

Abstract
Desosamine is a 3-(dimethylamino)-3,4,6-trideoxyhexose found in certain macrolide antibiotics such as the commonly prescribed erythromycin. Six enzymes are required for its biosynthesis in Streptomyces venezuelae. The focus of this article is DesV, which catalyzes the PLP-dependent replacement of a 3-keto group with an amino functionality in the fifth step of the pathway. For this study the three-dimensional structures of both the internal aldimine and the ketimine intermediate with glutamate were determined to 2.05 Å resolution. DesV is a homodimer with each subunit containing 12 α-helical regions and 12 β-strands that together form three layers of sheet. The structure of the internal aldimine demonstrates that the PLP-cofactor is held in place by residues contributed from both subunits (Asp 164 and Gln 167 from Subunit I and Tyr 221 and Asn 235 from Subunit II). When the ketimine intermediate is present in the active site, the loop defined by Gln 225 to Ser 228 from Subunit II closes down upon the active site. The structure of DesV is similar to another sugar-modifying enzyme referred to as PseC. This enzyme is involved in the biosynthesis of pseudaminic acid, which is a sialic acid-like nonulosonate found in the flagellin of Helicobacter pylori. In the case of PseC, however, the amino group is transferred to the C-4 rather than the C-3 position. Details concerning the structural analysis of DesV and a comparison of its molecular architecture to that of PseC are presented.
Keywords: PLP-dependent enzyme, desosamine, transaminase, molecular structure, deoxysugars, macrolide antibiotics, aspartate aminotransferase, X-ray crystallography
Antibiotics have dramatically changed the course of human existence, as is evidenced by the increased life expectancy following the introduction of penicillin to the general population in the 1940s. The occurrence of antibiotic resistant strains of bacteria, however, has become an increasingly pernicious problem. Indeed, the National Institute of Allergy and Infectious Diseases fact sheet states that tuberculosis, gonorrhea, malaria, and even childhood ear infections are becoming more difficult to treat due to bacterial resistance. The Centers for Disease Control and Prevention (CDC) estimate that approximately two million hospital patients will acquire an infection each year and of those, nearly 90,000 will die. It is, indeed, a cause for alarm when, according to the CDC, more than 70% of bacteria that cause nosocomial infections are resistant to at least one of the commonly prescribed antibiotics. Furthermore, strains of Staphylococcus aureus are emerging that are resistant to the “last resort” drugs, methicillin and vancomycin.
The focus of this article is DesV, a PLP-dependent transaminase that is involved in the biosynthesis of the unusual sugar desosamine, which is found in macrolide antibiotics such as erythromycin and related compounds. Macrolides constitute a well-established class of antimicrobial agents demonstrating excellent clinical activity against Gram-positive bacteria. Interestingly, the efficacy of these drugs is often reduced or eliminated when their respective sugar moieties are modified or absent (Weymouth-Wilson 1997). It is believed that sugar groups, in addition to playing roles in solubility and permeability, may function as recognition elements that are essential to the mechanism of action of the respective drugs. Because of their biological role, there is growing research interest in developing therapeutics with altered sugar appendages for the specific purpose of producing new pharmacologically useful compounds.
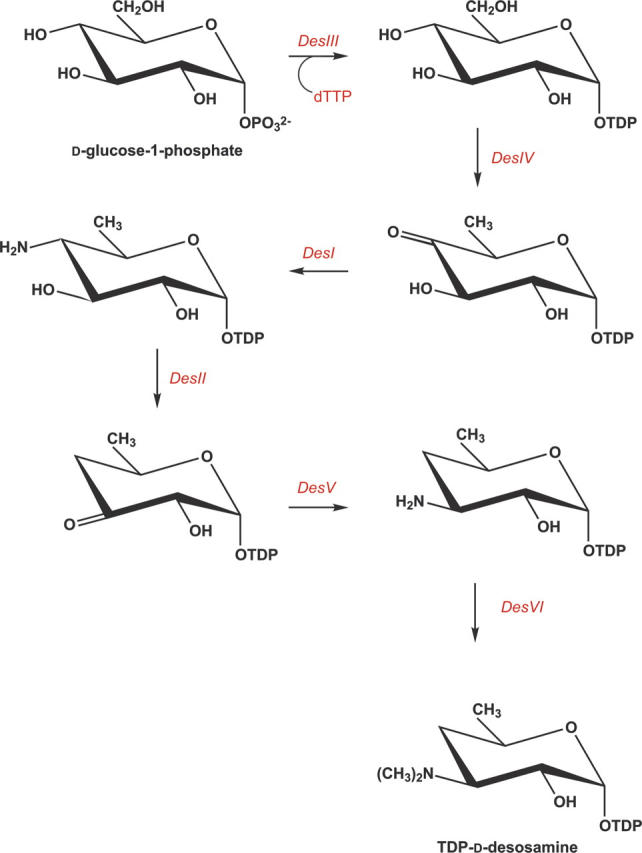
From past studies it is known that six enzymes are required for the biosynthesis of desosamine in Streptomyces venezuelae. The roles of these enzymes are indicated in Scheme 1 (Szu et al. 2005). DesV, which catalyzes the fifth step in the pathway, belongs to the aspartate aminotransferase family. Enzymes in this class are predominantly homodimers (Eliot and Kirsch 2004) and typically contain two key features: a PLP cofactor attached to a lysine via a Schiff base linkage and an aspartate residue that promotes protonation of the PLP pyridine ring nitrogen, thereby enhancing the cofactor's electron sink properties (Paiardini et al. 2004).
Scheme 1.
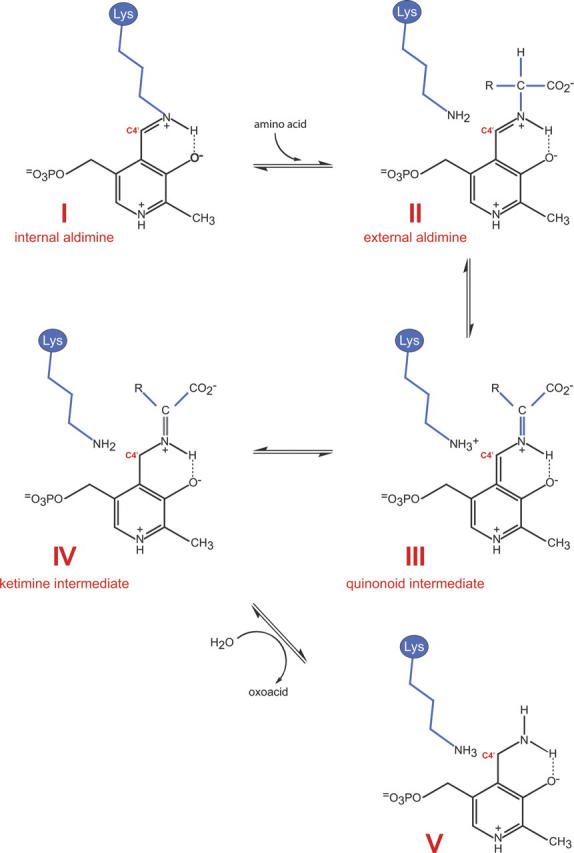
The catalytic reactions for many PLP-dependent aminotransferases have been well characterized (Eliot and Kirsch 2004). Briefly, catalysis is known to proceed via a ping pong mechanism in which the enzyme oscillates between PLP- and PMP-bound forms. In the absence of substrate, the PLP cofactor forms a Schiff base with the ɛ-amino group of a conserved active site lysine, labeled I, in Scheme 2. This is referred to as the internal aldimine. The α-amino group of an incoming amino acid (generally glutamate) attacks the C4′ atom of PLP, displacing the lysine and yielding the external aldimine (II). The conjugated ring system of PLP serves as an electron sink, which weakens the bonds about the α-carbon of the amino acid. Aminotransferases specifically orient the α-carbon/hydrogen bond perpendicular to the PLP π-system, thereby favoring this bond for cleavage. The resulting resonance stabilized carbanion is referred to as a quininoid intermediate (III in Scheme 2). Proton extraction is mediated by the active site lysine, which subsequently serves as the catalytic acid to protonate C4′, yielding a ketimine intermediate (IV). Subsequent attack by an activated water molecule at the α-carbon of the intermediate leads to the formation of PMP and an oxoacid (V) (Hayashi 1995). At this stage, PMP is ready to react with ketones such as keto sugars, and the process is reversed, resulting in regeneration of the PLP-lysine internal aldimine and the transaminated product.
Scheme 2.
Here we describe the three-dimensional structure of DesV from Streptomyces venezuelae. For this analysis complexes of both the internal aldimine form of the enzyme and the trapped ketimine intermediate were determined. The molecular architecture of DesV is similar to that observed for PseC, an aminotransferase involved in the biosynthesis of pseudaminic acid (Schoenhofen et al. 2006a). Details concerning the structure of DesV and a comparison with PseC are presented.
Results
The crystals of DesV employed in this investigation contained two dimers in the asymmetric unit. For the sake of simplicity, the following discussion refers only to the first dimer in the X-ray coordinate data set. The initial structure determined for DesV was of the form containing the internal aldimine. A ribbon representation of the dimer is presented in Figure 1A. As can be seen, the subunit:subunit interface is extensive, with a total buried surface of ∼5400 Å2, and is formed primarily by Lys 16 to Leu 40, which are located in the first α-helix of the subunit, Asn 65 to Met 68, which reside at the N terminus of the third α-helix, Tyr 93 to Leu 98, which are positioned at the N terminus of the fourth α-helix, and Tyr 190 to Asp 200, Tyr 221 to Gln 242, and Leu 315 to Gly 330, which are mostly located in random coil regions. The active sites are separated by ∼20 Å.
Figure 1.
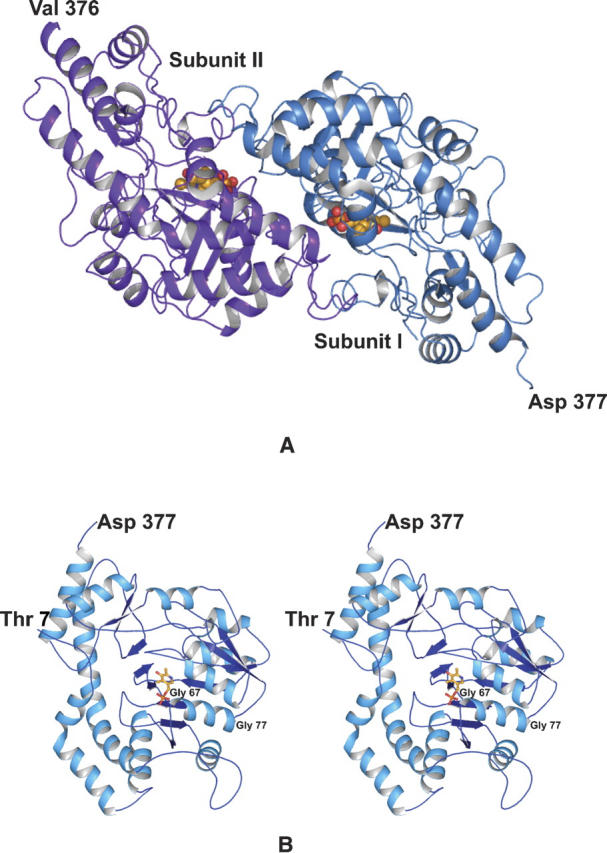
Overall structure of DesV. The crystals of DesV utilized in this investigation contained two dimers in the asymmetric unit. One of these is shown as a ribbon representation in (A). The local twofold rotational axis relating one subunit to the other is perpendicular to the plane of the paper. A stereo-representation of Subunit I is displayed in (B). The PLP cofactor is depicted in a stick representation. The first several residues at the N terminus are not visible in the electron density map.
A ribbon representation for Subunit I of the dimer is presented in Figure 1B. Each subunit contains 12 major α-helical regions and 12 β-strands, which form three layers of sheet (a two-stranded antiparallel, a three-stranded antiparallel, and a seven-stranded mixed sheet). Note that the helix, delineated by Gly 67 to Gly 77, is situated such that the positive end of its helix dipole moment projects toward the phosphoryl group of the PLP. In each subunit, Tyr 318 adopts the cis-conformation with its side chain pointing in toward the active site region. The polypeptide chains for the two subunits of the dimer are nearly identical such that their α-carbons superimpose with a root-mean-square deviation of 0.26 Å.
A close-up view of the electron density corresponding to the internal aldimine is presented in Figure 2A. As can be seen the density is well ordered for the cofactor. Shown in Figure 2B are those residues located within ∼3.6 Å of the internal aldimine. Two residues from Subunit II, namely Tyr 221 and Asn 235, participate in hydrogen bonding interactions with the phosphoryl group of the cofactor. Additional interactions that anchor the phosphoryl group into the active site occur via the backbone amide group of Met 68, Oγ of Ser 188, an ordered water molecule, and an ethylene glycol molecule. N1 and O3′ of the pyridoxal ring lie within hydrogen bonding distance to Asp 164 and Gln 167, respectively. Asp 164 is the conserved carboxylate among the aspartate aminotransferases that presumably promotes the protonation of N1, thereby enhancing the electron sink properties of the PLP pyridine ring.
Figure 2.
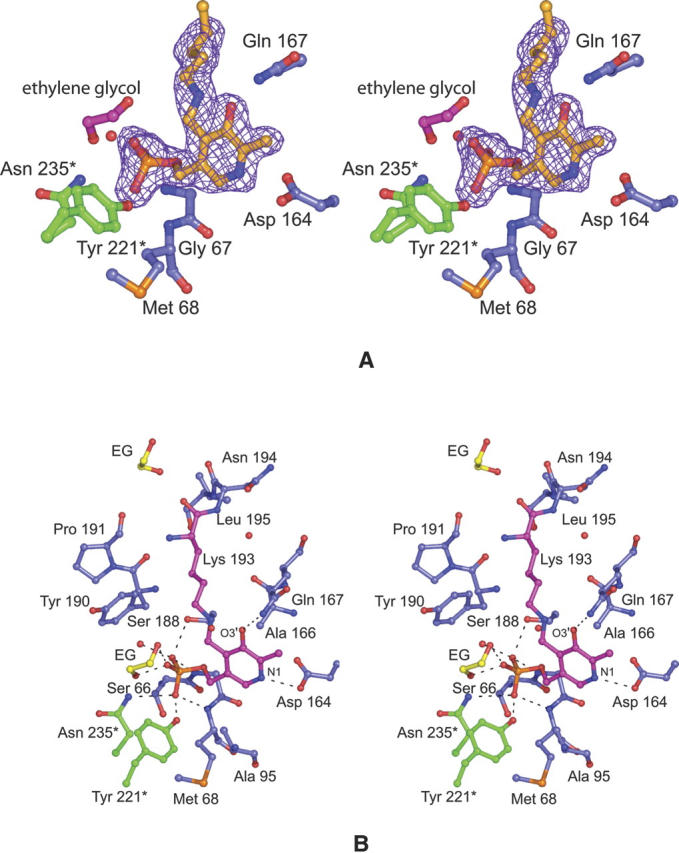
The structure of the DesV internal aldimine intermediate. Electron density corresponding to the internal aldimine is presented in (A). The map was calculated with coefficients of the form (Fo − Fc), where Fo was the native structure factor amplitude and Fc was the calculated structure factor amplitude with the atoms corresponding to the lysine side chain and the cofactor excluded from the calculation. The contouring level was 2σ. A close-up view of the hydrogen-bonding pattern surrounding the internal aldimine is given in (B). Potential hydrogen bonds between the PLP (in pink bonds) and the protein are indicated by the dashed lines. Those residues belonging to Subunit I are highlighted in blue bonds whereas those contributed by Subunit II are shown in green bonds. The ethylene glycol molecule observed binding in the active site pocket is abbreviated as “EG.” Ordered water molecules are depicted as red spheres.
For the second structure determined in this study, crystals of DesV were grown in the presence of 40 mM glutamate, which resulted in the formation of the ketimine intermediate, as evidenced by the electron density presented in Figure 3A. The glutamate nitrogen of the intermediate lies ∼1 Å out of the plane of the pyridoxal ring. A close-up view of the hydrogen bonding pattern around the ketimine intermediate is presented in Figure 3B. Again, as in the case of the internal aldimine structure, both subunits in the dimer contribute to the formation of the binding pocket. Tyr 318 from Subunit I and Tyr 227 from Subunit II anchor the α-carboxylate group of the intermediate to the protein whereas Arg 237 from Subunit II forms a salt bridge with its side-chain carboxylate. The backbone amide group of Gly 67 and the side-chain hydroxyl of Ser 188 from Subunit I participate in hydrogen bonding interactions with one of the phosphoryl oxygens of the cofactor. Tyr 221 and Asn 235 from Subunit II provide additional hydrogen bonding interactions with the cofactor phosphate. Both the side chains of Gln 167 and Lys 193 lie within 3 Å of O3′ of the cofactor. The distance between O3′ of the cofactor and the glutamate nitrogen is 2.6 Å. Three ordered water molecules lie within 3.6 Å of the ketimine intermediate.
Figure 3.
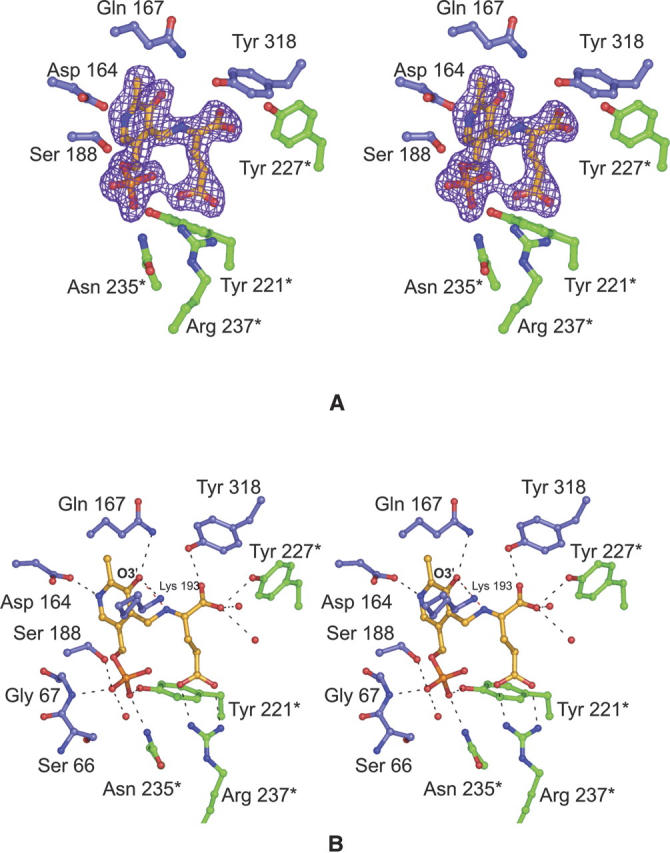
The structure of the DesV ketimine intermediate. Electron density corresponding to the ketimine intermediate is presented in (A). The map was calculated as described in the legend to Figure 2. A close-up view of the hydrogen-bonding pattern surrounding the ketimine intermediate is presented in (B). The color-coding is the same as described for Figure 2.
The structures of DesV with the internal aldimine versus the ketimine intermediate are very similar. The α-carbon traces for the dimers superimpose with a root-mean-square deviation of 0.39 Å. As highlighted in Figure 4, several residues move closer to the active site when the ketimine intermediate is formed. These include Tyr 318 from Subunit I and Tyr 227 from Subunit II, both of which participate in hydrogen bonding interactions with the α-carboxylate group of the ketimine intermediate. The glutamate side chain of the ketimine intermediate occupies the position originally filled by the ethylene glycol molecule in the internal aldimine structure.
Figure 4.

Superposition of the regions surrounding the internal aldimine versus the ketimine intermediate. The model corresponding to the internal aldimine is displayed in gold whereas that for the ketimine intermediate is depicted in green. Residues contributed by the second subunit are denoted by an asterisk. Note the movement of the loop beginning with Gln 225* toward the active site when the ketimine intermediate is bound.
Discussion
Deoxyaminosugars, such as desosamine, are synthesized by a variety of microorganisms and are found, for example, in lipopolysaccharides, in extracellular polysaccharides, or in antibiotics. The pathways leading to the production of such sugars invariably contain PLP-dependent enzymes. To date, three X-ray crystallographic structures of sugar-modifying aminotransferases have been determined: (1) ArnB from Salmonella typhimurium, a 4-amino-4-deoxy-L-arabinose lipopolysaccharide-modifying enzyme (Noland et al. 2002); (2) PglE, an enzyme responsible for amino transfer in the production of UDP-2,4-diacetamido-2,4,6,trideoxy-α-D-glucopyranose in Campylobacter jejuni (Schoenhofen et al. 2006b); and (3) PseC, an aminotransferase from Helicobacter pylori that is involved in the biosynthesis of pseudaminic acid (Schoenhofen et al. 2006a). The reactions catalyzed by these enzymes are presented in Scheme 3. All of these enzymes catalyze an amination at C-4 of the sugar, in marked contrast to DesV, which acts at C-3 of the hexose. As might be expected, the overall three-dimensional folds for these enzymes are similar. For example, the α-carbons for the DesV and PseC subunits superimpose with a root-mean-square deviation of ∼1.3 Å for 340 structurally equivalent α-carbons. A superposition of these two structures is given in Figure 5A. The two proteins only differ significantly in one loop (Gln 225–Ser 228) where in DesV there is a four-residue deletion. This loop provides part of the active site for a neighboring molecule in the dimer as discussed further below.
Scheme 3.
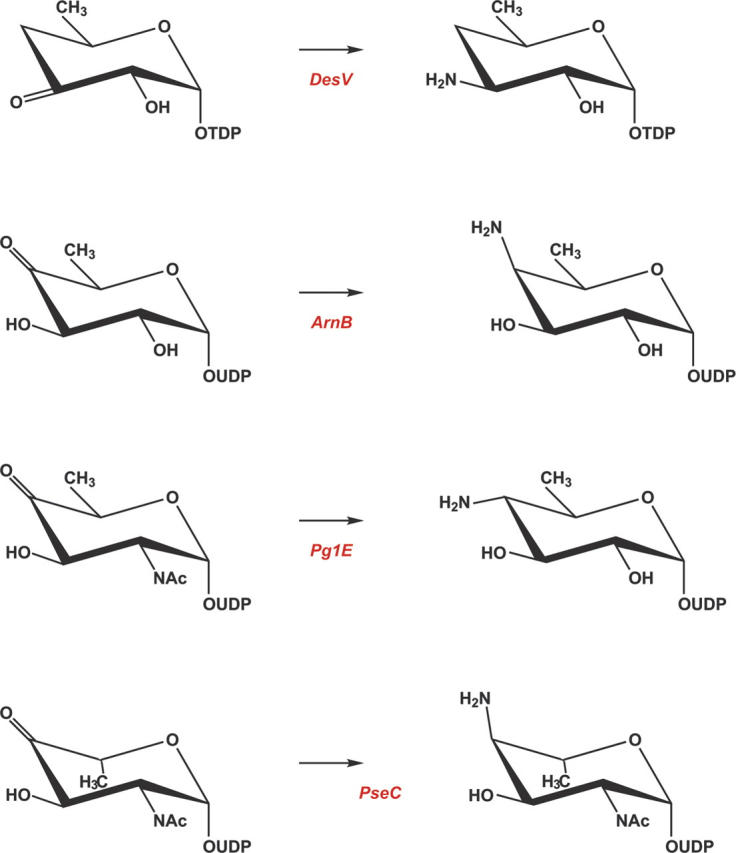
Figure 5.
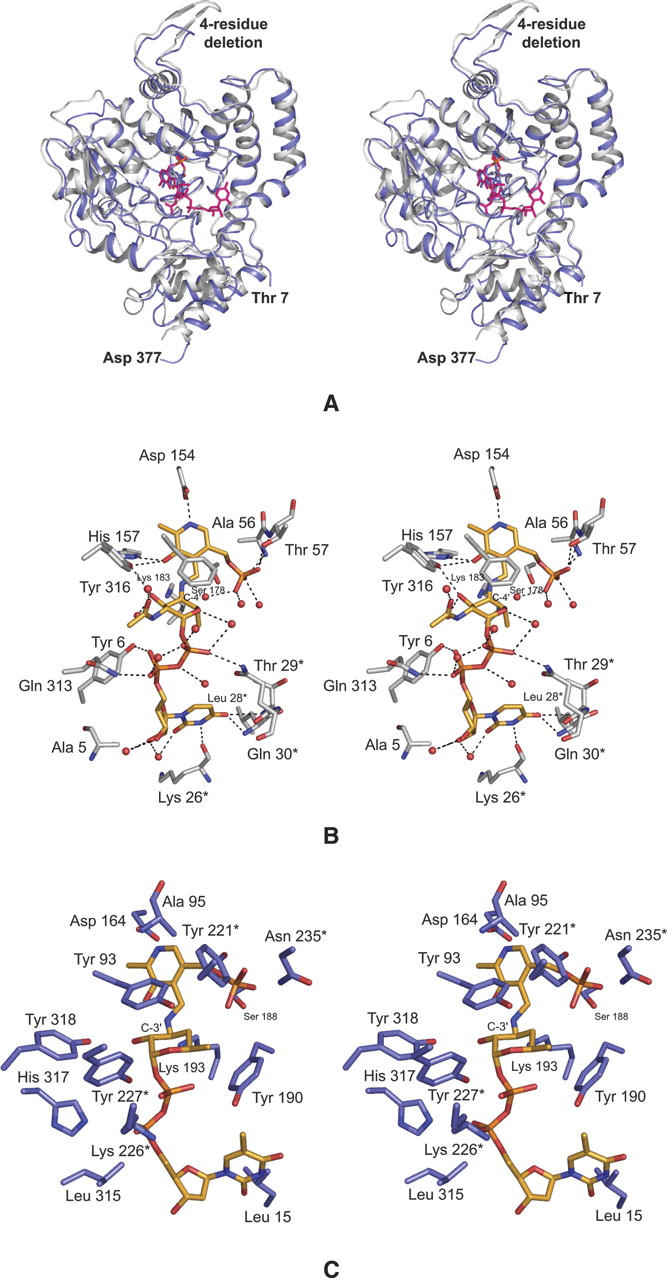
Comparison of DesV with PseC. The overall folds of the two proteins are very similar as highlighted in (A). The ribbon representations for PseC and DesV are presented in white and blue, respectively. X-ray coordinates for PseC were obtained from the protein data bank (accession number 2FNU). A close-up view of the active site for PseC with a bound sugar ligand is depicted in (B). Potential hydrogen bonds are indicated by the dashed lines. Residues contributed by the second subunit are marked by an asterisk. On the basis of that observed for PseC, a model of the external aldimine for DesV with its sugar substrate was built as shown in (C).
At present, the only sugar-modifying aminotransferase whose structure has been solved with a bound sugar ligand is PseC. Specifically, the structure was determined in the presence of its product, UDP-4-amino-4,6-dideoxy-β-L-AltNAc, that reacted in the crystalline lattice to form the external aldimine. As illustrated in Figure 5A, the uridine moiety of the product extends toward the solvent while the sugar is buried. A close-up view of the hydrogen bonding pattern surrounding the UDP-sugar ligand in PseC is presented in Figure 5B. The uracil ring is anchored to the protein via hydrogen bonds with O of Lys 26, N of Leu 28, and N of Thr 29, all of which are contributed by the second subunit of the dimer. These interactions could easily occur in DesV as well, given that the two proteins are very similar in this region. In PseC, there are no direct hydrogen bonds between the protein and the ligand ribose, but the phosphoryl moieties interact with the side chains of Tyr 6 and Gln 313 from Subunit I and Gln 30 from Subunit II (Fig. 5B). These types of interactions cannot occur in DesV given that Tyr 6 structurally corresponds to Phe 12 in DesV and the homologous residues for Gln 313 and Gln 30 in PseC are not present. In PseC, the bulky N-acetyl group of the sugar resides in a large cavity, where it does not directly interact with the protein via hydrogen bonds. This large cavity is not present in DesV because of the above mentioned four-residue deletion, which results in Lys 266 and Tyr 227 from Subunit II projecting into this space. Finally, Oη of Tyr 316 in PseC hydrogen bonds to the C-3′ hydroxyl group of the sugar. This residue is conserved in DesV as Tyr 318, and both adopt the cis-conformation.
Other than its amino acid sequence, little is known regarding the biochemistry of DesV and specifically its catalytic mechanism. It is known, however, that while PseC transfers the amino group axially onto C-4′, DesV adds the amino group equatorially to C-3′. Given the manner in which the sugar ligand binds to PseC, we have modeled the DesV substrate (dTDP-3-keto-4,6-dideoxyglucose) into its active site as an external aldimine (Fig. 5C). This modeling was done by first anchoring the nucleoside portion of the substrate into a similar position as observed in PseC. Subsequently, rotations were made about the phosphoryl backbone of the ligand to place C-3′ of the sugar next to the nitrogen of PMP and the C-2′ hydroxyl near Oη of Tyr 318. As a result of these rotations, the α- and β-phosphorus atoms are displaced by ∼2.5 Å toward the interior of the DesV active site pocket. The only significant steric clashes between the ligand and the protein occur with the side chains of Lys 226 and Tyr 227 from Subunit II and the phosphoryl groups of the substrate. Recall, however, that it is this region that clamps down upon the active site when the ketimine intermediate is bound (Fig. 4). Thus there is flexibility in this part of the polypeptide chain. From our model we would predict that Tyr 318 plays a key role in sugar binding by providing a hydrogen bond to the C-2′ hydroxyl of the sugar. Strikingly, the sugar moiety in DesV is surrounded by a ring of aromatic residues provided by Tyr 93, Tyr 190, and Tyr 318 from Subunit I and Tyr 221 and Tyr 227 from Subunit II. To more fully address the issue of substrate binding in DesV, the preparation of suitable substrate analogs is presently in progress.
From a chemical viewpoint, the in vitro synthesis of nucleotide-linked sugars is a complicated and arduous process at best. But, nature has developed a large repertoire of enzymes for the specific and efficient biosynthesis of unique carbohydrates. Recent advances have yielded in vivo production of nonnative antibacterial agents via the modification of carbohydrate biosynthetic pathways (Melancon et al. 2005). However, this technology has been limited by the paucity of knowledge concerning the required enzymes. The DesV structure described here represents another step toward elucidating the molecular architectures of the enzymes involved in desosamine production. By understanding the structures and functions of these enzymes it will eventually be possible to design modified proteins with altered substrate specificities, thereby yielding new “designer” nucleotide-linked sugars. These will have important ramifications for the development of new therapeutics in the future.
Materials and Methods
Molecular cloning of the DesV gene
S. venezuelae was purchased from ATCC (ATCC no. 15,439), grown overnight at 26°C in ISP Medium 1, and genomic DNA isolated according to standard procedures. The DesV coding sequence (Xue et al. 1998) was PCR amplified with Platinum Pfx DNA polymerase (Invitrogen). The PCR primers were designed to enable product insertion into and excision from the Promega pGEM-T vector (forward primer with NdeI site, 5′-AAAACATATGAGCAGCCGCGCCGAGACCC; reverse primer with XhoI site, 5′-AAAACTCGAGCTAGGCCTGGTCGACCCGCTCG, University of Wisconsin Biotechnology Center). PCR products were purified using a QIAquick PCR purification kit (Qiagen). Following the addition of 3′-A overhangs using Taq Polymerase (Promega), the product was ligated into a pGEM-T vector and used to transform competent Escherichia coli DH5α cells. Transformants containing inserts were screened by XhoI and NdeI fragment size (Promega) and insert sequence (Big Dye, University of Wisconsin Biotechnology Center). The pGEM-DesV vector was digested with XhoI and NdeI and the DNA fragments were separated by agarose gel electrophoresis and purified with a Qiagen QIAquick gel extraction kit. The DesV coding sequence was subsequently ligated into the expression vector pET28b(+) (modified according to Thoden and Holden 2005). This vector was used to transform competent Rosetta(DE3) cells. pET28b(+)-DesV encodes the full-length enzyme and an N-terminal His tag with the following amino acid sequence: MGSSHHHHHHSSENLYFQGH.
Protein expression and purification
Rosetta(DE3) cells containing pET28b(+)-DesV were grown in LB media at 37°C to an OD600 of ∼0.6. The culture was cooled on ice for 15 min, and incubation was continued at 16°C for 24 h in the presence of 1 mM IPTG. Cells were harvested by centrifugation at 1500g for 20 min, and the cell paste was frozen in liquid nitrogen. Frozen cells were resuspended in 10 mM imidazole, 150 mM NaCl, 50 mM sodium phosphate (pH 8.0), and 1 mM PLP and lysed on ice by six cycles of sonication (30 sec of sonication followed by 15 min of cooling). The remaining purification steps were conducted at 4°C. Cellular debris was removed by centrifugation at 40,000g for 45 min, and the supernatant was passed through a Ni-NTA column (Qiagen) equilibrated in the lysis buffer without PLP. DesV was eluted with a linear gradient of increasing imidazole concentration. Protein purity was judged by SDS-PAGE. The protein was dialyzed against storage buffer (150 mM NaCl, 10 mM HEPES at pH 7.5), concentrated to 50 mg/mL, and flash-frozen as pellets in liquid nitrogen. The His-tag was not removed prior to crystallization trials.
To express selenomethionine substituted DesV for MAD phasing, cells were grown on M9 media supplemented with 5 mg/L vitamin B1 at 37°C until an optical density of ∼0.9 was reached at 600 nm. Lysine, threonine, and phenylalanine were added to 100 mg/L, and leucine, isoleucine, valine, and selenomethionine were added to 50 mg/L. The culture was cooled on ice for 30 min. DesV expression was then induced by the addition of IPTG to 1 mM and incubation was continued at 16°C for 24 h. Selenomethionine-labeled DesV was purified as described for the wild-type enzyme. Five selenomethionines per DesV monomer were detected by mass spectrometry (University of Wisconsin Biotechnology Center).
Crystallization of DesV
Initial crystallization conditions of the wild-type protein were surveyed via the hanging drop method of vapor diffusion using a sparse matrix screen designed in the laboratory. Crystals were observed growing from poly(ethylene glycol) 3400 solutions. Diffraction quality crystals were grown via batch methods by first mixing equal volumes of DesV (50 mg/mL in storage buffer with or without 100 mM glutamate) with a precipitant solution containing 24% poly(ethylene glycol) 3400, 2% ethylene glycol, 100 mM MES (pH 6.0) and incubating at room temperature for 24–48 h. Precipitate was removed via centrifugation at 17,000g for 2 min, and 20–30 μL droplets of the supernatant were transferred into vacuum-greased batch plates and streak-seeded with previously prepared DesV microcrystals. The DesV crystals generally achieved maximum dimensions of 0.8 mm × 0.8 mm × 0.1 mm in 2 wk and belonged to the space group P21 with two dimers in the asymmetric unit. The unit cell dimensions for the crystals grown in the absence of glutamate were a = 71.8 Å, b = 143.8 Å, c = 81.1 Å, and β = 106.9°. The unit cell dimensions for the crystals grown in the presence of glutamate were a = 59.7 Å, b = 143.4 Å, c = 92.6 Å, and β = 108.5°. The selenomethionine-labeled protein crystals were grown and handled in a manner similar to that described above.
Crystals of the wild-type protein were equilibrated into a cryoprotectant solution (14% poly[ethylene glycol] 3400, 20% ethylene glycol, 500 mM NaCl, 50 mM MES at pH 6.0) in 10 increments, starting from an artificial mother liquor containing 12% poly(ethylene glycol) 3400, 2% ethylene glycol, 75 mM NaCl, 50 mM MES (pH 6.0). For those crystals grown in the presence of glutamate, 50 mM of the ligand were included in both the synthetic mother liquor and the cryoprotectant solutions. The selenomethionine-labeled protein crystals were handled in the same manner. All of the crystals were flash-cooled at 100 K in a nitrogen stream generated by an Oxford Cobra system.
Structural analysis of DesV
X-ray data sets from crystals of the selenomethionine-labeled protein were collected at the Advanced Photon Source, Argonne National Laboratory, Structural Biology Center, 19-BM beamline on a 3 × 3 tiled “SBC3” CCD detector to 2.1 Å. These data were processed with HKL2000 (Otwinowski and Minor 1997) and internally scaled with SCALEPACK (Otwinowski and Minor 1997). X-ray data sets from the wild-type crystals (in the presence or absence of glutamate) were collected “in-house” at 100 K to 2.05 Å resolution using a Bruker AXS Platinum 135 CCD detector equipped with Montel optics. CuKα radiation was generated by a Rigaku RU200 X-ray generator operated at 50 kV and 90 mA. These data were processed with SAINT (Bruker AXS) and internally scaled with SADABS (Bruker AXS). Relevant X-ray data collection statistics for both the synchrotron and in house data are presented in Table 1.
Table 1.
X-ray data collection statistics

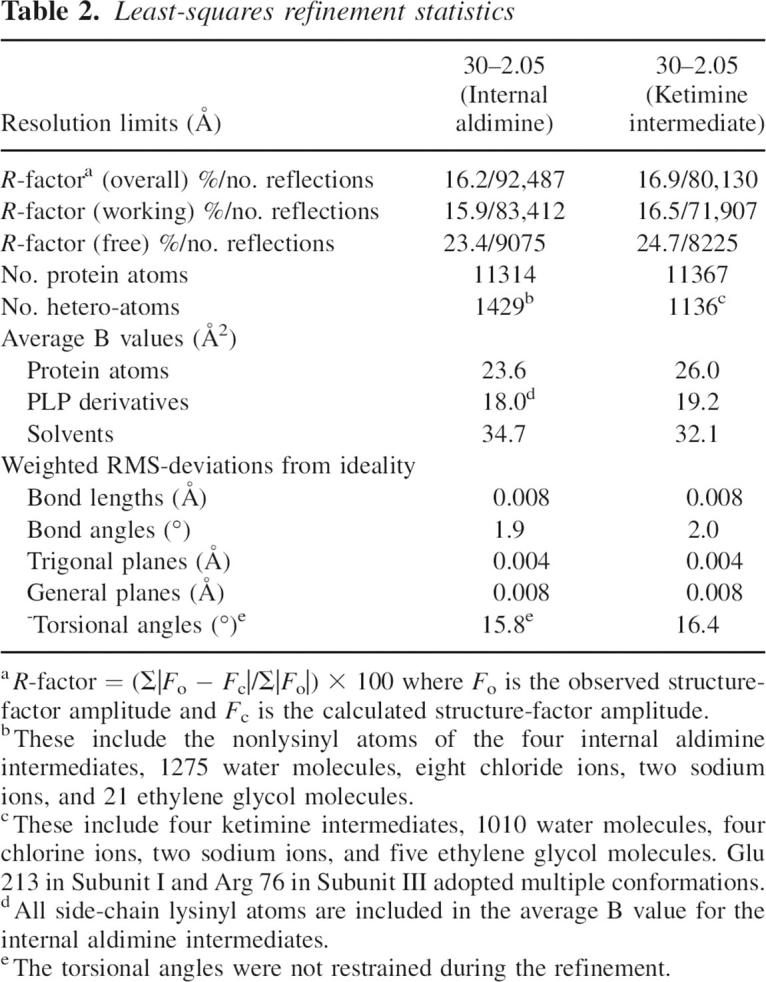
The structure of DesV was solved via MAD phasing using the software package SOLVE to locate the positions of the selenium atoms in the asymmetric unit and to generate initial protein phases (Terwilliger and Berendzen 1999). Density modification and molecular averaging with RESOLVE (Terwilliger 2000) resulted in a readily interpretable electron density map. A model for a single subunit of DesV was manually built into the averaged map using TURBO. This was then used as a search model to solve the structure of the wild-type protein (in either the presence or absence of glutamate) by molecular replacement with the software package PHASER (McCoy et al. 2005). Initial coordinates from the molecular replacement solutions were refined with REFMAC (Murshudov et al. 1997). With over 1400 residues in the asymmetric unit, the goal of the subsequent model building process was to lower the R-factor as much as possible using an “averaged” subunit before finally rebuilding in the asymmetric unit. To expedite this process, the electron densities corresponding to the four subunits in the asymmetric unit were averaged with the program DM (Cowtan and Main 1998) and a single subunit adjusted on the basis of an averaged electron density map. This “averaged” model was placed back into the four positions in the asymmetric unit and refined with the software package TNT (Tronrud et al. 1987). Following refinement, maps using coefficients of either the form (2Fo − Fc) or (Fo − Fc) were calculated, and all four subunits were then adjusted in the asymmetric unit. The matrices required for the averaging process were calculated with the software package LSQKAB (Kabsch 1976). Relevant refinements statistics are presented in Table 2.
Table 2.
Least-squares refinement statistics
For the DesV model without glutamate, 88.7%, 11.3%, and 0.1% of the residues lie in the “most favored,” “additionally allowed,” and “generously allowed” regions of the Ramachandran plot. Likewise, the DesV model in complex with glutamate refined well, with 88.1%, 11.6%, and 0.3% of the residues lying in the “most favored,” “additionally allowed,” and “generously allowed” regions.
Acknowledgments
This research was supported by a grant from the NIH (DK47814 to H.M.H.). X-ray coordinates have been deposited in the Protein Data Bank (20GE and 20GA).
Footnotes
Reprint requests to: Hazel M. Holden, 433 Babcock Drive, Department of Biochemistry, University of Wisconsin, Madison, Madison, WI 53706, USA; e-mail: hazel_holden@biochem.wisc.edu; fax: (608) 262-1319.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.062711007.
References
- Cowtan K. and Main, P. 1998. Miscellaneous algorithms for density modification. Acta Crystallogr. D Biol. Crystallogr. 54: 487–493. [DOI] [PubMed] [Google Scholar]
- Eliot A.C. and Kirsch, J.F. 2004. Pyridoxal phosphate enzymes: Mechanistic, structural, and evolutionary considerations. Annu. Rev. Biochem. 73: 383–415. [DOI] [PubMed] [Google Scholar]
- Hayashi H. 1995. Pyridoxal enzymes: Mechanistic diversity and uniformity. J. Biochem. 118: 463–473. [DOI] [PubMed] [Google Scholar]
- Kabsch W. 1976. A solution for the best rotation to relate sets of vectors. Acta Crystallogr. A 32: 922–923. [Google Scholar]
- McCoy A.J., Grosse-Kunstleve, R.W., Storoni, L.C., and Read, R.J. 2005. Likelihood-enhanced fast translation functions. Acta Crystallogr. D Biol. Crystallogr. 61: 458–464. [DOI] [PubMed] [Google Scholar]
- Melancon C.E., Yu, W.L., and Liu, H.W. 2005. TDP-mycaminose biosynthetic pathway revised and conversion of desosamine pathway to mycaminose pathway with one gene. J. Am. Chem. Soc. 127: 12240–12241. [DOI] [PubMed] [Google Scholar]
- Murshudov G.N., Vagin, A.A., and Dodson, E.J. 1997. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53: 240–255. [DOI] [PubMed] [Google Scholar]
- Noland B.W., Newman, J.M., Hendle, J., Badger, J., Christopher, J.A., Tresser, J., Buchanan, M.D., Wright, T.A., Rutter, M.E., Sanderson, W.E., et al. 2002. Structural studies of Salmonella typhimurium ArnB (PmrH) aminotransferase: A 4-amino-4-deoxy-L-arabinose lipopolysaccharide-modifying enzyme. Structure 10: 1569–1580. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z. and Minor, W. 1997. Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 276: 307–326. [DOI] [PubMed] [Google Scholar]
- Paiardini A., Bossa, F., and Pascarella, S. 2004. Evolutionarily conserved regions and hydrophobic contacts at the superfamily level: The case of the fold-type I, pyridoxal-5′-phosphate-dependent enzymes. Protein Sci. 13: 2992–3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoenhofen I.C., Lunin, V.V., Julien, J.P., Li, Y., Ajamian, E., Matte, A., Cygler, M., Brisson, J.R., Aubry, A., Logan, S.M., et al. 2006a. Structural and functional characterization of PseC, an aminotransferase involved in the biosynthesis of pseudaminic acid, an essential flagellar modification in Helicobacter pylori. J. Biol. Chem. 281: 8907–8916. [DOI] [PubMed] [Google Scholar]
- Schoenhofen I.C., McNally, D.J., Vinogradov, E., Whitfield, D., Young, N.M., Dick, S., Wakarchuk, W.W., Brisson, J.R., and Logan, S.M. 2006b. Functional characterization of dehydratase/aminotransferase pairs from Helicobacter and Campylobacter: Enzymes distinguishing the pseudaminic acid and bacillosamine biosynthetic pathways. J. Biol. Chem. 281: 723–732. [DOI] [PubMed] [Google Scholar]
- Szu P.H., He, X., Zhao, L., and Liu, H.W. 2005. Biosynthesis of TDP-D-desosamine: Identification of a strategy for C4 deoxygenation. Angew. Chem. Int. Ed. Engl. 44: 6742–6746. [DOI] [PubMed] [Google Scholar]
- Terwilliger T.C. 2000. Maximum-likelihood density modification. Acta Crystallogr. D Biol. Crystallogr. 56: 965–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terwilliger T.C. and Berendzen, J. 1999. Automated MAD and MIR structure solution. Acta Crystallogr. D Biol. Crystallogr. 55: 849–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thoden J.B. and Holden, H.M. 2005. The molecular architecture of human N-acetylgalactosamine kinase. J. Biol. Chem. 280: 32784–32791. [DOI] [PubMed] [Google Scholar]
- Tronrud D.E., Ten Eyck, L.F., and Matthews, B.W. 1987. An efficient general-purpose least-squares refinement program for macromolecular structures. Acta Crystallogr. A 43: 489–501. [Google Scholar]
- Weymouth-Wilson A.C. 1997. The role of carbohydrates in biologically active natural products. Nat. Prod. Rep. 14: 99–110. [DOI] [PubMed] [Google Scholar]
- Xue Y., Zhao, L., Liu, H.W., and Sherman, D.H. 1998. A gene cluster for macrolide antibiotic biosynthesis in Streptomyces venezuelae: Architecture of metabolic diversity. Proc. Natl. Acad. Sci. 95: 12111–12116. [DOI] [PMC free article] [PubMed] [Google Scholar]