

Abstract
Streptococcus pneumoniae, a ubiquitous gram-positive pathogen with an alarming, steadily evolving resistance to frontline antimicrobials, poses a severe global health threat both in the community and in the clinic. The recent discovery that diphosphomevalonate (DPM), an essential intermediate in the isoprenoid biosynthetic pathway, potently and allosterically inhibits S. pneumoniae mevalonate kinase (SpMK) without affecting the human isozyme established a new target and lead compound for antimicrobial design. Here we present the crystal structure of the first S. pneumoniae mevalonate kinase, at a resolution of 2.5 Å and in complex with DPM·Mg2+ in the active-site cleft. Structural comparison of SpMK with other members of the GHMP kinase family reveals that DPM functions as a partial bisubstrate analog (mevalonate linked to the pyrophosphoryl moiety of ATP) in that it elicits a ternary-complexlike form of the enzyme, except for localized disordering in a region that would otherwise interact with the missing portion of the nucleotide. Features of the SpMK-binding pockets are discussed in the context of established mechanistic findings and inherited human diseases linked to MK deficiency.
Keywords: mevalonate kinase, GHMP kinase, allosteric inhibitor, antimicrobial target, diphosphomevalonate, Streptococcus pnuemoniae, crystal structure, mevalonate deficiency
Streptococcus pneumoniae is the most common pathogen associated with community-acquired respiratory tract infections in the United States and a leading causative agent in fatal cases of meningitis or bacteremia that afflict young (<2 yr old) and elderly (>65 yr old) populations worldwide (Bridy-Pappas et al. 2005). The isoprenoid biosynthetic pathway is an increasingly attractive focus for pharmaceutical intervention in the treatment of pneumococcal disease (Rohdich et al. 2004, 2005). The end product of the pathway, isopentenyl-diphosphate, is the 5-carbon biosynthetic building block of isoprenoids, a large class of compounds that is essential to the survival of virtually all organisms, including S. pneumoniae. Disruption of the individual genes encoding HMG-CoA reductase and HMG-CoA synthase, which produce mevalonate, prevents growth of S. pneumoniae in the rat lung and results in a growth requirement for levels of mevalonate that far exceed those found in serum (Popjak et al. 1979; Wilding et al. 2000). The recent discovery that diphosphomevalonate, an intermediate in the mevalonate pathway, potently, allosterically down-regulates the activity of the S. pneumoniae mevalonate kinase without inhibiting the human enzyme provides an opportunity to develop a new class of antimicrobials that are capable of killing the bacterium without detriment to the host.
This report presents the crystal structure of mevalonate kinase from S. pneumoniae with the allosteric inhbitor, DPM, bound to the enzyme. The structure reveals a SpMK dimer with one molecule of DPM bound at each of the two active-site clefts. Structural comparisons of SpMK with other members of the GHMP kinase superfamily suggest that DPM binds as expected for a partial bisubstrate analog—mevalonate linked to the β,γ-pyrophosphoryl group of ATP. The comparisons also suggest structural divergence between eubacterial and eukaryotic MKs that might ultimately facilitate structure-based antibacterial drug design. Critical active-site residues that contact the mevalonate and pyrophosphate substructures of the inhibitor are discussed with regard to their functional relevance in human disease and their contributions to catalysis.
Results and Discussion
The overall fold
Examination of the crystal packing in the SpMK lattice reveals a homodimeric assembly that is consistent with the native molecular weight determined by analytical gel filtration (Andreassi et al. 2004). Similar to other dimeric GHMP kinases, tight packing of weakly conserved, interface residues in SpMK produces a unique geometric assembly from the prototypical GHMP monomer fold (Zhou et al. 2000; Wada et al. 2003). The dimer resembles a flattened cube with overall dimensions of 60 Å × 53 Å × 31 Å, and possesses a compact 2858 Å2 dimer interface that sits on a central, crystallographic twofold axis (Fig. 1A). The monomeric unit consists of two closely associated N- and C-terminal α + β domains, each comprising approximately half of the primary sequence. A structure-based sequence alignment of SpMK and other members of the GHMP kinase family is shown in Figure 1B, which highlights the motifs, secondary structural elements, and residues that are discussed throughout the text (Fig. 1B). The N-terminal domain is composed of two 310-helices (α1,3) and an elongated, antiparallel, six-stranded β-sheet (β1–6) that partially encloses an arclike four-helix bundle (α2,4,5,6) (Fig. 1A). The C-terminal domain is organized into a four-stranded antiparallel β-sheet (β7–10) flanked by two α helices (α10,11) and the helical bundle. Three adjacent helical elements (α7–9) coalesce to form a sharp apex that overhangs a β-platform composed of the last four strands of the N-terminal β-sheet (β1,2,5,6).
Figure 1.
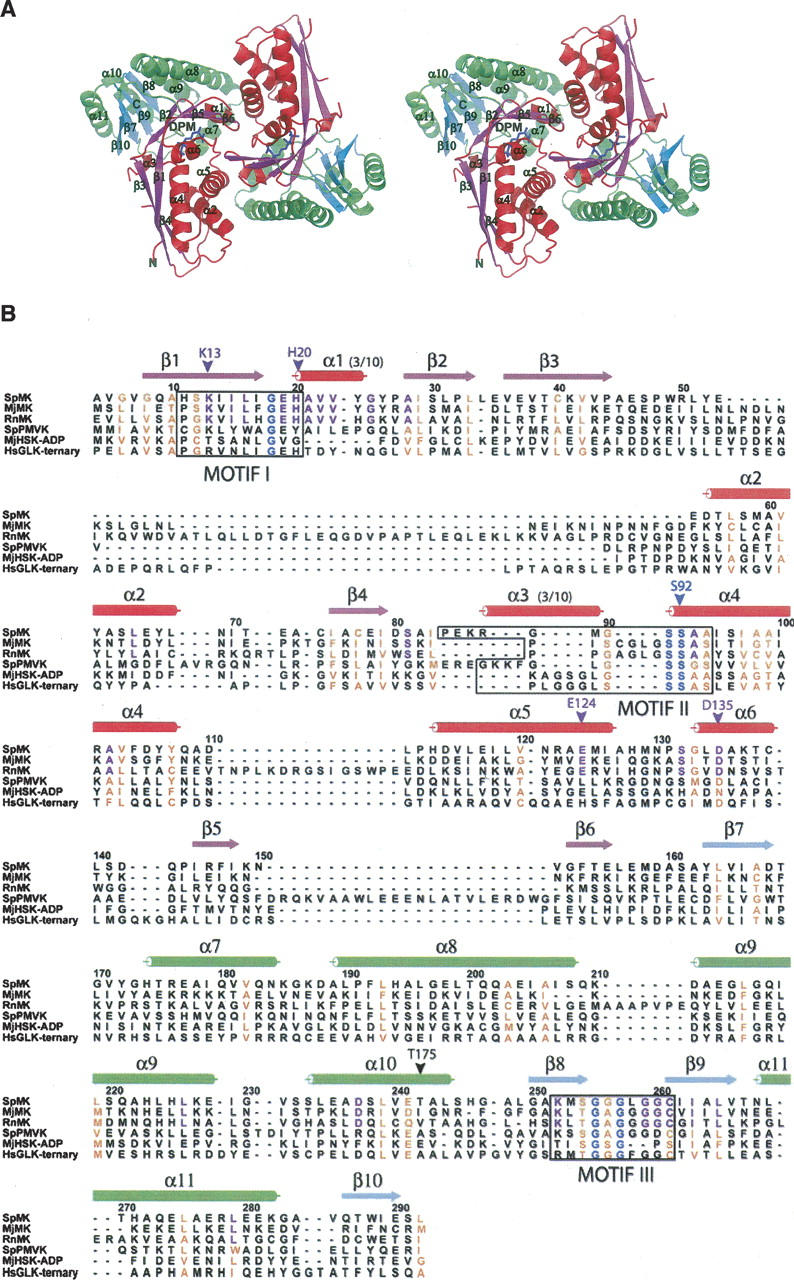
The overall fold and DPM-binding pocket of SpMK. (A) Stereo representation of the SpMK dimer. Color scheme: β-sheets and flanking α-helices are colored red and magenta or green and cyan in the N- and C-terminal domains, respectively. DPM is colored blue. (B) Structure-based sequence alignment of SpMK with MK orthologs from Rattus norvegicus (RnMK) and Methanococcus jannaschii (MjMK), and GHMP kinases including the Streptococcus pneumoniae PMK apoprotein (SpPMVK), ADP-bound M. jannaschii HSK (MjHSK-ADP), and the Homo sapiens GLK ternary complex (HsGLK-ternary). Sequences are numbered according to the SpMK primary sequence. Conserved GHMP and MK motifs are boxed. Invariant residues within the GHMP superfamily and mevalonate kinase family are colored blue and purple, respectively. Conserved positions are colored orange. Panels (A) and (B) were produced using PyMOL (DeLano 2002) and ALSCRIPT (Barton 1993), respectively.
Monomers are arranged head to tail about a twofold axis, generating two C/N-terminal contact surfaces and an elongated central pore that separates the interdomain clefts that comprise the DPM-binding sites (Fig. 1A, detailed below). The central pore is 20 Å deep and narrows from a 209-Å2 opening on the protein exterior to 42 Å2 on its innermost surface. The N-terminal dimerization interface is formed by interactions between a loop (α5/6) and two helices (α1 and α5); the C-terminal domain interface is defined by the interactions of two loops (α1/β2 and β5/β6) situated at the apex of the domain. A helical hairpin that generates a slightly acute angle between α7 and α8 within the C-terminal domain enables additional, interhelical interactions with α2 and α5 near the surface of the interface.
The DPM-binding site
Given that DPM binds tightly to SpMK (Kd = 500 nM) with a stoichiometry of 1 DPM per dimer, that its binding causes changes in the fluorescence of the protein (which is not observed upon binding of ATP and/or mevalonate), and that DPM is a noncompetitive inhibitor versus either ATP or mevalonate, we were surprised to find inhibitor density (Fig. 2A) exclusively at the active site of each monomer (Andreassi et al. 2004). The only crystals obtained from more than 3000 crystallization conditions (including attempts to drive DPM from the active site of SpMK using high [tens of millimolar] concentrations of mevalonate and AMPPNP) were those in which DPM was bound at the active site of the enzyme. The structural findings predict a stoichiometry of two DPM per dimer and that DPM will compete with mevalonate and ATP individually. Thus, the structural implications are at odds with the thermodynamic and kinetic studies of DPM binding. It is not surprising that DPM binds at the active site, and very weak binding would have been missed in the solution-phase studies. It is plausible that the crystal packing interactions have “trapped” a form of the DPM·SpMK complex that rarely presents in solution. This interpretation, which reconciles the crystallographic and solution-phase data sets, is consistent with the atypical ATP-binding loop conformation seen in the DPM-bound complex (see below).
Figure 2.
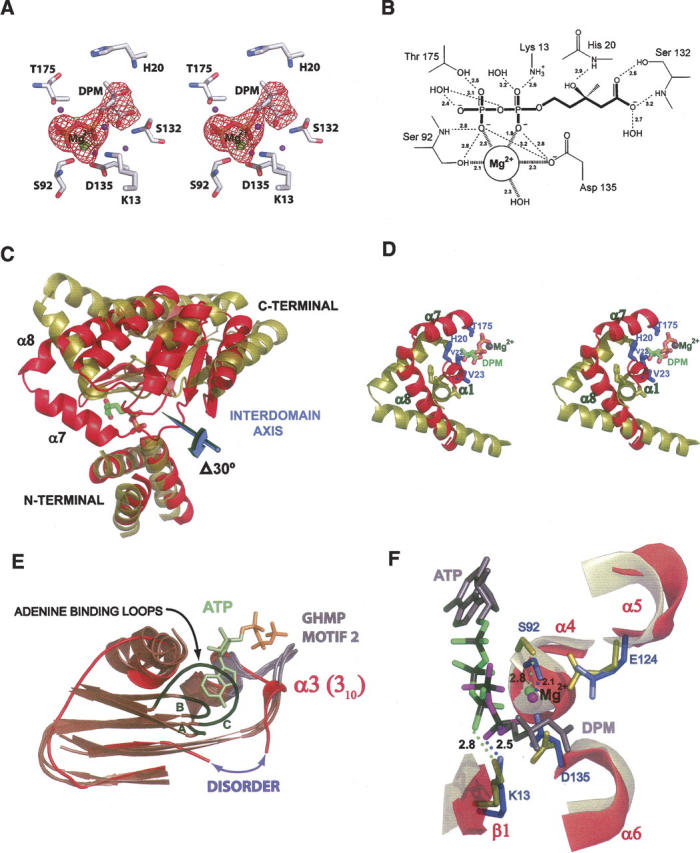
Distinguishing structural features of DPM-bound SpMK. (A) Stereo stick representation of the refined SpMK active site. A σA-weighted Fo − Fc electron density map calculated in CNS for the final model lacking DPM and Mg2+ is contoured at 4.0 σ and colored in red. Water molecules are represented as purple spheres. (B) Schematic diagram depicting electrostatic interactions of DPM as dotted lines and their interatomic distances (Å). (C) Ribbon representation of the N-terminal superposition of SpMK (red) and RnMK (deep olive). DPM is colored green (carbon), red (oxygen), blue (nitrogen), and yellow (phosphorus). The magnitude of the domain rotation and the position of the interdomain axis required to define the conformational difference between RnMK (red) and SpMK (gold) is indicated. (D) A stereo ribbon-and-stick representation of localized, conformational differences in the mevalonate-binding pocket using the superposed coordinates shown in panel (C). DPM ligands and the Mg2+ cofactor are colored blue and dark gray, respectively. (E) A ribbon-and-stick representation of the superposition of the N-terminal ATP-binding pocket in SpMK (red) with other GHMP superfamily members (brown) as follows: the MjMK apoprotein, ATP-bound RnMK, and HsGLK-ternary. The conserved ATP-binding loops are shown as gray and black ribbons, respectively. The ATP-binding loops from MjMK, RnMK, and HsGLK are labeled A, B, and C, respectively. The disordered stretch in SpMK corresponds to a gap in the polypeptide model and is designated with a purple double arrow. The representative ATP molecule (adenosine and triphophate colored olive and orange, respectively) is the RnMK substrate. (F) A ribbon-and-stick representation of the ATP-binding pocket using the superposed coordinates shown in panel (C). The Mg2+ cofactors and polyphosphate oxygens of ATP and DPM are colored green and magenta, respectively. The remainder of the substrates are colored gray (backbone) or black (phosphorus). Conserved SpMK residues that contact DPM, ATP, or Mg2+ are labeled and colored blue. Conserved residues that are located at corresponding positions in the RnMK structure are colored deep olive. Atomic distances are in Å. Panel (B) was produced with ChemDraw (Rubenstein 1987). Panels (A, C–F) were produced using PyMOL (DeLano 2002).
On the issue of where else DPM might bind to allosterically regulate turnover of SpMK, it is interesting to note that the thermodynamic and initial-rate experiments reveal that the binding and catalytic functions of each of the two active sites of the dimer appear to be affected identically by the binding of a single molecule of DPM, suggesting an allosteric-binding pocket that is symmetrically disposed with respect to the active sites (Andreassi et al. 2004). One such site is the central cavity of the enzyme, which appears sufficiently large to accommodate a single molecule of DPM. Further structural studies are required to resolve this important issue.
DPM is situated near the dimer interface in a hydrophobic, interdomain cleft that widens from 7 Å to 16 Å as it approaches the solvent exterior. The mevalonate portion of the inhibitor is cradled in a depression formed by a β-platform (β5/6), the N termini of the α4/6-helices, and a short 310-helix (α1) (Fig. 1A). The pyrophosphoryl group of DPM is positioned at the amino terminus of the α4-helix and lies within its positive dipole. The innermost region of the cleft, which is covered along 40% of its length by residues located in the loop between β7 and α7 and the first two turns of the α7-helix, forms a lid that partially shields DPM from solvent and contributes additional stabilizing contacts to both the mevalonate and pyrophosphoryl moieties. The DPM-binding site also contains a Mg2+ ion that is coordinated by two nonbridging oxygen atoms of the DPM pyrophosphoryl group, a water molecule, and side-chain oxygens of Asp135 and Ser92 (Fig. 2A,B). The amide proton of Ser92, an invariant residue among GHMP kinases, forms a hydrogen bond with the β-phosphoryl oxygen of DPM. The importance of proper active-site coordination of Mg2+ is underscored by the 105-fold decrease in catalytic efficiency that results from substitution of a hydrogen atom for the Ser92 hydroxyl group that is coordinated to Mg2+ in the human MVK (Cho et al. 2001).
The conformation of DPM-bound SpMK
The “closed” juxtaposition of the N- and C-terminal domains in the DPM-bound SpMK structure resembles that seen in ternary complexes of other GHMP kinase family members and differs markedly from the apo- and nucleotide-bound binary-MK structures. Thus, active-site closure appears to be linked either to mevalonate binding or to binding of the second substrate into the active site. Comparison of the Sp and rat mevalonate kinase structures reveals that the empty mevalonate-binding pocket in the rat structure is open and poised to accept the ligand (Fu et al. 2002). Movement from the open to closed form, as assessed by conformational modeling (described below), involves a hingelike movement of the N- and C-terminal domains (Fig. 2C) that positions the α1 and α7 helices of SpMK over the binding cleft, thereby shielding the substrate from solvent and placing the conserved mevalonate-binding residues (His20 [Motif I], Val22 and Val23 [α1], and Thr175 and Ile179 [α7]) into contact with DPM (Fig. 2D). Notably, similar conformational changes are seen when comparing liganded and nonliganded forms of other members of the GHMP kinase superfamily (Krishna et al. 2001). To gain further insight into the structural changes that produce the domain shift, SpMK was aligned individually with other GHMP kinase structures using an algorithm that allows the SpMK domains to move to achieve optimal superposition (FATCAT) (Ye and Godzik 2003). The native and FATCAT-generated SpMK structures were then compared with DynDom (Hayward and Berendsen 1998) to quantitatively assess the structural change associated with this transition. The closure is described well by a 30° rigid-body rotation that pivots about an interdomain hinge centered between Leu156 and Glu157 (Fig. 2C).
The ATP-binding pocket
The N termini of GHMP kinase superfamily members harbor an ATP-binding consensus sequence known as Motif II (-PXGXGLGSSAA-), which forms a large, ordered oxyanion hole that acts as an electropositive nest for the triphosphate tail of ATP (Figs. 1B, 2E). A survey of the superfamily structures reveals that the Motif II loop conformations of the binary (ATP-bound) and unliganded complexes are virtually identical, suggesting that the loop does not rearrange upon nucleotide binding (Fig. 2E). In the SpMK structure, the XGX subsection of Motif II (i.e., E85KR) exhibits side-chain disorder and was modeled as alanine. Furthermore, the structure is completely disordered over a small stretch of residues (SAIP84) that lie immediately upstream of Motif II in a conserved β-strand that interacts with the adenine ring of ATP in other family members. It is interesting to note that the disordered serine residue (Ser81) is invariant in the MK family and interacts directly with adenine N6. Thus, it appears that the absence of the nucleotidyl moiety results in a disordering of the residues that would otherwise interact with it. Remarkably, the E85KRGMGS segment of Motif II, which presents a geometrically well-defined set of peptidic hydrogens to the nucleotide triphosphate in other GHMP kinases, is organized in SpMK into a 310-helix that has retracted to the roof of the ATP-binding cleft (Fig. 2E). The final three residues of the SpMK ATP-binding loop (S92AA) faithfully assume the standard GHMP kinase Motif II fold, and position the catalytically essential Ser92 hydroxyl in contact with the Mg2+ cofactor in a fashion identical to that observed in the rat MK structure.
Situated in the N-terminal domains of SpMK·DPM and RnMK·ATP is a conserved core of residues that interact with the ATP tripolyphosphate chain and its associated Mg2+ ion. The Cα-backbones of these structures superpose well (RMSD = 0.68) and reveal that the Mg2+-polyphosphate chains of ATP and DPM overlap and exhibit nearly identical interactions with the conserved N-terminal nucleotide-binding core (Fig. 2F). Notably, the α-phosphoryl group of DPM seems to interact with a subsection of the binding pocket (in particular, the highly conserved Lys13, which otherwise interacts with the γ-phosphoryl group of ATP; Andreassi and Leyh 2004; Potter et al. 1997). Thus, the pyrophosphoryl group of DPM elicits conserved interactions normally associated with the β,γ portion of the tripolyphosphate chain of ATP, which, along with the ternary-complexlike configuration of the DPM structure, supports that DPM is acting as a bisubstrate analog.
Human disease relevance
A histidine residue located at an invariant position of the Motif I consensus in the MK (His20) and GLK (His44) members of the GHMP kinase superfamily is mutated in several heritable human diseases whose origins trace to substantial losses in binding affinity for the nonnucleotidic substrate of either MK (hyperimmunoglobulinaemia D and periodic fever syndrome; Houten et al. 1999) or GLK (galactosemia). The structure of the ATP·GLK·galactose complex has been determined, and the interaction of the conserved histidine (His44) with galactose has been described (Thoden et al. 2005); however, the structure of mevalonate bound to MK is not yet available. In the current DPM-bound MK structure, DPM is positioned precisely as expected for a γ-phosphoryl group acceptor in a GHMP kinase; thus, it appears to provide an excellent model for understanding the interactions between the acceptor and the disease-linked His20 in SpMK. His20, a unique Ramachandran outlier, presents in a loop between β1 and the α1 310-helix in a conformation that both recognizes the C3-hydroxyl of DPM (through a hydrogen bond to the His20 amide proton) and interacts directly with the C3-methyl group that is situated in a hydrophobic pocket defined primarily by the His20 side chain (Fig. 2A,D). Interestingly, His44 (GLK) and His20 (MK) contribute virtually identical atomic contacts to the similar substructures of otherwise dissimilar substrates (i.e., the C6-hydroxyl and pyranose-ether fragment of galactose versus the C3-hydroxyl and C3-methyl of DPM).
Residue Thr175 in SpMK is conserved across kingdoms in HSK and MK homologs and exists as a naturally occurring mutation in human MK that results in mevalonate deficiency (Hinson et al. 1999). Thr175 is located at the N-terminal edge of the α7-helix, or “mobile helix” (Andreassi and Leyh 2004), whose movement places this residue in contact with both substrates upon formation of the ternary complex (Krishna et al. 2001). In SpMK, the Thr175 hydroxyl hydrogen interacts directly with the nonbridging, β-phosphate oxygen of DPM and less directly with the bridging oxygen, and the Cγ-methyl of Thr243 is in van der Waals contact with the isopentanyl moiety of mevalonate (Fig. 2A,D). Similarly, the Thr183 equivalent in the HSK ternary complex contributes a hydrogen bond to the β-phosphate of the ATP analog, AMPPNP, and the Cγ-methyl interacts loosely with homoserine. Initial-rate studies demonstrate a clear role for the conserved threonine in the steady-state substrate recognition and turnover of MK—the Thr243Ala mutation in the human isozyme increases Km for both mevalonate and ATP (40- and fivefold, respectively) and causes a twofold decrease in kcat (Cho et al. 2001).
Materials and Methods
S. pneumoniae mevalonate kinase (SpMK) and diphosphomevalonate (DPM) were prepared as previously described (Pilloff et al. 2003; Andreassi et al. 2004). Crystals of SpMK in complex with DPM were prepared at ambient temperature by mixing 2 μL SpMK at 6.0 mg/mL (0.4 mM) in 50 mM HEPES (pH 7.5), 50 mM KCl, 1 mM DTT, 10 mm AMPPNP, 12 mM MgCl2, and 0.5 mM DPM with an equal volume of 40% (v/v) polyethylene glycol 400, 200 mM sodium formate as a sitting drop under silicon oil. Attempts to determine the crystal structure by the method of molecular replacement were not successful, and thus the method of single-wavelength anomalous diffraction (SAD) was used. A single SpMK crystal was equilibrated with several grains of trimethyllead acetate (TMLA) over a 2-d period at ambient temperature. The crystal was cooled to 125 K in liquid nitrogen, and X-ray diffraction data were measured to 2.8 Å resolution using an in-house Rigaku R-Axis IV++ image plate detector and RU-H3R rotating copper anode X-ray generator equipped with Osmic Blue optics and operating at 50 kV and 100 mA. The data were reduced with HKL (Otwinowski and Minor 1997). An additional SpMK crystal (underivatized) was used to measure native data to 2.5 Å resolution (Table 1).
Table 1.
Data collection and refinement statistics
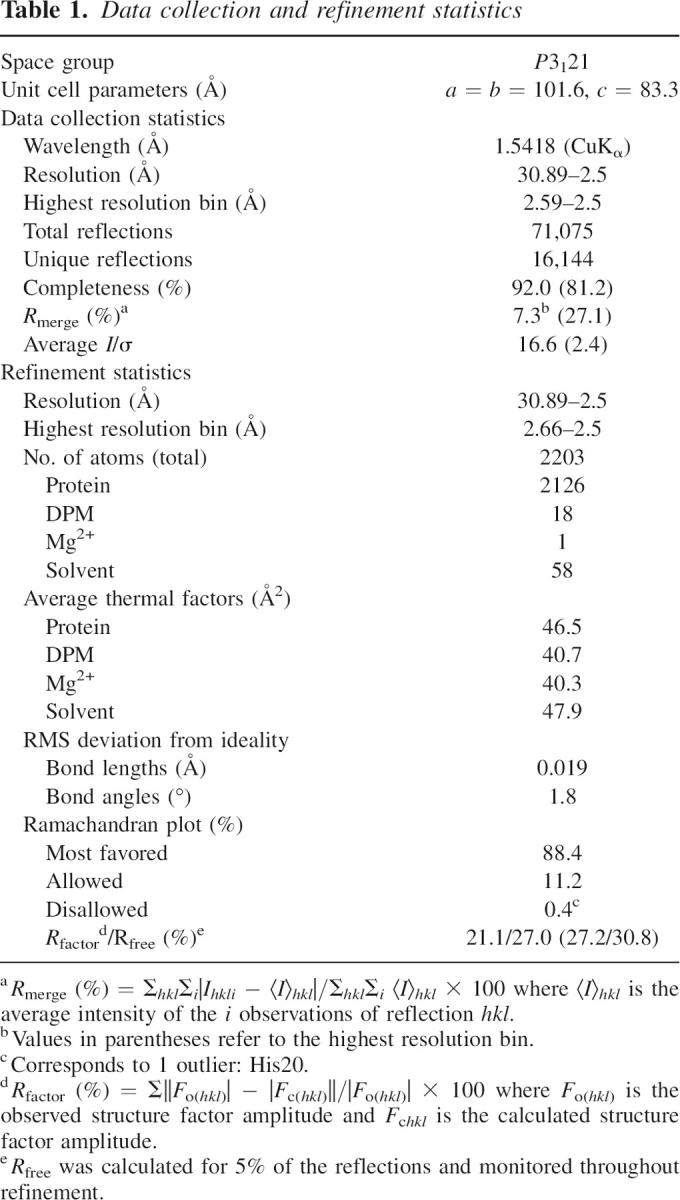
The SOLVE/RESOLVE program package was used to locate four TMLA sites and yielded a figure of merit of 0.30 for an initial 2.8 Å SAD phase determination (Terwilliger 2003). The solvent flattened map was improved by free atom insertion with Arp/wArp, and a crude initial model was autobuilt into this map with MAID (Levitt 2001; Morris et al. 2003). Manual refitting with O was followed by a single cycle of simulated annealing refinement in CNS (Jones et al. 1991; Brunger et al. 1998). The resulting model was then subjected to rigid body refinement against the higher resolution native data set, against which all further cycles of restrained parameter refinement and refitting were performed. The atomic model of SPMK corresponded to a single subunit of the dimeric enzyme present in the asymmetric unit (residues 4–80 and 85–292 of the 292-residue polypeptide), one molecule of DPM, one Mg2+ ion, and 58 water molecules. Although ATP was included in the crystallization experiment, no electron density corresponding to ATP was apparent. The atomic model of SpMK in complex with DPM and Mg2+ was refined to a final crystallographic R-factor of 21.1% (R-free = 27.0%) at 2.5 Å resolution (Table 1).
Data deposition
The atomic coordinates for mevalonate kinase in complex with disphosphomevalonate have been deposited with PDB accession code 2OI2.
Acknowledgments
This work was supported in part by National Institutes of Health grants AI068989 and GM054469 to T.S.L. S.L.R. was supported by the National Institutes of Health grant AI42154. M.W.V. was supported by grant AI33696 to John S. Blanchard.
Footnotes
Reprint requests to: Thomas S. Leyh, Albert Einstein College of Medicine, 305 Forchheimer Building, Department of Biochemistry, Bronx, NY 10461-1602, USA; e-mail: leyh@aecom.yu.edu; fax: (718) 430-8565.
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.072755707.
References
- Andreassi J.L. and Leyh, T.S. 2004. Molecular functions of conserved aspects of the GHMP kinase family. Biochemistry 43: 14594–14601. [DOI] [PubMed] [Google Scholar]
- Andreassi J.L., Dabovic, K., and Leyh, T.S. 2004. Streptococcus pneumoniae isoprenoid biosynthesis is downregulated by diphosphomevalonate: An antimicrobial target. Biochemistry 43: 16461–16466. [DOI] [PubMed] [Google Scholar]
- Barton G.J. 1993. ALSCRIPT: A tool to format multiple sequence alignments. Protein Eng. 6: 37–40. [DOI] [PubMed] [Google Scholar]
- Bridy-Pappas A.E., Margolis, M.B., Center, K.J., and Isaacman, D.J. 2005. Streptococcus pneumoniae: Description of the pathogen, disease epidemiology, treatment, and prevention. Pharmacotherapy 25: 1193–1212. [DOI] [PubMed] [Google Scholar]
- Brunger A.T., Adams, P.D., Clore, G.M., DeLano, W.L., Gros, P., Grosse-Kunstleve, R.W., Jiang, J.S., Kuszewski, J., Nilges, M., Pannu, N.S., et al. 1998. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 54: 905–921. [DOI] [PubMed] [Google Scholar]
- Cho Y.K., Rios, S.E., Kim, J.J., and Miziorko, H.M. 2001. Investigation of invariant serine/threonine residues in mevalonate kinase. Tests of the functional significance of a proposed substrate binding motif and a site implicated in human inherited disease. J. Biol. Chem. 276: 12573–12578. [DOI] [PubMed] [Google Scholar]
- DeLano W.L. 2002. The PyMOL molecular graphics system. Delano Scientific, San Carlos, CA.
- Fu Z., Wang, M., Potter, D., Miziorko, H.M., and Kim, J.J. 2002. The structure of a binary complex between a mammalian mevalonate kinase and ATP: Insights into the reaction mechanism and human inherited disease. J. Biol. Chem. 277: 18134–18142. [DOI] [PubMed] [Google Scholar]
- Hayward S. and Berendsen, H.J. 1998. Systematic analysis of domain motions in proteins from conformational change: New results on citrate synthase and T4 lysozyme. Proteins 30: 144–154. [PubMed] [Google Scholar]
- Hinson D.D., Ross, R.M., Krisans, S., Shaw, J.L., Kozich, V., Rolland, M.O., Divry, P., Mancini, J., Hoffmann, G.F., and Gibson, K.M. 1999. Identification of a mutation cluster in mevalonate kinase deficiency, including a new mutation in a patient of Mennonite ancestry. Am. J. Hum. Genet. 65: 327–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houten S.M., Kuis, W., Duran, M., de Koning, T.J., van Royen-Kerkhof, A., Romeijn, G.J., Frenkel, J., Dorland, L., de Barse, M.M., Huijbers, W.A., et al. 1999. Mutations in MVK, encoding mevalonate kinase, cause hyperimmunoglobulinaemia D and periodic fever syndrome. Nat. Genet. 22: 175–177. [DOI] [PubMed] [Google Scholar]
- Jones T.A., Zou, J.Y., Cowan, S.W., and Kjeldgaard, M. 1991. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A 47: 110–119. [DOI] [PubMed] [Google Scholar]
- Krishna S.S., Zhou, T., Daugherty, M., Osterman, A., and Zhang, H. 2001. Structural basis for the catalysis and substrate specificity of homoserine kinase. Biochemistry 40: 10810–10818. [DOI] [PubMed] [Google Scholar]
- Levitt D.G. 2001. A new software routine that automates the fitting of protein X-ray crystallographic electron-density maps. Acta Crystallogr. D Biol. Crystallogr. 57: 1013–1019. [DOI] [PubMed] [Google Scholar]
- Morris R.J., Perrakis, A., and Lamzin, V.S. 2003. ARP/wARP and automatic interpretation of protein electron density maps. Methods Enzymol. 374: 229–244. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z. and Minor, W. 1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276: 307–326. [DOI] [PubMed] [Google Scholar]
- Pilloff D., Dabovic, K., Romanowski, M.J., Bonanno, J.B., Doherty, M., Burley, S.K., and Leyh, T.S. 2003. The kinetic mechanism of phosphomevalonate kinase. J. Biol. Chem. 278: 4510–4515. [DOI] [PubMed] [Google Scholar]
- Popjak G., Boehm, G., Parker, T.S., Edmond, J., Edwards, P.A., and Fogelman, A.M. 1979. Determination of mevalonate in blood plasma in man and rat. Mevalonate “tolerance” tests in man. J. Lipid Res. 20: 716–728. [PubMed] [Google Scholar]
- Potter D., Wojnar, J.M., Narasimhan, C., and Miziorko, H.M. 1997. Identification and functional characterization of an active-site lysine in mevalonate kinase. J. Biol. Chem. 272: 5741–5746. [DOI] [PubMed] [Google Scholar]
- Rohdich F., Bacher, A., and Eisenreich, W. 2004. Perspectives in anti-infective drug design. The late steps in the biosynthesis of the universal terpenoid precursors, isopentenyl diphosphate and dimethylallyl diphosphate. Bioorg. Chem. 32: 292–308. [DOI] [PubMed] [Google Scholar]
- Rohdich F., Bacher, A., and Eisenreich, W. 2005. Isoprenoid biosynthetic pathways as anti-infective drug targets. Biochem. Soc. Trans. 33: 785–791. [DOI] [PubMed] [Google Scholar]
- Rubenstein S. 1987. ChemDraw. Cambridge Scientific Computing Inc, Cambridge, MA.
- Terwilliger T.C. 2003. SOLVE and RESOLVE: Automated structure solution and density modification. Methods Enzymol. 374: 22–37. [DOI] [PubMed] [Google Scholar]
- Thoden J.B., Timson, D.J., Reece, R.J., and Holden, H.M. 2005. Molecular structure of human galactokinase: Implications for type II galactosemia. J. Biol. Chem. 280: 9662–9670. [DOI] [PubMed] [Google Scholar]
- Wada T., Kuzuyama, T., Satoh, S., Kuramitsu, S., Yokoyama, S., Unzai, S., Tame, J.R., and Park, S.Y. 2003. Crystal structure of 4-(cytidine 5′-diphospho)-2-C-methyl-D-erythritol kinase, an enzyme in the non-mevalonate pathway of isoprenoid synthesis. J. Biol. Chem. 278: 30022–30027. [DOI] [PubMed] [Google Scholar]
- Wilding E.I., Brown, J.R., Bryant, A.P., Chalker, A.F., Holmes, D.J., Ingraham, K.A., Iordanescu, S., So, C.Y., Rosenberg, M., and Gwynn, M.N. 2000. Identification, evolution, and essentiality of the mevalonate pathway for isopentenyl diphosphate biosynthesis in gram-positive cocci. J. Bacteriol. 182: 4319–4327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Y. and Godzik, A. 2003. Flexible structure alignment by chaining aligned fragment pairs allowing twists. Bioinformatics Suppl 2 19: II246–II255. [DOI] [PubMed] [Google Scholar]
- Zhou T., Daugherty, M., Grishin, N.V., Osterman, A.L., and Zhang, H. 2000. Structure and mechanism of homoserine kinase: Prototype for the GHMP kinase superfamily. Structure 8: 1247–1257. [DOI] [PubMed] [Google Scholar]