

Abstract
Small proteins are generally observed to fold in an apparent two-state manner. Recently, however, more sensitive techniques have demonstrated that even seemingly single-domain proteins are actually made up of smaller subdomains. T4 lysozyme is one such protein. We explored the relative autonomy of its two individual subdomains and their contribution to the overall stability of T4 lysozyme by examining a circular permutation (CP13*) that relocates the N-terminal A-helix, creating subdomains that are contiguous in sequence. By determining the high-resolution structure of CP13* and characterizing its energy landscape using native state hydrogen exchange (NSHX), we show that connectivity between the subdomains is an important determinant of the energetic cooperativity but not structural integrity of the protein. The circular permutation results in a protein more easily able to populate a partially unfolded form in which the C-terminal subdomain is folded and the N-terminal subdomain is unfolded. We also created a fragment model of this intermediate and demonstrate using X-ray crystallography that its structure is identical to the corresponding residues in the full-length protein with the exception of a small network of hydrophobic interactions. In sum, we conclude that the C-terminal subdomain dominates the energetics of T4 lysozyme folding, and the A-helix serves an important role in coupling the two subdomains.
Keywords: native state hydrogen exchange, X-ray crystallography, T4 lysozyme, protein folding, intermediates
A fundamental question in biology is how a given amino acid sequence is able to fold into a unique, functional three-dimensional conformation. Large multidomain proteins are thought to fold in a hierarchical manner, with the individual domains folding independently from one another (Jaenicke 1999). These proteins demonstrate multiple equilibrium unfolding transitions (Creighton 1992; Privalov 1996), and can be thought of as being comprised of individual modules, an important concept in protein evolution (Doolittle 1995; Orengo et al. 1997).
The folding of single domains is also complicated. For most small proteins, only a single equilibrium unfolding transition is observed, and the process can be described using a two state model (U ⇆ N) (Creighton 1992). Recently, however, more sensitive techniques such as equilibrium native state hydrogen exchange (Krishna et al. 2004) and computational modeling (Fischer and Marqusee 2000; Hondoh et al. 2006) have identified regions within domains that may be capable of autonomous folding. Kinetic studies of the folding process have also shown that many small proteins fold through intermediate conformations (Jackson 1998; Baldwin and Rose 1999; Bilsel and Matthews 2006).
Together, these kinetic and equilibrium studies clearly indicate that, like multidomain proteins, certain protein domains are also comprised of smaller modules of structure. Unfortunately, the high energy and/or transient nature of such partially folded states make such intermediates extremely difficult to study. Protein dissection (formally cutting the protein into smaller pieces) (Peng and Kim 1994; Wu et al. 1994; Fontana et al. 2004) and protein engineering (selectively modifying certain regions of a protein) (Fersht 1998; Zhou et al. 2005) provide strategies to circumvent these difficulties and examine these basic modules of structure.
T4 lysozyme (T4L*) (* indicates a cysteine-free T4L) (Matsumura and Matthews 1989) is an excellent model protein for investigating such experimentally concealed modules of protein architecture. The structure and stability of this small protein (164 amino acids) have been extremely well characterized (Matthews 1995, 1996). At equilibrium, T4L* shows apparent two-state behavior (Elwell and Schellman 1975); structurally, however, it is comprised of two subdomains, an α/β N-terminal lobe and an all-α C-terminal lobe that also contains the N-terminal helix (Weaver and Matthews 1987). Several studies have suggested some independence between the subdomains: Various crystal structures show a hinge bending between the two regions of up to 50° (Zhang et al. 1995), and the relative orientation of the lobes changes by ∼8 Å upon substrate binding (Anderson et al. 1993; McHaourab et al. 1997). Protein dissection studies have demonstrated that the C-terminal lobe can fold autonomously; however, the isolated N-terminal lobe appears to be unstructured (Llinas and Marqusee 1998). Further evidence that these two lobes can be uncoupled comes from native state hydrogen exchange (NSHX) experiments, which imply the existence of a rarely populated partially unfolded form (PUF) under native conditions in which the C-terminal subdomain remains folded while the N-terminal subdomain unfolds (Llinas et al. 1999). In spite of this relative independence, the apparent two-state behavior at equilibrium and the lack of autonomous folding in the N-terminal lobe suggest that the lobes cannot be considered as true independent “domains” but, rather, subdomains that depend on one another for function and stability.
The C-terminal subdomain is discontinuous in protein sequence: In addition to the C-terminal region of the protein, it also contains the first 13 residues of the protein (helix A). In order to investigate the role of this discontinuity, a circular permutation of the protein, CP13*, was made. CP13* consists of a starting methionine followed by residues 13–164, a Ser(Gly)4Ala linker, and residues 1–12 (see Fig. 1A) such that the residues forming the subdomains are contiguous. CP13* is known to fold cooperatively into an active protein (Llinas and Marqusee 1998).
Figure 1.

Structures of CP13*, xT4L*, and LCTF. (A) The structure of LCTF (yellow) superimposes well with the corresponding structure in CP13* (blue). In none of the three structures was the linker region visible. (B) Schematic representation of the three variants: T4L*, CP13*, and LCTF. (Blue) The N-terminal subdomain; (green) the C-terminal subdomain; and (pale blue) the linker region. (C) The structure of CP13* (blue) superimposes well with the xT4L* control (purple).
Here, we explored the relative autonomy of the individual subdomains in T4 lysozyme by determining the high-resolution crystal structure of CP13*, characterizing its energy landscape using native state hydrogen exchange (NSHX), and determining the stability and high-resolution structure of a newly engineered fragment of the C-terminal subdomain. The structure of CP13* matches that of T4L*, and the hydrogen exchange studies demonstrate that, compared to the wild-type sequence, the energetics of the C-terminal subdomain in CP13* are more independent and uncoupled from the rest of the protein. The structure of the isolated C-terminal fragment is indistinguishable from the corresponding region of the intact protein. Together, these studies imply that: (1) The first 13 residues (A-helix) are important for energetic but not structural coupling between the two domains of the protein, and (2) the partially unfolded conformation of the protein detected by hydrogen exchange can be viewed simply as a native-like fragment of the protein. The accompanying paper (Cellitti et al. 2007) presents kinetic data that suggest this native-like intermediate resides on the folded side of the major transition state and results in an unfolding intermediate detected under denaturing conditions. The results presented here highlight the importance of the location of the N and C termini to the cooperativity of protein folding and open new questions as to the molecular basis for coupling between subdomains.
Results
Crystal structure of CP13*
To interpret the changes in the energetic landscapes between the wild-type T4L* and the circular permutant, high-resolution crystal structures were obtained for both CP13* and an extended version of T4L* (xT4L*) (Zhang et al. 1993) that contains the six additional C-terminal residues used as a linker for the circular permutant (1.9 Å and 1.8 Å resolution, respectively) (Fig. 1B; Table 1). Residue numbers for all variants are referred to based on their T4* sequence; that is, while the residues of helix A would be sequentially at the C terminus in CP13*, they are still referred to as residues 1–12 (Fig. 1A).
Table 1.
Crystallographic and refinement statistics for CP13*, LCTF, and xT4l*

The structures of CP13* and xT4L* are nearly superimposable. The RMSD is <1.0 Å for virtually all atoms, and there are no major changes in solvent-accessible surface area. The largest variations are found in residues having long side chains such as Arg and Lys. The Cα RMSD between the structures is 0.32 Å, and the backbone RMSD is 0.33 Å. Solvent-accessible surface area calculations for CP13* and xT4L* show almost no change for the main chain, while some side chains in both the N- and C-terminal subdomains have small changes of no more than 8.0 Å2. When compared to the T4L* structure (pdb 4lzm), the Cα RMSD is 0.44 Å for CP13* and 0.29 Å2 for xT4L.
There are some structural perturbations detected along the C-helix (connecting the two subdomains) of CP13* as compared to xT4L* (variable φ-ψ orientations) and, as expected, at the newly introduced chain break. The new C-terminal residue (Gly 12) and the newly introduced initiator Met (preceding Leu 13) are not visible in the electron density of CP13*. Neither CP13* nor xT4L* shows any ordered structure for the linker. Importantly, this linker does not seem to introduce any structural perturbations to the rest of the protein. Both structures superimpose well with the T4L* structure in the regions before and after the linker. Indeed, the N terminus of helix A is fully visible in CP13*, whereas it is normally frayed in the context of T4L*. It is interesting to note that the active-site residue, Glu 11, which is only one residue from the new C terminus in CP13*, is in the exact same geometry as in the wild-type WT* structure, consistent with the noted similarity in activity (Llinas and Marqusee 1998).
NSHX on CP13*
Native state hydrogen exchange (NSHX), hydrogen exchange as a function of denaturant (GdmCl) monitored by NMR, was carried out on CP13*. In order to interpret these data, we assigned the 1H and 15N chemical shifts of the backbone amide protons. In total, 140 amide protons were assigned using HNCS, HNCACB, 3D NOESY, and HSQC spectra. The incomplete regions of the assignments include most of helix A and residues in helix C. Not surprisingly, chemical shift changes between CP13* and WT indicate that the most significant changes are for those residues in the A- and C-helices.
Hydrogen exchange data were compiled for 88 amides distributed throughout the sequence of CP13* (Table 2). Compared to wild type, there are 29 fewer amide probes (amides whose exchange is slow enough to monitor by NMR), with the majority of these residing in the N-terminal subdomain. The most likely basis for the loss of these probes is a drop in protection such that they are now below the 2.5 kcal/mol experimental detection limit for ΔGHX. No probes were observed in the linker region, presumably for the same reason. Additionally, many N-terminal probes were lost at higher denaturant concentrations where their exchange rates were again faster than the detection limit. As expected from the similar crystal structures, residues protected in CP13* are also protected in the wild-type protein (Llinas et al. 1999). The only exception to this is Asp 2, which was likely subject to fraying in the T4L*, while in CP13* it is stabilized by the presence of the linker. Figure 2 shows the exchange free energies as a function of denaturant for representative residues, demonstrating exchange by both local fluctuations and unfolding events (Chamberlain et al. 1996), as described below.
Table 2.
Native state hydrogen exchange data for CP13*
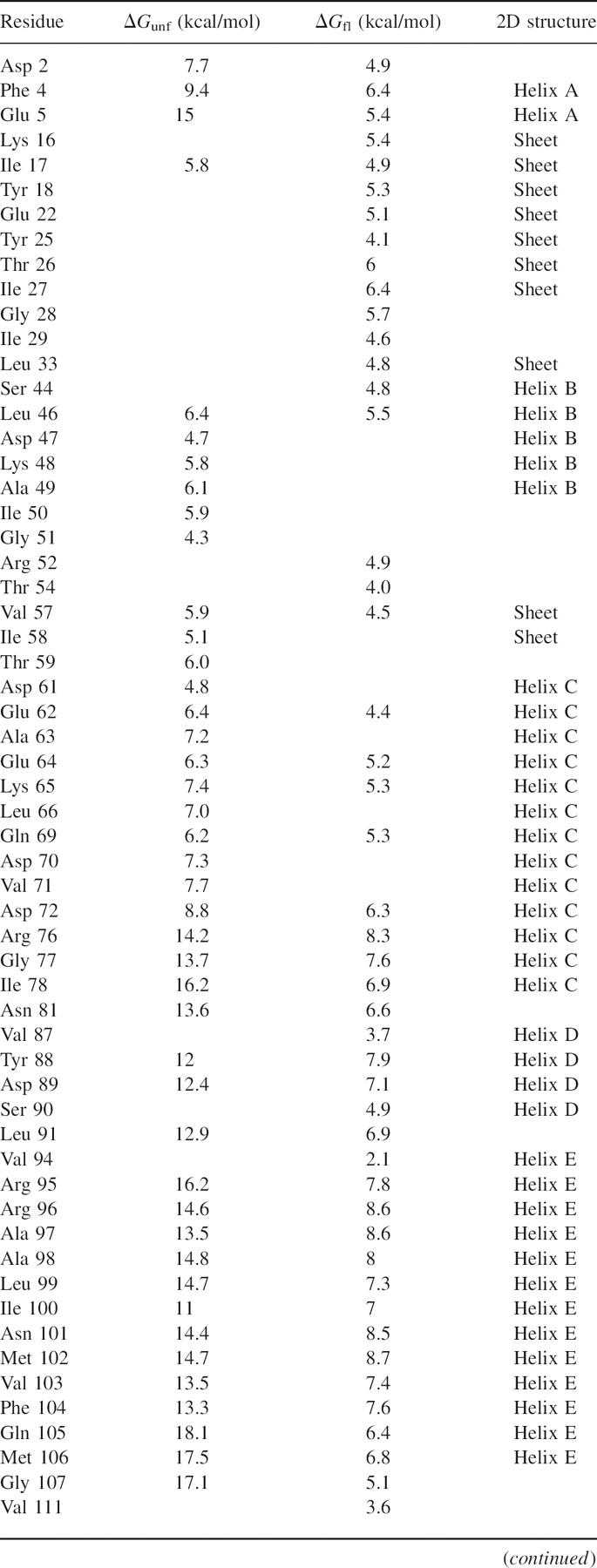

Figure 2.

Hydrogen exchange free energy as a function of GdmCl. Data for seven probes representing residues from both subdomains: N-terminal residues (•) Tyr 18 and (○) Ala 49 exhibit exchange only through subglobal unfolding, while C-terminal residues (▪) Glu 5, (□) Gly 77, (♦) Arg 96, (⋄) Arg 125, and (▴) Gly 156 exhibit exchange through local fluctuations and global unfolding.
Exchange due to local fluctuations
The majority of the probes in CP13* (68 of 88) exchange via local fluctuation mechanisms at low denaturant concentrations, similar to what was observed for the wild-type protein (Llinas et al. 1999). For the vast majority of amides demonstrating local fluctuations in both CP13* and T4L*, the local fluctuation energies (ΔGfl) are identical (Fig. 3), suggesting that the mechanism of exchange is not only independent of denaturant but also of the global stability of the protein. There is a small subset of residues for which the ΔGfls have been significantly altered: Glu 5 and Leu 121 have lower ΔGfls, while Phe 4 and Asp 2 have higher ΔGfls. (The ΔGfl for Asp 2 is, by definition, higher since it is now observable.) The linker effect may also explain the increased ΔGfl of Phe 4, although this argument cannot explain the reduced ΔGfl of Glu 5.
Figure 3.

Comparison of ΔGfl between CP13* and T4L*. The majority of residues show the same local exchange free energies in the wild-type and permuted proteins.
In the previous NSHX study on T4L* (Llinas et al. 1999), data were incomplete for 16 residues at low denaturant concentrations (sites corresponding to the globally unfolding core), preventing definitive classification as locally exchanging sites. These residues correspond to sites in the globally unfolding core of the protein. In CP13*, data were collected for these residues, conclusively demonstrating exchange by local fluctuation.
Exchange due to unfolding
Hydrogen exchange due to unfolding events (those that are denaturant dependent) was measured for 64 amide probes in CP13*. The distribution of extrapolated unfolding energies (ΔGunf) shows a broad range of energies (Fig. 4A,B). As in T4L*, it is clear that there are two clusters of amide protons in CP13*. The average stability of probes residing in the C-terminal subdomain of CP13* is 13.9 (±1.7) kcal/mol, as compared to 14.7 (±1.8) kcal/mol for the wild type, while that for the N-terminal subdomain is 6.0 (±1.0) kcal/mol in CP13* and 8.3 (±1.7) kcal/mol in wild type. For both proteins, the average C-terminal subdomain stability reflects the global stability of the protein under the conditions of the experiment (Llinas and Marqusee 1998).
Figure 4.
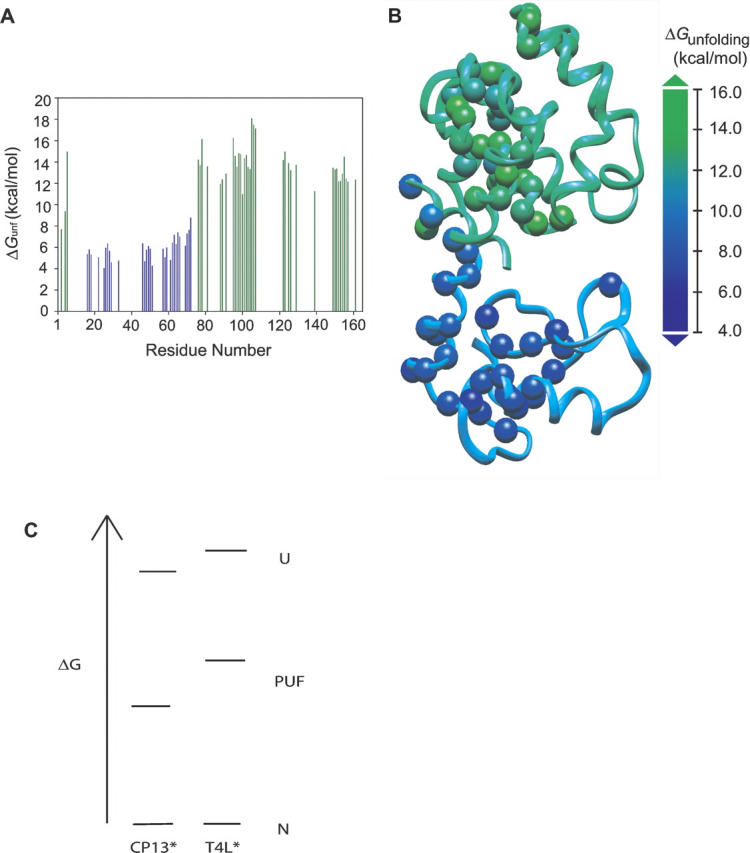
The unfolding free energies for residues in CP13*. (A) C-terminal residues (green) are dramatically more stable than N-terminal residues (blue). (B) The C-terminal subdomain is more stable than the N-terminal subdomain. Spheres represent residues that were probes in the NSHX experiment. (C) Boltzmann diagram of the native, PUF, and unfolded conformations for CP13* and WT*.
The differences in the average stabilities of each subdomain highlight the effect of permutation: The wild-type protein is more cooperative in that the energy of the N-terminal subdomain is more tightly coupled to the whole protein. The difference in energy between the PUF and the globally unfolded state is 6.4 kcal/mol (14.7–8.3) in T4L*, whereas in CP13* it is 7.9 kcal/mol (13.9–6.0). In other words, the permutation has a larger destabilizing effect on the N-terminal subdomain than the C-terminal subdomain or the whole protein (Fig. 4C).
A more stable C-terminal fragment
Prior to any NSHX studies on T4 lysozyme, a protein dissection study identified autonomous folding in a fragment of the C-terminal subdomain (residues 75–164, Ser[Gly]4Ala-1–12) (Llinas and Marqusee 1998), which serves as a good model for the intermediate detected by NSHX. We further refined the boundaries of this fragment using the program RAFT (Fischer and Marqusee 2000), which optimizes for intrafragment versus interfragment contacts. RAFT suggested a subdomain break at residue 60, rather than 76, thereby including most of the helix connecting the two lobes (helix C). This longer C-terminal fragment (LCTF), corresponding to the initiator Met, followed by residues 60–164, the Ser(Gly)4Ala linker, and residues 1–12, was generated as previously described (Llinas and Marqusee 1998). LCTF is remarkably stable (Table 3; Fig. 5), significantly more stable than the previous shorter fragment (7.6 kcal/mol vs. 2.1 kcal/mol). LCTF was shown to be monomeric in solution by analytical ultracentrifuge (data not shown) and a lack of concentration dependence to the stability measurements.
Table 3.
Equilibrium denaturation results for the three variants of T4 lysozyme

Figure 5.

Chemical denaturation of T4 lysozyme variants. (▴) LCTF data; (○) CP13*; (•) T4L*. See also Table 3.
The crystal structure of LCTF at 0.84 Å resolution was solved by molecular replacement using the corresponding region in the wild-type protein and refined to an Rfree of 10.87% (Fig. 1C; Table 1). Residues 69–162 and 1–12 were modeled into the electron density. The Ser(Gly)4Ala linker was only visible as scattered, uninterpretable density, and the Asn-Leu corresponding to the C terminus of the wild-type enzyme (163–164 in WT) was poorly ordered. Backbones of the core residues in LCTF align extremely well with the corresponding residues in the T4L* protein. The initial nine residues of helix C (the start of the fragment), however, are unwound, presumably because of missing interactions with the N-domain. The Cα RMSD between the corresponding residues of CP13* and LCTF (excluding the linker and the unwound nine N-terminal residues) is 0.38 Å, and the backbone RMSD is 0.43 Å. Except for Arg and Lys residues on the surface and residues at the interface between the N- and C-terminal subdomains, nearly all side chains are in the same conformation as in the wild-type protein. A striking exception was noted for a small set of side chains near the interface between the N- and C-terminal subdomains (Fig. 6). In the full-length protein, Phe 104 is central in a network of hydrophobic interactions between Ile 29 and Phe 67. Ile 29 is obviously not present in LCTF, which begins at residue 60. In LCTF, a dramatic rearrangement of three residues results in a small nonnative set of hydrophobic interactions centered around Phe 104. The side chain of Phe 67 swings out, while those of Leu 66 and Phe 4 swing in to form a new nonnative set of interactions that presumably replaces the stabilizing interactions provided by Ile 29. Remarkably, this alternative packing results in a very local perturbation, as the helix continues in a native-like structure at residue 68. This alternative packing arrangement is distinct from the linked conformational change that has been observed in this region associated with large-magnitude hinge-bending motions (Zhang et al. 1995).
Figure 6.

Overlay of side chains from hydrophobic core residues. (Green) LCTF; (blue) T4L. In LCTF, the absence of Ile 29 results in a striking rearrangement of the side chains of Phe 4, Phe 67, and Leu 66.
Discussion
T4L*, like most small monomeric proteins, exhibits apparent two-state equilibrium unfolding behavior. The equilibrium energy landscape determined by NSHX, however, reveals two separate modules: a stable C-terminal subdomain and a less stable N-terminal subdomain. These two subdomains are directly coupled by two structural features: (1) the A-helix (residues 1–13), which is sequentially contiguous with the N-terminal subdomain but structurally part of the C-terminal subdomain; and (2) the long C-helix that spans the hinge region between the two subdomains.
The circular permutant studied here (CP13*) removes one of these connections—the contiguity between the A-helix and the N-terminal subdomain. The X-ray structure of CP13* demonstrates that the protein's structure appears unaffected: Helix A forms the same core interactions with the C-domain without any apparent direct connections to the N-domain. Recently, the crystal structure for a similar circular permutant of T4L* was reported and also revealed a native-like structure (Sagermann et al. 2004). This circular permutant begins at residue 12 and has only a three-amino-acid linker, (Gly)2Ala. Unlike our linker, this shorter linker is ordered in the crystal structure, and the Ala interacts directly with the rest of the protein. Despite this difference in linker composition, the stabilities of both permutants are very similar to that of T4L*. Therefore, discontinuity of the C-terminal subdomain is clearly not required to maintain the fold of T4L*.
Our NSHX data reveal an energy landscape of CP13* that is qualitatively similar to that of T4L*: The protein shows two regions of different stability corresponding to the N- and C-terminal subdomains. Quantitatively, however, the loss of the direct attachment to the N-terminal subdomain in CP13* results in a loss of cooperativity between the two lobes. It is energetically easier to unfold the N-terminal subdomain in the context of CP13*, creating a partially unfolded form 7.9 kcal/mol more stable than the unfolded state, versus 6.4 kcal/mol for the same PUF in T4L*. Together, the T4L* and CP13* NSHX studies suggest a model for a high-energy intermediate consisting of a folded C-terminal subdomain (including the N-terminal helix) and an unfolded N-terminal subdomain. Indeed, the engineered fragment corresponding to the C-terminal subdomain (LCTF) forms a cooperatively folded structure with stability similar to that expected from the NSHX experiments (7.5 kcal/mol), and therefore serves as an excellent model for the equilibrium intermediate detected by NSHX. The structure encoded by this protein fragment is well within the variation displayed within this region by the many structures available for T4 lysozyme, suggesting that the C-terminal region is able to adopt a native-like fold in the absence of the rest of the protein. The crystals of the LCTF subdomain diffracted to <0.85 Å, emphasizing that this sequence alone is able to encode the T4 lysozyme C-terminal lobe. Thus, the model for the high-energy intermediate is a completely folded C-terminal lobe and a disordered N-terminal lobe.
Previous studies on a fragment modeling the C-terminal subdomain resulted in a much less stable and less well-folded protein. The original model (CTF) is identical to LCTF except it started with residue 75 instead of residue 60. While CTF was folded in isolation and was shown to have helical structure, it was unclear whether or not this fragment formed a native-like fold. Furthermore, it formed only a marginally stable structure (ΔG = 2.1 kcal/mol). Extending the fragment to begin at residue 60 in LCTF dramatically increased the stability of this domain (ΔG = 7.6 kcal/mol). This is particularly noticeable in light of the fact that helix formation does not begin until around residue 65 in LCTF. Thus, the stable folding of just 10 residues results in an increase in stability of 5.5 kcal/mol. This added stability may arise from the ability to form the small nonnative set of hydrophobic interactions involving Leu 66 and Phe 67, and the ability to form a stable somewhat isolated helix with the expected fraying at the ends. This nonnative set of interactions suggests that Ile 29 is playing an important role in the coupling between the two subdomains in the full-length protein. It is intriguing to think that this non-native set of interactions is also formed in the hidden intermediate of the full-length protein.
For most proteins, the high-energy intermediates detected at equilibrium using NSHX mirror those detected transiently during kinetic refolding experiments (Chamberlain and Marqusee 2000). T4 lysozyme is a well-known example where this is not the case. The refolding kinetics of T4L* are biphasic, with the slower phase attributed to proline isomerization-limited folding (Lu and Dahlquist 1992). The chevron plot, or denaturant dependence, of the faster of these two observable phases shows a rollover in refolding, which is interpreted as reflecting the formation of an early intermediate within the dead time of the stopped-flow mixing device (Parker and Marqusee 1999). A pulse-labeling hydrogen exchange study demonstrated protection for residues in both the N- and C-terminal subdomains within 12 msec (Lu and Dahlquist 1992), suggesting a folding intermediate clearly different from the model proposed here. Moreover, the protection factors for this early folding intermediate were marginal and inconsistent with the 5–7 kcal/mol stable intermediate as suggested by these studies. Since the NSHX experiment reveals information about accessible intermediates without regard to their position relative to the folding transition state, the intermediate modeled here may well fall on the native side of the barrier. It is unclear why, given the behavior of LCTF, residues in both the N- and C-terminal subdomains are involved in the early folding intermediate, while the C-terminal subdomain is not observed as an intermediate during the folding process.
Conclusions
Proteins with multiple subdomains are often discontinuous, with some element of structure noncontiguous in sequence (Thornton and Sibanda 1983). In the case of T4L*, that element is the A-helix. This helix is crucial both for the overall stability as well as the energetic coupling of the subdomains. Repositioning of the helix, as in CP13*, selectively destabilizes the N-terminal subdomain, and a fragment consisting of the C-terminal half of this protein folds very well in isolation. Nonetheless, the difference in stability between CP13* (11.2 kcal/mol) and LCTF (7.6 kcal/mol) suggests that coupling of the N- and C-terminal subdomains through helix C makes a large stabilizing contribution. Controlling the cooperativity between such subdomains allows proteins to fine tune their ability to sample such partially folded conformations, which, in turn, plays an important role in the function and folding of many proteins.
Materials and Methods
Plasmid construction and protein purification
The genes for T4L* and CP13* were amplified by PCR, and the NdeI/KpnI-containing product was digested and ligated into pET27K, a derivative of pET27 (Novagen) with an introduced KpnI site in the MCS. WT* and CP13* proteins were then overexpressed and purified as previously described (Llinas and Marqusee 1998) with the exception that the expression strain used here was Rosetta/pLacI (Novagen).
The extended C-terminal fragment (LCTF) was made as described previously (Llinas and Marqusee 1998), but with a 5′-primer designed to amplify the gene with a start AUG before the codon corresponding to residue 60. Overexpressed LCTF is soluble and was purified as described for T4L*.
Circular dichroism spectroscopy and denaturation melts
Far-UV CD experiments were carried out on an Aviv 62DS spectrophotometer equipped with a Peltier-temperature-controlled sample holder. Denaturation experiments were carried out by monitoring the CD signal at 222 nm as a function of guanidinium chloride (GdmCl), urea concentration, or temperature. Samples for urea melts consisted of 2 μM protein in 20 mM potassium phosphate at pH 6.0 and 100 mM NaCl. Samples for GdmCl melts were in 50 mM potassium phosphate at pH 5.25. Denaturation experiments were carried out at 25°C with samples at the varying concentrations of denaturant and given at least 24 h to equilibrate. All unfolding data were fit using a two-state model and linear extrapolation using the program SigmaPlot 8.0 for Windows (Systat Software Inc.).
Crystallization
CP13*
Crystals were grown at 4°C by the hanging-drop method. Drops consisted of 2.5 μL of protein (18.0 mg/mL) and 2.5 μL of mother liquor (0.9 M K2HPO4, 1.1 M NaH2PO4 at pH 6.9, 0.25 M NaCl, and 49.0 mM oxidized β-mercaptoethanol). One crystal grew after 9 mo. Prior to data collection, the crystal was soaked for 15 min in 20% glycerol, 0.9 M K2HPO4, 1.1 M NaH2PO4 (pH 6.8), and 0.5 M NaCl, followed by 30 min in 40% glycerol, 0.9 M K2HPO4, 1.1 M NaH2PO4 (pH 6.8), and 0.5 M NaCl, and flash-frozen in liquid nitrogen.
xT4L*
Crystals were grown at 4°C by the hanging-drop method. Drops consisted of 3.0 μL of protein (14.6 mg/mL) and 3.0 μL of mother liquor (0.9 M K2HPO4, 1.1 M NaH2PO4 at pH 6.6, 0.25 M NaCl, 49.0 mM oxidized β-mercaptoethanol). Many crystals were obtained after 1 wk. Cryoprotection was similar to that for CP13* but at pH 6.6.
LCTF
Crystals were grown by the hanging-drop method. Drops consisted of 2 μL of protein (10.4 mg/mL in 10 mM HEPES at pH 7.5, 50 mM NaCl) and 2 μL of mother liquor (100 mM NaCacodylate at pH 5.0, 200 mM NaAcetate, and 26% PEG 8000). Crystals were briefly transferred to well solution plus 14% xylitol and flash-frozen in liquid nitrogen.
Data collection and refinement
Diffraction data for xT4L* and CP13* were collected from a single crystal each on an Raxis IIC detector. The data were processed and scaled using HKL2000 (xT4L*) (Otwinowski and Minor 1997) or Elves (CP13*) (Holton and Alber 2004). The structures were solved by molecular replacement using Molrep (Vagin and Teplyakov 2000) with WT* as the search model (PDB accession code 1l63) and rebuilt by ARP/wARP (Perrakis et al. 1999). The models were refined using REFMAC5 (Murshudov et al. 1997) with manual rebuilding in Coot. All residues fall into the preferred or allowed regions of the Ramachandran plot.
The data for LCTF were collected from a single crystal at the Advanced Light Source beamline 8.3.1 (Lawrence Berkeley National Laboratory) using an ADSC Q210 detector, at a wavelength of 0.8266 Å. Diffraction was observed to at least 0.75 Å for individual crystals, but owing to hardware limitations, we collected complete data to 0.84 Å. Two separate wedges of data were recorded, one with brief (0.5 sec) exposures to measure low-resolution reflections, and the second with 40-sec exposures to obtain maximum resolution. The data were processed with the HKL2000 (Otwinowski and Minor 1997) suite, and the structure was solved by molecular replacement using the equivalent parts of the highest-resolution T4 lysozyme structure (PDB accession code 1sx7). Initial coordinates were rebuilt automatically by ARP/wARP (Perrakis et al. 1999) at 1.3 Å resolution, and refined manually using REFMAC (Murshudov et al. 1997) and O (Jones et al. 1991) with isotropic temperature factors and no hydrogens. The high-resolution cutoff was slowly increased as anisotropic B-factors, riding hydrogens, and disordered waters were added. Starting at 1.0 Å resolution, refinement was performed with SHELXL (Sheldrick and Schneider 1997), and occupancies of alternate conformers were added as parameters. The final round used full-matrix least-squares minimization for the full resolution range. The initial methionine was modeled as oxidized in two different stereoisomers. All residues fall into the preferred or allowed regions of the Ramachandran plot. All structure figures and superpositions were generated with PyMOL (DeLano Scientific; http://pymol.org).
Hydrogen–deuterium exchange
The exchange experiment was performed and analyzed as described previously (Llinas et al. 1999). The data were collected over a 3-mo period.
Accession numbers
The coordinates and structure factors for the CP13*, xT4L*, and LCTF models have been deposited in the Protein Data Bank with accession numbers 2o4w, 2o79, and 2o7a, respectively.
Note added in proof
Recently, Kato and coworkers engineered a similar model for the isolated C-terminal subdomain by replacing residues 17–58 of the N-terminal subdomain with five glycines. NMR analyses suggest that this engineered model of the folding intermediate also forms a native-like structure (Kato et al. 2007).
Acknowledgments
We thank the entire Marqusee laboratory staff for helpful discussions and comments; and James Holton, George Meigs, and Jane Tanamachi for support at ALS 8.3.1. We are especially indebted to James Holton for assistance with data processing, and Mark Sales, Ho-Leung Ng, and Harry Powell for crystallography advice. BL8.3.1 was funded by the National Science Foundation, the University of California, and Henry Wheeler. This work was supported by NIH grants GM50945 to S.M. and GM48958 to T.A.
Footnotes
Reprint requests to: Susan Marqusee, Department of Molecular and Cell Biology and QB3 Institute–Berkeley, University of California, Berkeley, Berkeley, California 94720-3206, USA; e-mail: marqusee@berkeley.edu; fax: (510) 643-9290.
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.062628607.
References
- Anderson D.E., Lu, J., McIntosh, L.P., and Dahlquist, F.W. 1993. The folding, stability and dynamics of T4 lysozyme: A perspective using nuclear magnetic resonance. In NMR of Proteins (eds. G.M. Clore and A.M. Gronenborn), pp. 258–304. McMillan Press, London.
- Baldwin R.L. and Rose, G.D. 1999. Is protein folding hierarchic? II. Folding intermediates and transition states. Trends Biochem. Sci. 24: 77–83. [DOI] [PubMed] [Google Scholar]
- Bilsel O. and Matthews, C.R. 2006. Molecular dimensions and their distributions in early folding intermediates. Curr. Opin. Struct. Biol. 16: 86–93. [DOI] [PubMed] [Google Scholar]
- Cellitti J., Bernstein, R., and Marqusee, S. 2007. Exploring subdomain cooperativity in T4 lysozyme II: Uncovering the C-terminal subdomain as a hidden intermediate in the kinetic folding pathway. Protein Sci. (this issue). [DOI] [PMC free article] [PubMed]
- Chamberlain A.K. and Marqusee, S. 2000. Comparison of equilibrium and kinetic approaches for determining protein folding mechanisms. Adv. Protein Chem. 53: 283–328. [DOI] [PubMed] [Google Scholar]
- Chamberlain A.K., Handel, T.M., and Marqusee, S. 1996. Detection of rare partially folded molecules in equilibrium with the native conformation of RNaseH. Nat. Struct. Biol. 3: 782–787. [DOI] [PubMed] [Google Scholar]
- Creighton T.E. 1992. Protein folding. W.H. Freeman, New York.
- Doolittle R.F. 1995. The multiplicity of domains in proteins. Annu. Rev. Biochem. 64: 287–314. [DOI] [PubMed] [Google Scholar]
- Elwell M. and Schellman, J. 1975. Phage T4 lysozyme. Physical properties and reversible unfolding. Biochim. Biophys. Acta 386: 309–323. [DOI] [PubMed] [Google Scholar]
- Fersht A.R. 1998. Structure and mechanism in protein science: A guide to enzyme catalysis and protein folding. W.H. Freeman, New York.
- Fischer K.F. and Marqusee, S. 2000. A rapid test for identification of autonomous folding units in proteins. J. Mol. Biol. 302: 701–712. [DOI] [PubMed] [Google Scholar]
- Fontana A., de Laureto, P.P., Spolaore, B., Frare, E., Picotti, P., and Zambonin, M. 2004. Probing protein structure by limited proteolysis. Acta Biochim. Pol. 51: 299–321. [PubMed] [Google Scholar]
- Holton J. and Alber, T. 2004. Automated protein crystal structure determination using ELVES. Proc. Natl. Acad. Sci. 101: 1537–1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hondoh T., Kato, A., Yokoyama, S., and Kuroda, Y. 2006. Computer-aided NMR assay for detecting natively folded structural domains. Protein Sci. 15: 871–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson S.E. 1998. How do small single-domain proteins fold? Fold. Des. 3: R81–R91. [DOI] [PubMed] [Google Scholar]
- Jaenicke R. 1999. Stability and folding of domain proteins. Prog. Biophys. Mol. Biol. 71: 155–241. [DOI] [PubMed] [Google Scholar]
- Jones T.A., Zou, J.Y., Cowan, S.W., and Kjeldgaard, M. 1991. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A 47: 110–119. [DOI] [PubMed] [Google Scholar]
- Kato H., Feng, H., and Bai, Y. 2007. The folding pathway of T4 lysozyme: The high-resolution structure and folding of a hidden intermediate. J. Mol. Biol. 365: 870–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishna M.M., Hoang, L., Lin, Y., and Englander, S.W. 2004. Hydrogen exchange methods to study protein folding. Methods 34: 51–64. [DOI] [PubMed] [Google Scholar]
- Llinas M. and Marqusee, S. 1998. Subdomain interactions as a determinant in the folding and stability of T4 lysozyme. Protein Sci. 7: 96–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llinas M., Gillespie, B., Dahlquist, F.W., and Marqusee, S. 1999. The energetics of T4 lysozyme reveal a hierarchy of conformations. Nat. Struct. Biol. 6: 1072–1078. [DOI] [PubMed] [Google Scholar]
- Lu J. and Dahlquist, F.W. 1992. Detection and characterization of an early folding intermediate of T4 lysozyme using pulsed hydrogen exchange and two-dimensional NMR. Biochemistry 31: 4749–4756. [DOI] [PubMed] [Google Scholar]
- Matsumura M. and Matthews, B.W. 1989. Control of enzyme activity by an engineered disulfide bond. Science 243: 792–794. [DOI] [PubMed] [Google Scholar]
- Matthews B.W. 1995. Studies on protein stability with T4 lysozyme. Adv. Protein Chem. 46: 249–278. [DOI] [PubMed] [Google Scholar]
- Matthews B.W. 1996. Structural and genetic analysis of the folding and function of T4 lysozyme. FASEB J. 10: 35–41. [DOI] [PubMed] [Google Scholar]
- McHaourab H.S., Oh, K.J., Fang, C.J., and Hubbell, W.L. 1997. Conformation of T4 lysozyme in solution. Hinge-bending motion and the substrate-induced conformational transition studied by site-directed spin labeling. Biochemistry 36: 307–316. [DOI] [PubMed] [Google Scholar]
- Murshudov G.N., Vagin, A.A., and Dodson, E.J. 1997. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53: 240–255. [DOI] [PubMed] [Google Scholar]
- Orengo C.A., Michie, A.D., Jones, S., Jones, D.T., Swindells, M.B., and Thornton, J.M. 1997. CATH—A hierarchic classification of protein domain structures. Structure 5: 1093–1108. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z. and Minor, W. 1997. Processing X-ray diffraction data collected in oscillation mode. In Methods in enzymology: Macromolecular crystallography, Part A (ed. R. Carter Jr), pp. 307–326. Academic Press, New York. [DOI] [PubMed]
- Parker M.J. and Marqusee, S. 1999. The cooperativity of burst phase reactions explored. J. Mol. Biol. 293: 1195–1210. [DOI] [PubMed] [Google Scholar]
- Peng Z.Y. and Kim, P.S. 1994. A protein dissection study of a molten globule. Biochemistry 33: 2136–2141. [DOI] [PubMed] [Google Scholar]
- Perrakis A., Morris, R.M., and Lamzin, V.S. 1999. Automated protein model building combined with iterative structure refinement. Nat. Struct. Biol. 6: 458–463. [DOI] [PubMed] [Google Scholar]
- Privalov P.L. 1996. Intermediate states in protein folding. J. Mol. Biol. 258: 707–725. [DOI] [PubMed] [Google Scholar]
- Sagermann M., Baase, W.A., Mooers, B.H., Gay, L., and Matthews, B.W. 2004. Relocation or duplication of the helix A sequence of T4 lysozyme causes only modest changes in structure but can increase or decrease the rate of folding. Biochemistry 43: 1296–1301. [DOI] [PubMed] [Google Scholar]
- Sheldrick G.M. and Schneider, T.R. 1997. SHELXL: High-resolution refinement. Methods Enzymol. 277: 319–343. [PubMed] [Google Scholar]
- Thornton J.M. and Sibanda, B.L. 1983. Amino and carboxy-terminal regions in globular proteins. J. Mol. Biol. 167: 443–460. [DOI] [PubMed] [Google Scholar]
- Vagin A. and Teplyakov, A. 2000. An approach to multi-copy search in molecular replacement. Acta Crystallogr. D Biol. Crystallogr. 56: 1622–1624. [DOI] [PubMed] [Google Scholar]
- Weaver L.H. and Matthews, B.W. 1987. Structure of bacteriophage T4 lysozyme refined at 1.7 Å resolution. J. Mol. Biol. 193: 189–199. [DOI] [PubMed] [Google Scholar]
- Wu L.C., Grandori, R., and Carey, J. 1994. Autonomous subdomains in protein folding. Protein Sci. 3: 369–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T., Berkelsen, E., Benvegnu, D., and Alter, T. 1993. Circular permutation of T4 lysozyme. Biochemistry 32: 12311–12318. [DOI] [PubMed] [Google Scholar]
- Zhang X.J., Wozniak, J.A., and Matthews, B.W. 1995. Protein flexibility and adaptability seen in 25 crystal forms of T4 lysozyme. J. Mol. Biol. 250: 527–552. [DOI] [PubMed] [Google Scholar]
- Zhou Z., Huang, Y., and Bai, Y. 2005. An on-pathway hidden intermediate and the early rate-limiting transition state of Rd-apocytochrome b562 characterized by protein engineering. J. Mol. Biol. 352: 757–764. [DOI] [PubMed] [Google Scholar]