

Abstract
Lasofoxifene is a new and potent selective estrogen receptor modulator (SERM). The structural basis of its interaction with the estrogen receptor has been investigated by crystallographic analysis of its complex with the ligand-binding domain of estrogen receptor α at a resolution of 2.0 Å. As with other SERMs, lasofoxifene diverts the receptor from its agonist-bound conformation by displacing the C-terminal AF-2 helix into the site at which the LXXLL motif of coactivator proteins would otherwise be able to bind. Lasofoxifene achieves this effect by occupying the space normally filled by residue Leu 540, as well as by modulating the conformation of residues of helix 11 (His 524, Leu 525). A well-defined salt bridge between lasofoxifene and Asp 351 suggests that charge neutralization in this region of the receptor may explain the some of the antiestrogenic effects of lasofoxifene. The results suggest general features of ERα/SERM recognition, and add a new dimension to efforts to rationalize differences between the biological activity profiles exhibited by these important pharmacological agents.
Keywords: structure, crystallography, nuclear receptor, SERM, estrogen
The estrogen receptor (ER) belongs to the nuclear hormone receptor superfamily, and exists as two isoforms, ERα and ERβ (Fawell et al. 1990; Kuiper et al. 1996; Mosselman et al. 1996). The ER mediates diverse biological effects in a range of tissues in response to a variety of endogenous, environmental, and pharmacological agents. Selective estrogen receptor modulators (SERMs) are an important group of pharmacological agents used primarily for treatment of breast cancer and osteoporosis. Their most remarkable property is to act as estrogens in some tissues but antiestrogens in others, an effect made possible by the role of coactivator and corepressor proteins that function by forming complexes with the ER and are differentially expressed in different tissues. The existence of the two isoforms, which have slightly different specificities, and the additional complexity brought to the picture by coregulatory proteins all contribute to making ER-mediated signaling a highly complex and subtle means of tissue-specific control of gene expression (Paech et al. 1997).
The two isoforms of human ER possess primary structures with 46% identity and similar domain architectures (Kuiper et al. 1996; Mosselman et al. 1996). An N-terminal activation function 1 (AF-1) domain is followed by a highly conserved zinc-finger-containing DNA-binding domain, a poorly conserved linker region, and finally a C-terminal ligand binding domain (LBD). The ER LBD contains 10–12 helices arranged in a compact, three-layer helical bundle. Its ligand-binding pocket resides in the interior of the domain, nearly completely sequestering agonist ligands such as estradiol from the solvent. The LBD also contains a region critical for transactivation, termed the activation function 2 region (AF-2) (Danielian et al. 1992), consisting primarily of the residues located in the C-terminal helix of the LBD. Structural studies have demonstrated that in the presence of agonist, the AF-2 helix docks into a pocket formed at the junction of helices 3, 5, and 11 (Shiau et al. 1998, 2002). In this conformation, the LBD can accept a peptide segment, the LXXLL motif or NR-box, from a variety of coactivator proteins including members of the p160 family of proteins, such as SRC-1 and GRIP1 (for review, see Edwards 1999). In the presence of an antagonist, however, the AF-2 helix undergoes a large conformational shift and occupies the LXXLL-binding cleft (Shiau et al. 1998, 2002; Pike et al. 1999, 2001). Surprisingly, the SERMs tamoxifen and raloxifene (Shang and Brown 2002) induce a conformation of AF-2 that is clearly the antagonist-bound form (Brzozowski et al. 1997; Shiau et al. 1998; Pike et al. 1999). In both of these SERMs, a large “pendant” side chain emanating from the core of the ligand occupies the space normally occupied by Leu 540, preventing the AF-2 helix from docking in its preferred (agonist) conformation, and shifting it to occupy the LXXLL-binding cleft, thereby preventing coactivator binding. Despite the similar effects of these two ligands on ER structure, they possess very different biological profiles—tamoxifen acts as a partial agonist in uterine tissue and a potent antagonist in breast tissue, while raloxifene acts as an antagonist in both tissue types (Shang and Brown 2002). The tissue-dependence of their respective biological activities has been shown to be dependent on the different concentrations of various coactivator proteins (Shang and Brown 2002).
Lasofoxifene {(5R,6S)-6-phenyl-5-[4-(2-pyrrolidin-1-yl-ethoxy)-phenyl]-5,6,7,8-tetrahydro-naphthalen-2-ol} is a member of the diaryltetrahydronaphthalene family (Fig. 1) and was discovered as part of a program to identify potent SERMs that possess bone efficacy, with improved bioavailability over raloxifene (Evista). The primary impediment to absorption of raloxifene has been reported to be glucuronidation in the gut; a pharmacophore model that predicts resistance to gut wall glucuronidation has been proposed for which the primary structural requirement is a nonplanar topology with steric bulk in close proximity to the plane of a fused bicyclic aromatic system (Rosati et al. 1998). Lasofoxifene satisfies this requirement, while retaining potent ERα-binding affinity (Rosati et al. 1998). Lasofoxifene possesses SERM activity, as it completely prevents bone loss in ovariectomized, orchidectomized, and aged rats and does not cause uterine hyperplasia (Ke et al. 1998, 2000, 2004). Emerging clinical data have confirmed lasofoxifene's efficacy in the prevention of bone loss and reduction of LDL cholesterol in postmenopausal women (Moffet 2002).
Figure 1.

Structure of lasofoxifene.
We have determined the structure of the human ERα LBD bound to the novel SERM lasofoxifene at a resolution of 2.0 Å. The structure reveals both the specific interactions between the protein and the ligand, and the conformation of the AF-2 domain. This study extends the knowledge of SERM–target interactions that is required to rationalize the complex activity of this important class of drugs.
Results
Description of the structure
The structure presented in this study consists of one ERα complexed with lasofoxifene and 161 water molecules. Of the 253 amino acid residues present in the crystallization experiment, 247 have been modeled; residues 301–305 (SKKNS), T553, and side-chain atoms of Leu 306, Arg 335, Met 437, Leu 462, Ser 463, Ser 464, Lys 531, Asn 532, Val 534, and Pro 552 have been omitted because of lack of electron density. Several regions of the structure could not be modeled with certainty, however, including the region Lys 416 to Met 421, Leu 462, and Tyr 526 to Leu 536. Nevertheless, electron density maps (SigmaA weighted, either 2mFodFc or mFodFc), calculated after omission of these residues and application of a random 0.5 Å shift to all coordinates and several cycles of refinement, suggested that the placement of these residues was correct. The high mobility of these residues is reflected in the very high B-factors for atoms in these regions (Bave = 63.6 Å2). In particular, residues Lys 416 to Met 421 appear to occupy several different conformations; the major conformation has been modeled with unit occupancy. Sparse, disconnected electron density at the 3X-RMSD level indicates, however, that some minor fraction occupies a conformation similar to that modeled in the ERα/tamoxifen structure (3ERT). However, modeling the structure as either conformation alone, or dual conformations with half occupancy, did not improve the model, as judged by the local electron density or more global indicators such as R or Rfree. Cysteine residues 417 and 530 are each within these poorly defined regions; modification of these residues by carboxymethylation has been determined previously to be important for maintaining homogeneity during the course of purification and crystallization. While it is possible that the modification of these two residues may be partially responsible for the disorder observed in the structure, this is unlikely because other ERα structures with S-carboxymethylation at these sites appear well-ordered (Shiau et al. 1998; Shiau et al. 2002). The remaining two Cys residues, Cys 381 and Cys 447, show no evidence of modification, in good agreement with biochemical results (Goldstein et al. 2001).
The ERα adopts the same three-layer “helical sandwich” described for ERα and other nuclear hormone receptor LBDs (Fig. 2). As seen in the crystal structures of ERα in complex with other SERMs or estrogen antagonists (Shiau et al. 1998, 2002; Pike et al. 1999, 2001), the C-terminal helix 12 is in the “antagonist-bound” conformation, and occupies the surface of the molecule that normally accepts the LXXLL motif of coactivator proteins such as SRC or TIF2 (see below).
Figure 2.
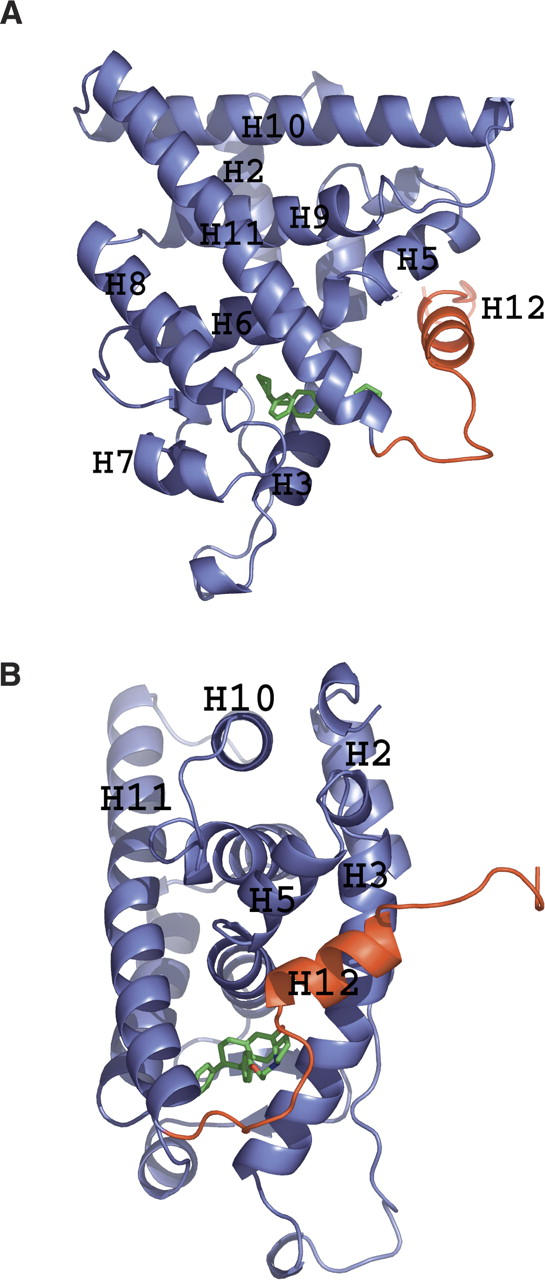
Ribbon diagram of the structure of ERα LBD showing the principal secondary structure elements. (A) The pendant side chain of bound lasofoxifene (green) displaces helix 12 (H12, red) from its normal location in the agonist-bound conformation of the domain. (B) Structure rotated by 90° around the vertical axis.
Description of lasofoxifene
Lasofoxifene was clearly identifiable in unbiased electron density maps (Fig. 3A; we have chosen to use standard IUPAC nomenclature for numbering, which differs from that used in Rosati et al. 1998). The structure explains why lasofoxifene is more active than its 5S enantiomer, as the 5R configuration places the “pendant” side chain along the same trajectory as that seen in tamoxifen (Shiau et al. 1998), raloxifene (Brzozowski et al. 1997), and ICI-164384 (Pike et al. 2001). The 5S enantiomer presumably does not bind in the same productive manner, and based on the current structure, would be predicted to direct the side chain in the opposite direction, toward the exterior of the protein between helix 3 and β-strand 2. The orientation of lasofoxifene is fixed by an H-bonding network involving the 2-OH of lasofoxifene, the guanidinium group of Arg 394, and the carboxylate of Glu 353 (Fig. 3A). The tetrahydronaphthalene core of lasofoxifene is sandwiched between the hydrophobic residues in the ERα ligand-binding pocket; Leu 346, Leu 391, Phe 404, and Met 421 form the “top” of the binding pocket, while Leu 384, Leu 387, and Met 388 form the “bottom” (Fig. 3A). The pendant side chain forms both hydrophobic and hydrophilic interactions with ERα residues as it protrudes from the ligand binding pocket out toward the surface of the protein (Fig. 3A). Trp 383, Thr 347, and Leu 525 all pack against the aryl and alkyl portions of the side chain, while the pyrrolodine nitrogen atom is in a position to form a nearly ideal H-bonding interaction with the side-chain carboxylate of Asp 351 (2.74 Å), indicating that this N is most likely protonated (Fig. 3B). The 6-phenyl group makes largely hydrophobic interactions with Met 343, Met 421, His 524, and Leu 525.
Figure 3.

Electron density contoured at 1XRMSD corresponding to lasofoxifene bound in the ligand-binding cavity of ERα LBD. (A) Hydrogen-bonding interactions (blue dashed lines) of the aromatic hydroxyl group of lasofoxifene with Glu 353 and Arg 394, and of the pyrrolidine N with Asp 351. Other packing interactions around the hydrophobic core of the molecule are labeled. (B) Electron density corresponding to the terminal pyrrolidine of lasofoxifene and Asp 351.
Discussion
Hydroxyl groups on the A-ring and D-ring of 17β-estradiol participate in interactions that orient and anchor the molecule in the ligand-binding pocket of ERα; that on the A-ring is involved in a hydrogen bond with Arg 394, and the hydroxyl on the D-ring with His 524. The four rings of the hormone are sequestered within the internal cavity of the LBD and stabilize the LBD in an “agonist-bound” conformation (Fig. 4A). Numerous crystal structures of ERα complexed with various SERMs have revealed a conserved mode of binding, the primary features of which involve (1) a hydrogen-bond acceptor that serves to “anchor” the ligand in the ERα ligand-binding pocket via a polar interaction with Arg 394; (2) a nearly planar “core” structure typically composed of a biaryl heterocycle, analogous to the A-ring and B-ring of 17β-estradiol; (3) a “pendant” side chain emanating from the “B-ring equivalent” of the biaryl structure (analogous to the B-ring of estradiol); and (4) a second substituent (typically aromatic) that fills the remainder (“C-ring and D-ring” equivalent) volume of the ligand-binding pocket. A variety of SERMs with biaryl core structures have been identified in recent years, including tetrahydroisoquinolines, benzothiophenes, chromanes, and dihydrobenzoxathiins (Brzozowski et al. 1997; Renaud et al. 2003, 2005; Kim et al. 2004; Blizzard et al. 2005; Tan et al. 2005). X-ray crystal structures of representatives of each of these scaffolds illustrate the generality of the Arg 394 H-bond “anchor” and planar, biaryl topology features of SERM recognition (Fig. 4B–E). Likewise, in each of these cases, a pendant side chain protrudes from one side of the core structure, threading its way through the body of the protein to ultimately hinder the proper positioning of the AF-2 helix in its “agonist” conformation. Typically, this side chain is stabilized by both hydrophobic interactions as well as a well-conserved H-bond between a terminal tertiary amine and Asp 351, indicating a shared proton between the charged Asp side chain and the otherwise neutral tertiary amine. Finally, the C-ring and D-ring equivalent volume of the ligand-binding pocket of ERα is filled in these examples by either a phenyl or phenolic substituent. Typically, the phenyl or phenolic substituent of each molecule adopts an orientation that is nearly orthogonal to the D-ring of estradiol (Fig. 4A). A notable exception is the SERM raloxifene (Brzozowski et al. 1997), which, presumably because of its five-membered heterocycle core (benzothiophene), directs its phenolic substituent along an altered trajectory, relative to other SERMS, while maintaining approximate coplanarity with the D-ring of estradiol (Fig. 4A,E). The conserved features of ligand binding in these diverse SERM core structures are mirrored by a high degree of similarity among the receptor structures themselves, with the most significant differences occurring primarily in the regions surrounding the D-ring pocket. While it is possible that some of the differences in protein structure in this region may be due to different forms of ERα (some forms used a Cys-Ser triple mutant to ameliorate problems with protein aggregation) (Renaud et al. 2003, 2005), it is likely that some of the structural differences observed in these compounds are due to the inherent plasticity of this region of the ERα LBD.
Figure 4.
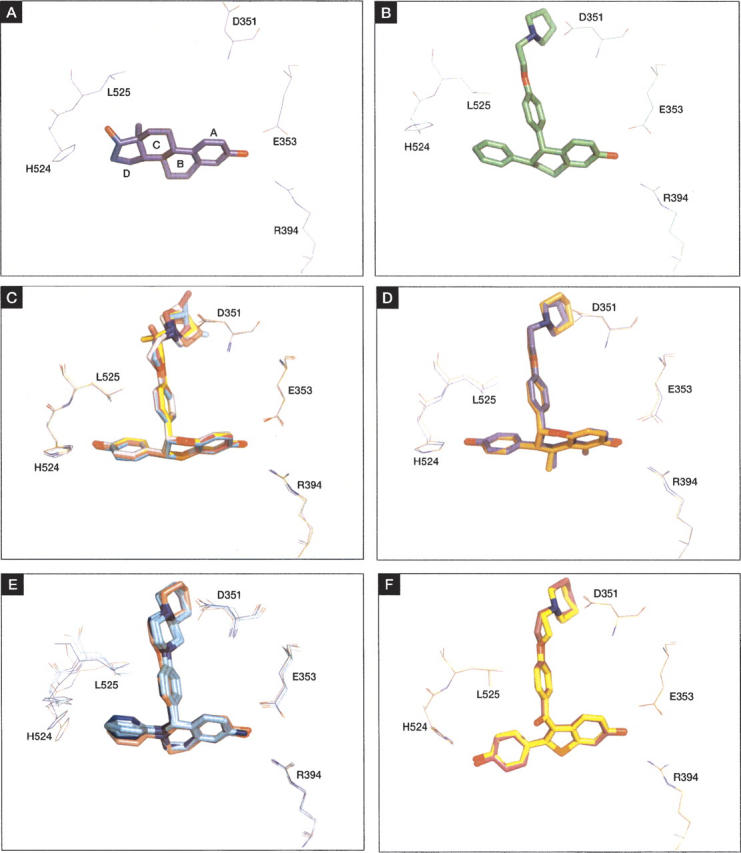
Representative SERM families (stick) with key interacting residues (Arg 394, Glu 353, Asp 351, His 524, and Leu 525). (A) 17β-Estradiol (PDB entry 1ERE). (B) Lasofoxifene. (C) Benzoxathiins (PDB entries 1XP1, 1XP6, 1XP9, 1XPC, 1SJ0). (D) Chromanes (PDB entries 1YIM, 1YIN). (E) Tetrahydroisoquinolines (PDB entries 1UOM, 1XQCa,b,c,d). (F) Raloxifene (PDB entry 1ERRa,b). All structures, including those with multiple molecules in the asymmetric unit (1XQC and 1ERR), were superimposed on the ERα/lasofoxifene structure for comparison.
The structure of ERα complexed with lasofoxifene is consistent with the general features of SERM–ERα recognition, including a nearly planar topology (the tetrahydronapthalene carbocycle) and a bidentate H-bonding interaction with Arg 394 and Glu 353 (via the terminal OH group; Fig. 4B). As in other SERM–ERα structures, the C-ring and D-ring volume of the ligand-binding pocket are filled by the phenyl side chain of lasofoxifene. The orientation of this phenyl ring is consistent with that of the tetrahydroisoquinolines, chromanes, and dihydrobenzoxathiins (Renaud et al. 2003, 2005; Kim et al. 2004; Blizzard et al. 2005), but is distinct from that of the benzothiophene structure of raloxifene (Brzozowski et al. 1997). The placement and orientation of this aromatic substituent in lasofoxifene forces His 524 into an alternative position (Fig. 4B). Simultaneously, a close contact between the pendant side chain oxygen atom and Leu 525 forces this residue into a different rotamer, disrupting van der Waals interactions between this residue and the helix 12 residue Leu 544. These two “trigger” residues, His 524 and Leu 525, are identical to those described by Shiau et al. (2002) in their description of passive antagonism in the ERβ/THC structure (Shiau et al. 2002). In the passive antagonism model, the shift of these residues to nonproductive conformations causes secondary shifts, notably in the positions of Met 528 and Val 533, and ultimately results in an unwinding of the C-terminal end of helix 11. As this region of ERα forms one side of the AF-2 helix-binding pocket, the net effect is to destabilize the agonist conformation of the AF-2 helix. It is tempting to speculate that a similar mechanism may be at work in the present system, disfavoring the agonist-bound conformation of the AF-2 helix through “indirect” interactions, and shifting the conformational equilibrium to the “antagonist-bound” form of the receptor.
In addition to these general features of SERM recognition, a large alkyl “pendant” side chain, terminating in a pyrollidine head group, threads its way from the ligand-binding pocket out toward the surface of the protein, where it directly interferes with the correct positioning of the AF-2 helix (Fig. 4B). As seen in the structure of raloxifene bound to ERα (Brzozowski et al. 1997), the pyrrolidine tertiary amine in lasofoxifene is clearly engaged in a nearly ideal H-bonding interaction with the side-chain carboxyl group of Asp 351 (Fig. 5A,B). This hydrogen bond is notably absent in the structure of 4-hydroxytamoxifen bound to ERα, where the distance between the tertiary amine and the carboxylate of Asp 351 (3.8 Å) is clearly too great to be consistent with an H-bond interaction (Fig. 5C). The structures of lasofoxifene and raloxifene bound to ERα suggest that the methylene carbons that complete the terminal azacycle facilitate this H-bond to Asp 351 by anchoring this portion of the pendant side chain against a hydrophobic pocket, where the nitrogen is now correctly positioned for interaction with the side-chain carboxylate. This interaction may serve to enhance the interaction of ERα with corepressor proteins by neutralizing the charge of Asp 351, thereby allowing for a possible repositioning of AF-2 against the otherwise hydrophobic face of helix 3. Such a repositioning of AF-2 has been demonstrated to be critical for corepressor recruitment in the case of PPARα bound to a potent antagonist (Xu et al. 2002), and Nettles and Greene (2005) have suggested a similar mechanism at work in ERα. In support of this mechanism, modifications to the pendant side chain of tamoxifen that alter the nature of the azacycle, while preserving the tertiary amine functionality, show dramatic differences in the level of transcriptional activation in cell-based assays (Dayan et al. 2006). Abolition of the negative charge at Asp 351, by mutation of this residue to Ala or Val, greatly reduced the propensity for transcriptional activation in tamoxifen derivatives, regardless of the chemical structure of the terminal azacycle (Dayan et al. 2006). Similarly, Webb et al. (2003) have reported differential effects of tamoxifen and raloxifene on recruitment of corepressor proteins. Tamoxifen and raloxifene have each been shown to display antiestrogenic properties in breast tissue (Fisher et al. 1996; Cummings et al. 1999); in contrast, tamoxifen, but not raloxifene, has demonstrated agonist-like properties in uterine tissue (Baker et al. 1998; Bernstein et al. 1999). This tissue-dependent biological activity profile has been rationalized by a model invoking the different relative stoichiometries of corepressors and coactivators such as NCOR and SRC-1 (Shang and Brown 2002). Taken together, these observations suggest that the tissue-specific effects of different SERMs are due in part to differential effects on corepressor interactions, and that these interactions are sensitive to the charge-neutralization capacity of the ligand in this region.
Figure 5.

Hydrogen-bonding interaction between Asp 351 and the tertiary amines of (A) lasofoxifene, (B) raloxifene, and (C) tamoxifen. (D–F) The same interaction against a surface representation of ERα. Note the deeper insertion of the closed azacycles of lasofoxifene and raloxifene into the hydrophobic pocket, relative to that of tamoxifen.
In summary, the crystal structure of ERα bound to the novel SERM lasofoxifene has revealed the specific interactions between the receptor and ligand. These interactions are consistent with the general nature of SERM recognition, as revealed through analysis of X-ray crystal structures of ERα/SERM complexes of widely differing structural classes. The nature of lasofoxifene's interaction with residues in the C-ring and D-ring region of the ERα-binding pocket suggests a more general role for passive antagonism in SERM activity, although this connection cannot be definitively established on the basis of the single structure presented here. Finally, the interaction of the terminal azacycle on the “pendant” side chain of lasofoxifene with Asp 351 closely mirrors that seen in the structure of ERα bound to raloxifene. The charge-neutralizing capacity of this salt bridge is consistent with the biological activity profiles of raloxifene and tamoxifen. It will be of great interest to extend this analysis to links between the tissue specificity of coactivator and corepressor expression and the biological activity profile of lasofoxifene.
Materials and Methods
Expression, purification, and crystallization of ERα/lasofoxifene
The ligand-binding domain of estrogen receptor-α (ERα), comprising residues 301–553 (SWISS-PROT P03372), was amplified by PCR and ligated into pET23b (Novagen) for overexpression in Escherichia coli. Cells containing the protein were harvested following overnight induction with 50 μM IPTG at 25°C. ERα was purified, carboxymethylated, and complexed with lasofoxifene as described (Goldstein et al. 2001). The resulting complex was concentrated to a final concentration of 8–10 mg/mL by diafiltration into buffer containing 20 mM Tris-HCl, 5 mM NaCl, 0.5 mM EDTA, and 5 mM DTT (pH 8). Crystals were grown by vapor diffusion using the hanging-drop method. Drops consisted of 2 μL of the complex mixed with 2 μL of reservoir solution containing 0.1 M Na HEPES (pH 6.7), 0.5 M NaCl, 6% ethylene glycol, and 10%–12% PEG 8000. Crystals grew within 1 wk at 22°C.
Data collection
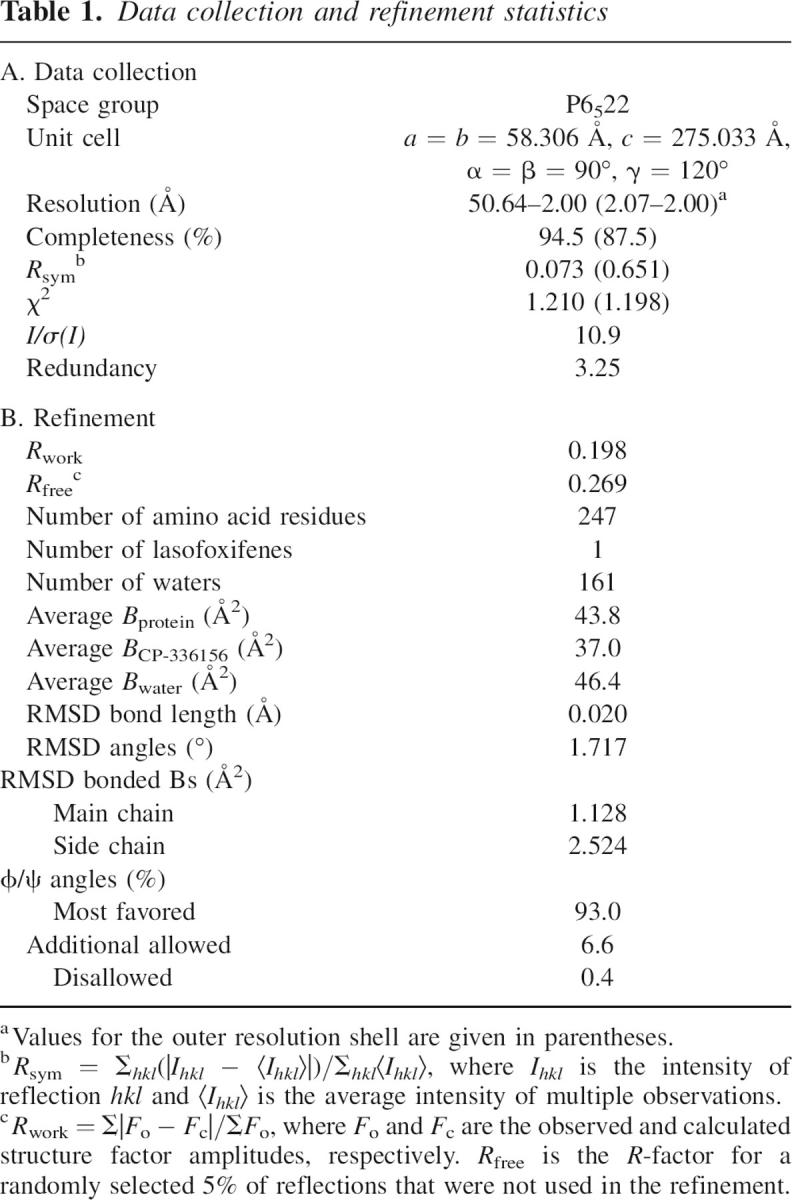
Crystals were harvested into a solution containing 90 mM Na HEPES (pH 6.7), 0.5 M NaCl, 12% PEG 8000, 5 mM DTT, and 24% ethylene glycol and were flash-cooled in a nitrogen gas stream at 100 K for data collection. Diffraction measurements to 2.0 Å were made at Cu Ka wavelength (1.5418 Å) on an RU-H2R rotating anode X-ray generator equipped with an Raxis IIC image-plate detector system. A total of 120° of data were collected in 1° increments, and reduced using the HKL suite of programs (Otwinowski and Minor 1997). Intensities were truncated to amplitudes by the method of French and Wilson, using the program TRUNCATE (French and Wilson 1978; Collaborative Computational Project Number 4 1994). Various file manipulations, including assignment of a test set of reflections, were carried out using programs from the CCP4 suite (Collaborative Computational Project Number 4 1994). Data statistics are reported in Table 1A.
Table 1.
Data collection and refinement statistics
Structure determination and refinement
The structure of the ERα/lasofoxifene complex was determined by the molecular replacement method using the coordinates of ERα from the published structure of ERα complexed with tamoxifen (PDB entry 3ERT) (Shiau et al. 1998). All calculations were carried out using the program AMoRe (Navaza 1994) with data from 10–2.5 Å for both cross-rotation function and translation function searches. A clear solution was found, and subjected to refinement against a maximum likelihood target function using the program REFMAC (Murshudov et al. 1997). The structure of lasofoxifene was modeled into unbiased electron density maps, and solvent molecules were placed either automatically using the program ARP/wARP (Lamzin and Wilson 1997), or manually in the program XFIT (McRee 1999). Minimal manual intervention was performed periodically using the program XFIT. The coordinates and structure factors have been deposited with the RCSB with accession code 2OUZ.
Footnotes
Reprint requests to: Felix F. Vajdos, Pfizer Inc., Eastern Point Road, Mail Stop 4039, Groton, CT 06340-8001, USA; e-mail: felix.vajdos@pfizer.com; fax: (860) 686-2095.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.062729207.
References
- Baker V.L., Draper, M., Paul, S., Allerheiligen, S., Glant, M., Shifren, J., and Jaffe, R.B. 1998. Reproductive endocrine and endometrial effects of raloxifene hydrochloride, a selective estrogen receptor modulator, in women with regular menstrual cycles. J. Clin. Endocrinol. Metab. 83: 6–13. [DOI] [PubMed] [Google Scholar]
- Bernstein L., Deapen, D., Cerhan, J.R., Schwartz, S.M., Liff, J., McGann-Maloney, E., Perlman, J.A., and Ford, L. 1999. Tamoxifen therapy for breast cancer and endometrial cancer risk. J. Natl. Cancer Inst. 91: 1654–1662. [DOI] [PubMed] [Google Scholar]
- Blizzard T.A., DiNinno, F., Morgan, J.D., Chen, H.Y., Wu, J.Y., Kim, S., Chan, W., Birzin, E.T., Yang, Y.T., Pai, L.-Y., et al. 2005. Estrogen receptor ligands. Part 9: Dihydrobenzoxathiin SERAMs with alkyl substituted pyrrolidine side chains and linkers. Bioorg. Med. Chem. Lett. 15: 107–113. [DOI] [PubMed] [Google Scholar]
- Brzozowski A.M., Pike, A.C.W., Dauter, Z., Hubbard, R.E., Bonn, T., Engstrom, O., Ohman, L., Greene, G.L., Gustafsson, J.A., and Carlquist, M. 1997. Molecular basis of agonism and antagonism in the oestrogen receptor. Nature 389: 753–758. [DOI] [PubMed] [Google Scholar]
- Collaborative Computational Project Number 4 1994. The CCP4 Suite: Programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50: 760–763. [DOI] [PubMed] [Google Scholar]
- Cummings S.R., Eckert, S., Krueger, K.A., Grady, D., Powles, T.J., Cauley, J.A., Norton, L., Nickelsen, T., Bjarnason, N.H., Morrow, M., et al. 1999. The effect of raloxifene on risk of breast cancer in postmenopausal women: Results from the MORE randomized trial. JAMA 281: 2189–2197. [DOI] [PubMed] [Google Scholar]
- Danielian P.S., White, R., Lees, J.A., and Parker, M.G. 1992. Identification of a conserved region required for hormone dependent transcriptional activation by steroid hormone receptors. EMBO J. 11: 1025–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dayan G., Lupien, M., Auger, A., Anghel, S.I., Rocha, W., Croisetiere, S., Katzenellenbogen, J.A., and Mader, S. 2006. Tamoxifen and raloxifene differ in their functional interactions with aspartate 351 of estrogen receptor α. Mol. Pharmacol. 70: 579–588. [DOI] [PubMed] [Google Scholar]
- Edwards D.P. 1999. Coregulatory proteins in nuclear hormone receptor action. Vitam. Horm. 55: 165–218. [DOI] [PubMed] [Google Scholar]
- Fawell S.E., Lees, J.A., White, R., and Parker, M.G. 1990. Characterization and colocalization of steroid binding and dimerization activities in the mouse estrogen receptor. Cell 60: 953–962. [DOI] [PubMed] [Google Scholar]
- Fisher B., Dignam, J., Bryant, J., DeCillis, A., Wickerham, D.L., Wolmark, N., Costantino, J., Redmond, C., Fisher, E.R., Bowman, D.M., et al. 1996. Five versus more than five years of tamoxifen therapy for breast cancer patients with negative lymph nodes and estrogen receptor-positive tumors. J. Nat. Cancer Inst. 88: 1529–1542. [DOI] [PubMed] [Google Scholar]
- French S. and Wilson, K. 1978. On the treatment of negative intensity observations. Acta Crystallogr. A 34: 517–525. [Google Scholar]
- Goldstein S.W., Bordner, J., Hoth, L.R., and Geoghegan, K.F. 2001. Chemical and biochemical issues related to X-ray crystallography of the ligand-binding domain of estrogen receptor α. Bioconjug. Chem. 12: 406–413. [DOI] [PubMed] [Google Scholar]
- Ke H.Z., Paralkar, V.M., Grasser, W.A., Crawford, D.T., Qi, H., Simmons, H.A., Pirie, C.M., Chidsey-Frink, K.L., Owen, T.A., Smock, S.L., et al. 1998. Effects of CP-336,156, a new, nonsteroidal estrogen agonist/antagonist, on bone, serum cholesterol, uterus and body composition in rat models. Endocrinology 139: 2068–2076. [DOI] [PubMed] [Google Scholar]
- Ke H.Z., Qi, H., Crawford, D.T., Chidsey-Frink, K.L., Simmons, H.A., and Thompson, D.D. 2000. Lasofoxifene (CP-336,156), a selective estrogen receptor modulator, prevents bone loss induced by aging and orchidectomy in the adult rat. Endocrinology 141: 1338–1344. [DOI] [PubMed] [Google Scholar]
- Ke H.Z., Foley, G.L., Simmons, H.A., Shen, V., and Thompson, D.D. 2004. Long-term treatment of lasofoxifene preserves bone mass and bone strength and does not adversely affect the uterus in ovariectomized rats. Endocrinology 145: 1996–2005. [DOI] [PubMed] [Google Scholar]
- Kim S., Wu, J.Y., Birzin, E.T., Frisch, K., Chan, W., Pai, L.-Y., Yang, Y.T., Mosley, R.T., Fitzgerald, P.M.D., Sharma, N., et al. 2004. Estrogen receptor ligands. II. Discovery of benzoxathiins as potent, selective estrogen receptor α modulators. J. Med. Chem. 47: 2171–2175. [DOI] [PubMed] [Google Scholar]
- Kuiper G.G.J.M., Enmark, E., Pelto-Huikko, M., Nilsson, S., and Gustafsson, J.A. 1996. Cloning of a novel estrogen receptor expressed in rat prostate and ovary. Proc. Natl. Acad. Sci. 93: 5925–5930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamzin V.S. and Wilson, K.S. 1997. Automated refinement for protein crystallography. Methods Enzymol. 277: 269–305. [DOI] [PubMed] [Google Scholar]
- McRee D.E. 1999. XtalView/Xfit—A versatile program for manipulating atomic coordinates and electron density. J. Struct. Biol. 125: 156–165. [DOI] [PubMed] [Google Scholar]
- Moffet A. 2002. Emerging data and experience with lasofoxifene: Highlights from clinical trials. Osteoporos. Int. 13: S151. [Google Scholar]
- Mosselman S., Polman, J., and Dijkema, R. 1996. ER β: Identification and characterization of a novel human estrogen receptor. FEBS Lett. 392: 49–53. [DOI] [PubMed] [Google Scholar]
- Murshudov G.N., Vagin, A.A., and Dodson, E.J. 1997. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53: 240–255. [DOI] [PubMed] [Google Scholar]
- Navaza J. 1994. AMoRe: An Automated package for molecular replacement. Acta Crystallogr. A 50: 157–163. [Google Scholar]
- Nettles K.W. and Greene, G.L. 2005. Ligand control of coregulator recruitment to nuclear receptors. Annu. Rev. Physiol. 67: 309–333. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z. and Minor, W. 1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276: 307–326. [DOI] [PubMed] [Google Scholar]
- Paech K., Webb, P., Kuiper, G.G.J.M., Nilsson, S., Gustafsson, J.A., Kushner, P.J., and Scanlan, T.S. 1997. Differential ligand activation of estrogen receptors ERα and ERβ at AP1 sites. Science 277: 1508–1510. [DOI] [PubMed] [Google Scholar]
- Pike A.C.W., Brzozowski, A.M., Hubbard, R.E., Bonn, T., Thorsell, A.G., Engstrom, O., Ljunggren, J., Gustafsson, J.K., and Carlquist, M. 1999. Structure of the ligand-binding domain of oestrogen receptor β in the presence of a partial agonist and a full antagonist. EMBO J. 18: 4608–4618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pike A.C.W., Brzozowski, A.M., Walton, J., Hubbard, R.E., Thorsell, A.G., Li, Y.L., Gustafsson, J.A., and Carlquist, M. 2001. Structural insights into the mode of action of a pure antiestrogen. Structure 9: 145–153. [DOI] [PubMed] [Google Scholar]
- Renaud J., Bischoff, S.F., Buhl, T., Floersheim, P., Fournier, B., Halleux, C., Kallen, J., Keller, H., Schlaeppi, J.-M., and Stark, W. 2003. Estrogen receptor modulators: Identification and structure–activity relationships of potent ERα-selective tetrahydroisoquinoline ligands. J. Med. Chem. 46: 2945–2957. [DOI] [PubMed] [Google Scholar]
- Renaud J., Bischoff, S.F., Buhl, T., Floersheim, P., Fournier, B., Geiser, M., Halleux, C., Kallen, J., Keller, H., and Ramage, P. 2005. Selective estrogen receptor modulators with conformationally restricted side chains. Synthesis and structure–activity relationship of ERα-selective tetrahydroisoquinoline ligands. J. Med. Chem. 48: 364–379. [DOI] [PubMed] [Google Scholar]
- Rosati R.L., Da Silva Jardine, P., Cameron, K.O., Thompson, D.D., Ke, H.Z., Toler, S.M., Brown, T.A., Pan, L.C., Ebbinghaus, C.F., Reinhold, A.R., et al. 1998. Discovery and preclinical pharmacology of a novel, potent, nonsteroidal estrogen receptor agonist/antagonist, CP-336156, a diaryltetrahydronaphthalene. J. Med. Chem. 41: 2928–2931. [DOI] [PubMed] [Google Scholar]
- Shang Y. and Brown, M. 2002. Molecular determinants for the tissue specificity of SERMs. Science 295: 2465–2468. [DOI] [PubMed] [Google Scholar]
- Shiau A.K., Barstad, D., Loria, P.M., Cheng, L., Kushner, P.J., Agard, D.A., and Greene, G.L. 1998. The structural basis of estrogen receptor/coactivator recognition and the antagonism of this interaction by tamoxifen. Cell 95: 927–937. [DOI] [PubMed] [Google Scholar]
- Shiau A.K., Barstad, D., Radek, J.T., Meyers, M.J., Nettles, K.W., Katzenellenbogen, B.S., Katzenellenbogen, J.A., Agard, D.A., and Greene, G.L. 2002. Structural characterization of a subtype-selective ligand reveals a novel mode of estrogen receptor antagonism. Nat. Struct. Biol. 9: 359–364. [DOI] [PubMed] [Google Scholar]
- Tan Q., Blizzard, T.A., Morgan, J.D., Birzin, E.T., Chan, W., Yang, Y.T., Pai, L.-Y., Hayes, E.C., DaSilva, C.A., Warrier, S., et al. 2005. Estrogen receptor ligands. Part 10: Chromanes: Old scaffolds for new SERAMs. Bioorg. Med. Chem. Lett. 15: 1675–1681. [DOI] [PubMed] [Google Scholar]
- Webb P., Nguyen, P., and Kushner, P.J. 2003. Differential SERM effects on corepressor binding dictate ERα activity in vivo. J. Biol. Chem. 278: 6912–6920. [DOI] [PubMed] [Google Scholar]
- Xu H.E., Stanley, T.B., Montana, V.G., Lambert, M.H., Shearer, B.G., Cobb, J.E., McKee, D.D., Galardi, C.M., Plunket, K.D., Nolte, R.T., et al. 2002. Structural basis for antagonist-mediated recruitment of nuclear co-repressors by PPARα. Nature 415: 813–817. [DOI] [PubMed] [Google Scholar]