plociniak M, Afroz R, Bassell GJ. Localization of FMRP-associated mRNA granules and requirement of microtubules for activity-dependent trafficking in hippocampal neurons. Genes Brain Behav. 2005;4:350–359. doi: 10.1111/j.1601-183X.2005.00128.x. [DOI] [PubMed] [Google Scholar]
- Antar LN, Li C, Zhang H, Carroll RC, Bassell GJ. Local functions for FMRP in axon growth cone motility and activity-dependent regulation of filopodia and spine synapses. Mol Cell Neurosci. 2006;32:37–48. doi: 10.1016/j.mcn.2006.02.001. [DOI] [PubMed] [Google Scholar]
- Aschrafi A, Cunningham BA, Edelman GM, Vanderklish PW. The fragile X mental retardation protein and group I metabotropic glutamate receptors regulate levels of mRNA granules in brain. Proc Natl Acad Sci USA. 2005;102:2180–2185. doi: 10.1073/pnas.0409803102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azkue JJ, Liu XG, Zimmermann M, Sandkuhler J. Induction of long-term potentiation of C fibre-evoked spinal field potentials requires recruitment of group I, but not group II/III metabotropic glutamate receptors. Pain. 2003;106:373–379. doi: 10.1016/j.pain.2003.08.007. [DOI] [PubMed] [Google Scholar]
- Bagni C, Greenough WT. From mRNP trafficking to spine dysmorphogenesis: the roots of fragile X syndrome. Nat Rev Neurosci. 2005;6:376–387. doi: 10.1038/nrn1667. [DOI] [PubMed] [Google Scholar]
- Banko JL, Hou L, Poulin F, Sonenberg N, Klann E. Regulation of eukaryotic initiation factor 4E by converging signaling pathways during metabotropic glutamate receptor-dependent long-term depression. J Neurosci. 2006;26:2167–2173. doi: 10.1523/JNEUROSCI.5196-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baranauskas G, Nistri A. Sensitization of pain pathways in the spinal cord: cellular mechanisms. Prog Neurobiol. 1998;54:349–365. doi: 10.1016/s0301-0082(97)00067-1. [DOI] [PubMed] [Google Scholar]
- Barbee SA, Estes PS, Cziko AM, Hillebrand J, Luedeman RA, Coller JM, Johnson N, Howlett IC, Geng C, Ueda R, Brand AH, Newbury SF, Wilhelm JE, Levine RB, Nakamura A, Parker R, Ramaswami M. Staufen- and FMRP-containing neuronal RNPs are structurally and functionally related to somatic P bodies. Neuron. 2006;52:997–1009. doi: 10.1016/j.neuron.2006.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardoni B, Davidovic L, Bensaid M, Khandjian EW. The fragile X syndrome: exploring its molecular basis and seeking a treatment. Expert Rev Mol Med. 2006;8:1–16. doi: 10.1017/S1462399406010751. [DOI] [PubMed] [Google Scholar]
- Bear MF, Huber KM, Warren ST. The mGluR theory of fragile X mental retardation. Trends Neurosci. 2004;27:370–377. doi: 10.1016/j.tins.2004.04.009. [DOI] [PubMed] [Google Scholar]
- Berry-Kravis E, Goetz CG, Leehey MA, Hagerman RJ, Zhang L, Li L, Nguyen D, Hall DA, Tartaglia N, Cogswell J, Tassone F, Hagerman PJ. Neuropathic features in fragile X premutation carriers. Am J Med Genet A. 2007;143:19–26. doi: 10.1002/ajmg.a.31559. [DOI] [PubMed] [Google Scholar]
- Bhave G, Karim F, Carlton SM, Gereau RWt. Peripheral group I metabotropic glutamate receptors modulate nociception in mice. Nat Neurosci. 2001;4:417–423. doi: 10.1038/86075. [DOI] [PubMed] [Google Scholar]
- Bourquin AF, Suveges M, Pertin M, Gilliard N, Sardy S, Davison AC, Spahn DR, Decosterd I. Assessment and analysis of mechanical allodynia-like behavior induced by spared nerve injury (SNI) in the mouse. Pain. 2006;122:14.e1–14.e14. doi: 10.1016/j.pain.2005.10.036. [DOI] [PubMed] [Google Scholar]
- Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods. 1994;53:55–63. doi: 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- Coderre TJ, Melzack R. The contribution of excitatory amino acids to central sensitization and persistent nociception after formalin-induced tissue injury. J Neurosci. 1992;12:3665–3670. doi: 10.1523/JNEUROSCI.12-09-03665.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coderre TJ, Vaccarino AL, Melzack R. Central nervous system plasticity in the tonic pain response to subcutaneous formalin injection. Brain Res. 1990;535:155–158. doi: 10.1016/0006-8993(90)91835-5. [DOI] [PubMed] [Google Scholar]
- The Dutch-Belgian Fragile X Consortium. Fmr1 knockout mice: a model to study fragile X mental retardation. Cell. 1994;78:23–33. [PubMed] [Google Scholar]
- Fisher K, Coderre TJ. The contribution of metabotropic glutamate receptors (mGluRs) to formalin-induced nociception. Pain. 1996;68:255–263. doi: 10.1016/s0304-3959(96)03212-5. [DOI] [PubMed] [Google Scholar]
- Hargreaves K, Dubner R, Brown F, Flores C, Joris J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain. 1988;32:77–88. doi: 10.1016/0304-3959(88)90026-7. [DOI] [PubMed] [Google Scholar]
- Heinke B, Sandkuhler J. Signal transduction pathways of group I metabotropic glutamate receptor-induced long-term depression at sensory spinal synapses. Pain. 2005;118:145–154. doi: 10.1016/j.pain.2005.08.004. [DOI] [PubMed] [Google Scholar]
- Herrero JF, Laird JM, Lopez-Garcia JA. Wind-up of spinal cord neurones and pain sensation: much ado about something? Prog Neurobiol. 2000;61:169–203. doi: 10.1016/s0301-0082(99)00051-9. [DOI] [PubMed] [Google Scholar]
- Hou L, Antion MD, Hu D, Spencer CM, Paylor R, Klann E. Dynamic translational and proteasomal regulation of fragile X mental retardation protein controls mGluR-dependent long-term depression. Neuron. 2006;51:441–454. doi: 10.1016/j.neuron.2006.07.005. [DOI] [PubMed] [Google Scholar]
- Hou WY, Shyu BC, Chen TM, Shieh JY, Sun WZ. Protein synthesis inhibitor cycloheximide dose-dependently decreases formalin-induced c-Fos protein and behavioral hyperalgesia in rats. Neurosci Lett. 1997;227:99–102. doi: 10.1016/s0304-3940(97)00321-2. [DOI] [PubMed] [Google Scholar]
- Hu NW, Zhang HM, Hu XD, Li MT, Zhang T, Zhou LJ, Liu XG. Protein synthesis inhibition blocks the late-phase LTP of C-fiber evoked field potentials in rat spinal dorsal horn. J Neurophysiol. 2003;89:2354–2359. doi: 10.1152/jn.01027.2002. [DOI] [PubMed] [Google Scholar]
- Huber KM, Gallagher SM, Warren ST, Bear MF. Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proc Natl Acad Sci USA. 2002;99:7746–7750. doi: 10.1073/pnas.122205699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda H, Stark J, Fischer H, Wagner M, Drdla R, Jager T, Sandkuhler J. Synaptic amplifier of inflammatory pain in the spinal dorsal horn. Science. 2006;312:1659–1662. doi: 10.1126/science.1127233. [DOI] [PubMed] [Google Scholar]
- Jacquemont S, Hagerman RJ, Leehey M, Grigsby J, Zhang L, Brunberg JA, Greco C, Des Portes V, Jardini T, Levine R, Berry-Kravis E, Brown WT, Schaeffer S, Kissel J, Tassone F, Hagerman PJ. Fragile X premutation tremor/ataxia syndrome: molecular, clinical, and neuroimaging correlates. Am J Hum Genet. 2003;72:869–878. doi: 10.1086/374321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji RR, Kohno T, Moore KA, Woolf CJ. Central sensitization and LTP: do pain and memory share similar mechanisms? Trends Neurosci. 2003;26:696–705. doi: 10.1016/j.tins.2003.09.017. [DOI] [PubMed] [Google Scholar]
- Jin P, Duan R, Qurashi A, Qin Y, Tian D, Rosser TC, Liu H, Feng Y, Warren ST. Pur alpha binds to rCGG repeats and modulates repeat-mediated neurodegeneration in a Drosophila model of fragile X tremor/ataxia syndrome. Neuron. 2007;55:556–564. doi: 10.1016/j.neuron.2007.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karim F, Wang CC, Gereau RWt. Metabotropic glutamate receptor subtypes 1 and 5 are activators of extracellular signal-regulated kinase signaling required for inflammatory pain in mice. J Neurosci. 2001;21:3771–3779. doi: 10.1523/JNEUROSCI.21-11-03771.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelleher RJ, III, Govindarajan A, Tonegawa S. Translational regulatory mechanisms in persistent forms of synaptic plasticity. Neuron. 2004;44:59–73. doi: 10.1016/j.neuron.2004.09.013. [DOI] [PubMed] [Google Scholar]
- Kim SJ, Thomas KS, Calejesan AA, Zhuo M. Macromolecular synthesis contributes to nociceptive response to subcutaneous formalin injection in mice. Neuropharmacology. 1998;37:1091–1093. doi: 10.1016/s0028-3908(98)00099-9. [DOI] [PubMed] [Google Scholar]
- Koekkoek SK, Yamaguchi K, Milojkovic BA, Dortland BR, Ruigrok TJ, Maex R, De Graaf W, Smit AE, VanderWerf F, Bakker CE, Willemsen R, Ikeda T, Kakizawa S, Onodera K, Nelson DL, Mientjes E, Joosten M, De Schutter E, Oostra BA, Ito M. Deletion of FMR1 in Purkinje cells enhances parallel fiber LTD, enlarges spines, and attenuates cerebellar eyelid conditioning in Fragile X syndrome. Neuron. 2005;47:339–352. doi: 10.1016/j.neuron.2005.07.005. [DOI] [PubMed] [Google Scholar]
- Li J, Pelletier MR, Perez Velazquez JL, Carlen PL. Reduced cortical synaptic plasticity and GluR1 expression associated with fragile X mental retardation protein deficiency. Mol Cell Neurosci. 2002;19:138–151. doi: 10.1006/mcne.2001.1085. [DOI] [PubMed] [Google Scholar]
- Martinez-Gomez J, Lopez-Garcia JA. Electrophysiological and pharmacological characterisation of ascending anterolateral axons in the in vitro mouse spinal cord. J Neurosci Methods. 2005;146:84–90. doi: 10.1016/j.jneumeth.2005.01.015. [DOI] [PubMed] [Google Scholar]
- Mazroui R, Huot ME, Tremblay S, Filion C, Labelle Y, Khandjian EW. Trapping of messenger RNA by Fragile X Mental Retardation protein into cytoplasmic granules induces translation repression. Hum Mol Genet. 2002;11:3007–3017. doi: 10.1093/hmg/11.24.3007. [DOI] [PubMed] [Google Scholar]
- Molliver DC, Wright DE, Leitner ML, Parsadanian AS, Doster K, Wen D, Yan Q, Snider WD. IB4-binding DRG neurons switch from NGF to GDNF dependence in early postnatal life. Neuron. 1997;19:849–861. doi: 10.1016/s0896-6273(00)80966-6. [DOI] [PubMed] [Google Scholar]
- Moretti P, Levenson JM, Battaglia F, Atkinson R, Teague R, Antalffy B, Armstrong D, Arancio O, Sweatt JD, Zoghbi HY. Learning and memory and synaptic plasticity are impaired in a mouse model of Rett syndrome. J Neurosci. 2006;26:319–327. doi: 10.1523/JNEUROSCI.2623-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muddashetty RS, Kelic S, Gross C, Xu M, Bassell GJ. Dysregulated metabotropic glutamate receptor-dependent translation of AMPA receptor and postsynaptic density-95 mRNAs at synapses in a mouse model of fragile X syndrome. J Neurosci. 2007;27:5338–5348. doi: 10.1523/JNEUROSCI.0937-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murashov AK, Chintalgattu V, Islamov RR, Lever TE, Pak ES, Sierpinski PL, Katwa LC, Van Scott MR. RNAi pathway is functional in peripheral nerve axons. FASEB J. 2007;21:656–670. doi: 10.1096/fj.06-6155com. [DOI] [PubMed] [Google Scholar]
- Nosyreva ED, Huber KM. Metabotropic receptor-dependent long-term depression persists in the absence of protein synthesis in the mouse model of fragile X syndrome. J Neurophysiol. 2006;95:3291–3295. doi: 10.1152/jn.01316.2005. [DOI] [PubMed] [Google Scholar]
- Osman OT, Loschen EL. Self-injurious behavior in the developmentally disabled: pharmacologic treatment. Psychopharmacol Bull. 1992;28:439–449. [PubMed] [Google Scholar]
- Percy AK. Rett syndrome. Current status and new vistas. Neurol Clin. 2002;20:1125–1141. doi: 10.1016/s0733-8619(02)00022-1. [DOI] [PubMed] [Google Scholar]
- Price DD, Hu JW, Dubner R, Gracely RH. Peripheral suppression of first pain and central summation of second pain evoked by noxious heat pulses. Pain. 1977;3:57–68. doi: 10.1016/0304-3959(77)90035-5. [DOI] [PubMed] [Google Scholar]
- Price DD, Hayes RL, Ruda M, Dubner R. Neural representation of cutaneous aftersensations by spinothalamic tract neurons. Fed Proc. 1978;37:2237–2239. [PubMed] [Google Scholar]
- Price TJ, Flores CM. Critical evaluation of the colocalization between calcitonin gene-related peptide, substance P, transient receptor potential vanilloid subfamily type 1 immunoreactivities, and isolectin B4 binding in primary afferent neurons of the rat and mouse. J Pain. 2007;8:263–272. doi: 10.1016/j.jpain.2006.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price TJ, Flores CM, Cervero F, Hargreaves KM. The RNA binding and transport proteins staufen and fragile X mental retardation protein are expressed by rat primary afferent neurons and localize to peripheral and central axons. Neuroscience. 2006;141:2107–2116. doi: 10.1016/j.neuroscience.2006.05.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter JD, Sonenberg N. Regulation of cap-dependent translation by eIF4E inhibitory proteins. Nature. 2005;433:477–480. doi: 10.1038/nature03205. [DOI] [PubMed] [Google Scholar]
- Sandkuhler J. Understanding LTP in pain pathways. Mol Pain. 2007;3:9. doi: 10.1186/1744-8069-3-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandkuhler J, Chen JG, Cheng G, Randic M. Low-frequency stimulation of afferent Adelta-fibers induces long-term depression at primary afferent synapses with substantia gelatinosa neurons in the rat. J Neurosci. 1997;17:6483–6491. doi: 10.1523/JNEUROSCI.17-16-06483.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuman EM, Dynes JL, Steward O. Synaptic regulation of translation of dendritic mRNAs. J Neurosci. 2006;26:7143–7146. doi: 10.1523/JNEUROSCI.1796-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofola OA, Jin P, Qin Y, Duan R, Liu H, de Haro M, Nelson DL, Botas J. RNA-binding proteins hnRNP A2/B1 and CUGBP1 suppress fragile X CGG premutation repeat-induced neurodegeneration in a Drosophila model of FXTAS. Neuron. 2007;55:565–571. doi: 10.1016/j.neuron.2007.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steward O. mRNA at synapses, synaptic plasticity, and memory consolidation. Neuron. 2002;36:338–340. doi: 10.1016/s0896-6273(02)01006-1. [DOI] [PubMed] [Google Scholar]
- Symons FJ, Danov SE. A prospective clinical analysis of pain behavior and self-injurious behavior. Pain. 2005;117:473–477. doi: 10.1016/j.pain.2005.07.010. [DOI] [PubMed] [Google Scholar]
- Symons FJ, Clark RD, Hatton DD, Skinner M, Bailey DB., Jr Self-injurious behavior in young boys with fragile X syndrome. Am J Med Genet A. 2003;118:115–121. doi: 10.1002/ajmg.a.10078. [DOI] [PubMed] [Google Scholar]
- Symons FJ, Thompson A, Rodriguez MC. Self-injurious behavior and the efficacy of naltrexone treatment: a quantitative synthesis. Ment Retard Dev Disabil Res Rev. 2004;10:193–200. doi: 10.1002/mrdd.20031. [DOI] [PubMed] [Google Scholar]
- Tassone F, Beilina A, Carosi C, Albertosi S, Bagni C, Li L, Glover K, Bentley D, Hagerman PJ. Elevated FMR1 mRNA in premutation carriers is due to increased transcription. RNA. 2007;13:555–562. doi: 10.1261/rna.280807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd PK, Mack KJ, Malter JS. The fragile X mental retardation protein is required for type-I metabotropic glutamate receptor-dependent translation of PSD-95. Proc Natl Acad Sci USA. 2003;100:14374–14378. doi: 10.1073/pnas.2336265100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma P, Chierzi S, Codd AM, Campbell DS, Meyer RL, Holt CE, Fawcett JW. Axonal protein synthesis and degradation are necessary for efficient growth cone regeneration. J Neurosci. 2005;25:331–342. doi: 10.1523/JNEUROSCI.3073-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiler IJ, Spangler CC, Klintsova AY, Grossman AW, Kim SH, Bertaina-Anglade V, Khaliq H, de Vries FE, Lambers FA, Hatia F, Base CK, Greenough WT. Fragile X mental retardation protein is necessary for neurotransmitter-activated protein translation at synapses. Proc Natl Acad Sci USA. 2004;101:17504–17509. doi: 10.1073/pnas.0407533101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weragoda RM, Ferrer E, Walters ET. Memory-like alterations in Aplysia axons after nerve injury or localized depolarization. J Neurosci. 2004;24:10393–10401. doi: 10.1523/JNEUROSCI.2329-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis D, Li KW, Zheng JQ, Chang JH, Smit A, Kelly T, Merianda TT, Sylvester J, van Minnen J, Twiss JL. Differential transport and local translation of cytoskeletal, injury-response, and neurodegeneration protein mRNAs in axons. J Neurosci. 2005;25:778–791. doi: 10.1523/JNEUROSCI.4235-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis DE, Twiss JL. The evolving roles of axonally synthesized proteins in regeneration. Curr Opin Neurobiol. 2006;16:111–118. doi: 10.1016/j.conb.2006.01.002. [DOI] [PubMed] [Google Scholar]
- Wilson BM, Cox CL. Absence of metabotropic glutamate receptor-mediated plasticity in the neocortex of fragile X mice. Proc Natl Acad Sci USA. 2007;104:2454–2459. doi: 10.1073/pnas.0610875104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu KY, Hengst U, Cox LJ, Macosko EZ, Jeromin A, Urquhart ER, Jaffrey SR. Local translation of RhoA regulates growth cone collapse. Nature. 2005;436:1020. doi: 10.1038/nature03885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youings SA, Murray A, Dennis N, Ennis S, Lewis C, McKechnie N, Pound M, Sharrock A, Jacobs P. FRAXA and FRAXE: the results of a five year survey. J Med Genet. 2000;37:415–421. doi: 10.1136/jmg.37.6.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao MG, Toyoda H, Ko SW, Ding HK, Wu LJ, Zhuo M. Deficits in trace fear memory and long-term potentiation in a mouse model for fragile X syndrome. J Neurosci. 2005;25:7385–7392. doi: 10.1523/JNEUROSCI.1520-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng JQ, Kelly TK, Chang B, Ryazantsev S, Rajasekaran AK, Martin KC, Twiss JL. A functional role for intra-axonal protein synthesis during axonal regeneration from adult sensory neurons. J Neurosci. 2001;21:9291–9303. doi: 10.1523/JNEUROSCI.21-23-09291.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu CZ, Wilson SG, Mikusa JP, Wismer CT, Gauvin DM, Lynch JJ, III, Wade CL, Decker MW, Honore P. Assessing the role of metabotropic glutamate receptor 5 in multiple nociceptive modalities. Eur J Pharmacol. 2004;506:107–118. doi: 10.1016/j.ejphar.2004.11.005. [DOI] [PubMed] [Google Scholar]
- Zwick M, Davis BM, Woodbury CJ, Burkett JN, Koerber HR, Simpson JF, Albers KM. Glial cell line-derived neurotrophic factor is a survival factor for isolectin B4-positive, but not vanilloid receptor 1-positive, neurons in the mouse. J Neurosci. 2002;22:4057–4065. doi: 10.1523/JNEUROSCI.22-10-04057.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]