

Abstract
Pathogenic members of the flavivirus family, including West Nile Virus (WNV) and Dengue Virus (DV), are growing global threats for which there are no specific treatments. The two-component flaviviral enzyme NS2B-NS3 cleaves the viral polyprotein precursor within the host cell, a process that is required for viral replication. Here, we report the crystal structure of WNV NS2B-NS3pro both in a substrate-free form and in complex with the trypsin inhibitor aprotinin/BPTI. We show that aprotinin binds in a substrate-mimetic fashion in which the productive conformation of the protease is fully formed, providing evidence for an “induced fit” mechanism of catalysis and allowing us to rationalize the distinct substrate specificities of WNV and DV proteases. We also show that the NS2B cofactor of WNV can adopt two very distinct conformations and that this is likely to be a general feature of flaviviral proteases, providing further opportunities for regulation. Finally, by comparing the flaviviral proteases with the more distantly related Hepatitis C virus, we provide insights into the evolution of the Flaviviridae fold. Our work should expedite the design of protease inhibitors to treat a range of flaviviral infections.
Keywords: protein structure/folding, conformational changes, enzymes, active sites, viral proteases, crystallography, enzyme inhibitors, mechanism—enzymes, protein structures—new
Flaviviruses, such as the closely related West Nile (WNV) and Dengue (DV) viruses, are members of a larger family called the Flaviviridae (Rice 1996; Burke and Monath 2001), which include the more distantly related hepaciviruses (e.g., Hepatitis C Virus [HCV]) and pestiviruses (e.g., Bovine virus diarrhea). WNV, DV, and HCV are recognized as major health threats that affect millions of people worldwide, with no specific countermeasures to treat them. WNV is transmitted to animals, including humans, by mosquito bites. It is encoded by a single-strand, positive-sense, 11-kb RNA genome, which serves as mRNA for synthesis of the polyprotein precursor and as a template for genome replication in the host cell. The genome encodes three structural proteins found in the mature virion (C, prM, and E) and seven “nonstructural” (i.e., not part of the virion architecture) proteins (NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS5) (Fig. 1; Burke and Monath 2001). Full-length NS3 is a bifunctional protein (Chambers et al. 1990). The N-terminal 175 residues comprise a chymotrypsin-like protease, “NS3pro,” while the C-terminal portion is a helicase (“NS3hel”). NS3pro cleaves internal linkages within NS2A and the capsid protein C, as well as the boundaries between NS2A/NS2B, NS2B/NS3, NS3/NS4A, NS4A/NS4B, and NS4B/NS5. Mutations in the NS3pro cleavage sites in the polyprotein precursor abolish viral infectivity (Speight et al. 1988), suggesting that small molecule inhibition of NS3pro may be an effective antiviral strategy.
Figure 1.
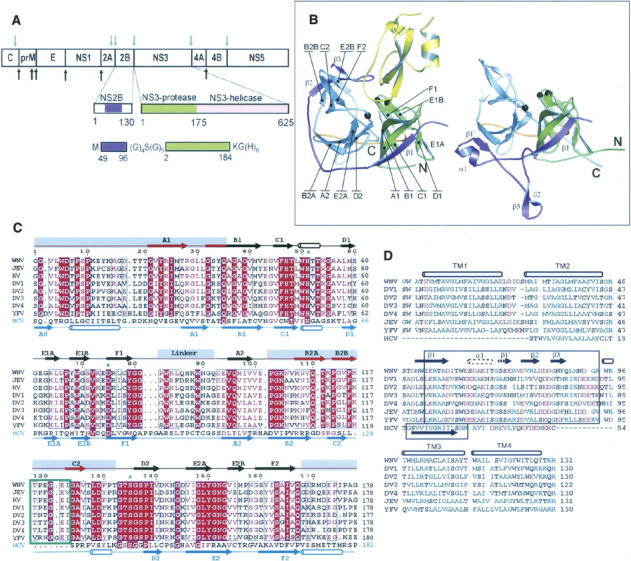
Structure and organization of the flaviviral NS2B-NS3 proteases. (A) Organization of the polyprotein precursor, showing cleavage points by the NS2B-NS3 protease (green arrows) and host cell proteases (black arrows), with detail of the NS2B and NS3 sequences, and (bottom) the fusion construct used in the present study. (B) Ribbon representations of (left) aprotinin-bound WNV NS2B-NS3pro and (right) aprotinin-free WNV NS2B-NS3pro with secondary structural elements and N and C termini indicated. NS3 is shown in green (N-terminal lobe), gray (C-terminal lobe), and orange (the linker between the two lobes); NS2B is in purple; aprotinin is in yellow. The catalytic triad is shown as black balls. (C) NS3 sequences of the flaviviruses: West Nile virus (WNV), Japanese Encephalitis virus (JEV), Kunjin virus (KV), Dengue virus serotypes 1–4 (DV1–4), Yellow Fever virus (YFV); and the hepacivirus, Hepatitis C (HCV). Red highlight indicates identity, red letters homology. Secondary structure elements above the sequences are for WNV NS2B-NS3pro; those below are for HCV NS3pro-NS4A (PDB entry 1JXP). Gray rectangles highlight regions where the folds of WNV NS2B-NS3pro and HCV NS3pro-NS4A differ. The seven-residue insertion unique to the flaviviruses is boxed in green. The structural elements of cofactor-free DV NS3pro (see Fig. 5) are identical to those of HCV NS3pro-NS4A, but the majority of its β-barrel-forming strands have incomplete H-bonding and are not classified as true sheets. (D) Sequences of flaviviral NS2B and HCV NS4A. Boxes indicate minimal cofactor segments required for activation of the NS3 protease in vitro. TM1–TM4 are predicted transmembrane regions. Hydrophobic and aromatic residues are in green, polar in black, acidic in red, and basic in blue. Secondary structure elements are for the WNV protease–aprotinin complex; the alternate elements (α1 and β1′) found in inhibitor-free WNV and DV NS2B-NS3pro are drawn with dotted lines.
The NS2B protein, which is located in the polypeptide precursor immediately upstream of the NS3pro domain, functions as the cofactor for NS3pro (Falgout et al. 1991). NS2B is conserved among the flaviviruses but not the hepaciviruses or pestiviruses. A 35–48 residue central portion is required for protease activity in vitro (Chambers et al. 1993; Wu et al. 2003), while N- and C-terminal flanking hydrophobic regions are predicted to anchor the NS2B-NS3 complex into the host ER membrane (Clum et al. 1997). Flaviviral NS2B-NS3pro can cleave the NS2B-NS3 linkage in cis (Chambers et al. 1993; Wu et al. 2003); moreover, the composition and length of the NS2B-NS3 linker are not important for protease activity toward external substrates, at least in vitro (Wu et al. 2003; Li et al. 2005).
DV and WNV NS2B-NS3 proteases are highly homologous (56% identity) and have similar but distinct substrate specificities. In common with mammalian trypsin, both proteases require a basic residue at the P1 position (Perona and Craik 1995; Wu et al. 2003; Li et al. 2005), which may explain the strong binding of the trypsin inhibitor, aprotinin (also known as bovine pancreatic trypsin inhibitor, BPTI), to flaviviral proteases (Li et al. 2005). Unlike trypsin, they also have clear preferences at the P2 and P1′ positions. HCV NS3pro has much lower similarity with the flaviviruses (e.g., 17% sequence identity with WNV NS3pro), although its overall structure and function are related. However, HCV NS3pro uses a distinct cofactor, NS4A, downstream from NS3, which lacks the C-terminal hydrophobic region of NS2B; moreover, only a 13-residue region of NS4A is required for protease activity in vitro (Lin et al. 1995).
Here, we report the crystal structure of WNV NS2B-NS3pro both in a substrate-free form and in complex with the inhibitor aprotinin/BPTI. We compare these structures with the recently determined structures of WNV in complex with a peptide-based inhibitor and a substrate-free form of DV NS2B-NS3pro (Erbel et al. 2006). This analysis allows us to rationalize for the first time the distinct substrate specificities of WNV and DV proteases, provides evidence for substrate “induced fit,” and leads us to propose that flaviviruses can adopt two conformations of the NS2B cofactor which could provide further opportunities for regulation. Finally, by comparing our structures with those of the HCV ortholog NS3pro-NS4A (Kim et al. 1996) and a cofactor-free DV NS3 (Murthy et al. 2000), we provide insights into Flaviviridae protease evolution.
Results
Crystallization and structure solution
We adopted the protocol previously developed to create a synthetic WNV construct in which the central 47 residues of NS2B are fused via a nine-residue flexible linker to the NS3pro sequence (Nall et al. 2004) (for simplicity, this construct is referred to as “NS2B-NS3pro”). The construct is capable of autolytic cleavage at the scissile bond formed between the first and second Gly residue of the linker. Following autoproteolysis, NS2B remains tightly associated with NS3. We also found that the linker mutation K96A significantly reduces autoproteolysis and that the lack of cleavage does not affect aprotinin binding or proteolytic activity toward exogenous substrates.
In order to crystallize the substrate-free enzyme, we mutated the histidine of the catalytic triad (His51) to alanine, since autoproteolysis of the wild-type enzyme inhibited crystal growth. Calorimetry, CD spectroscopy, and NMR analysis detected no structural differences between the mutant and wild-type enzymes. We crystallized the wild-type enzyme in the presence of the trypsin inhibitor aprotinin/BPTI, which we previously showed to be a potent inhibitor of WNV NS2B-NS3pro (Ki = 24 nM) (Shiryaev et al. 2006). Both approaches yielded crystals suitable for high-resolution structural analysis. SDS-PAGE of dissolved crystals showed that in both cases the enzyme was uncleaved. Attempts to solve the structures by molecular replacement, using the then-available HCV NS3pro-NS4A or (cofactor-free) DV NS3pro structures as templates, were unsuccessful. We therefore solved the WNV H51A structure using MAD phasing from SeMet crystals, and refined it to 1.8 Å resolution. The WNV NS2B-NS3pro–aprotinin complex was solved by molecular replacement, using the H51A mutant structure and aprotinin as search models, and refined to 2.3 Å resolution (Table 1).
Table 1.
Data collection and refinement statistics

Structure of aprotinin-bound WNV NS2B-NS3pro
WNV NS2B-NS3pro has a chymotrypsin-like fold with the active site located at the interface of the N- and C-terminal lobes (Fig. 1). The C-terminal part of the NS2B fragment (residues 64–96) forms a belt that wraps around NS3pro, ending in a β-hairpin (β2–β3) that augments the upper β-barrel and inserts its tip directly into the protease active site. The importance of this interaction is evidenced by the deleterious effects of mutation of NS2B residues Leu75 and Ile79 in DV type II (conserved hydrophobic residues in flaviviruses) (Niyomrattanakit et al. 2004), which lie on the inner surface of the invading cofactor β-hairpin, anchoring it to a hydrophobic cleft on NS3. The peptidic inhibitor-bound structure of WNV NS2B-NS3pro (Erbel et al. 2006) has a very similar overall structure (RMSD = 0.51 Å for 183 Cα atoms). However, in contrast to the peptidic inhibitor, aprotinin occupies all of the major specificity pockets of the protease (S2–S2′), and, moreover, induces a catalytically competent state with a fully formed oxyanion hole, allowing us to propose a complete rationale for the substrate specificity of the flaviviral proteases.
Organization of the flaviviral active site
The active site of chymotrypsin-like proteases includes three conserved elements: (1) A classic His–Asp–Ser catalytic triad (His51, Asp75, and Ser135 in WNV), whose precise arrangement in space is required to enhance the nucleophilicity of the serine hydroxyl group; (2) the “oxyanion hole,” which stabilizes the developing negative charge on the scissile peptide carbonyl oxygen in the transition state; and (3) the substrate binding β-strands E2 and B1, which help to position the substrate in the active site. In the ground state of the active protease, the Ser hydroxyl H-bonds to the His imidazole, which in turn H-bonds to the Asp carboxyl group. In our WNV NS2B-NS3pro-aprotinin complex (Fig. 2A), the catalytic triad adopts a productive geometry that is essentially identical to that observed in the bovine trypsin–aprotinin complex (PDB entry 2FTL) (Pasternak et al. 1999). Thus, the Ser135 OH–His51 Nɛ2 distance (2.8 Å) and the His51 Nδ1–Asp75 CO2− distance (2.7 Å) are indicative of strong H-bonds, as is the distance (2.8 Å) between the Ser135 hydroxyl oxygen and the (nominally) scissile peptide carbonyl of aprotinin.
Figure 2.
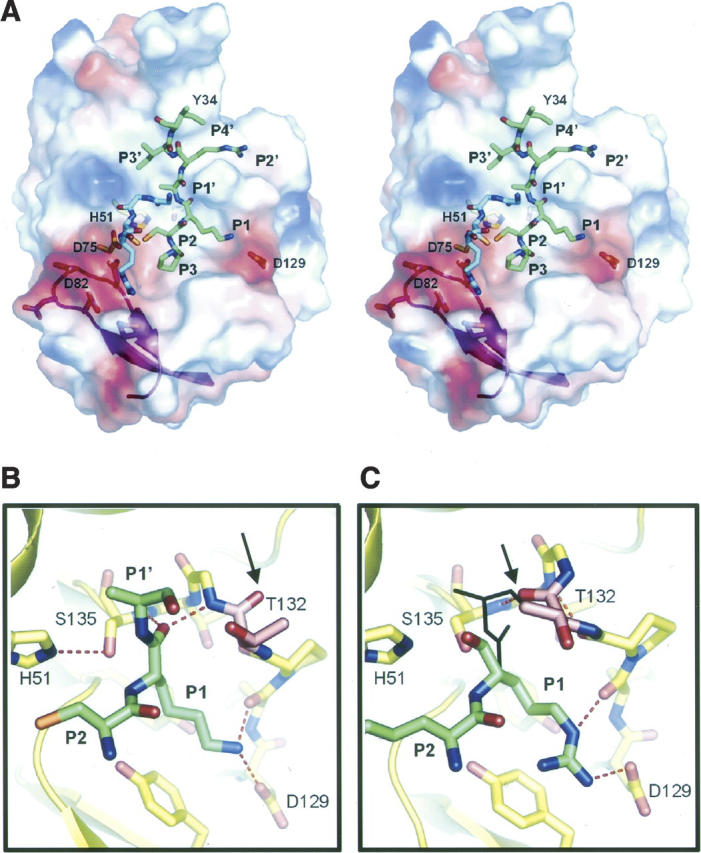
Structure of the NS2B-NS3–inhibitor interface and evidence for induced fit. (A) Stereo view of WNV NS2B-NS3pro surface with selected aprotinin residues. The green and blue sticks are two parts of aprotinin (residues 13PCKARII19 and 35GGCR39) that interact with the protease. The surface is colored by electrostatic potential (negative, red; positive, blue). The magenta ribbons show the invading β2–β3 hairpin of NS2B contributing to the active site. Selected residues of the protease are shown as sticks. (B,C) Comparison of the oxyanion hole conformations in the WNV NS2B-NS3pro complexed with (B) aprotinin and (C) a peptidic inhibitor (Erbel et al. 2006; PDB code 2FP7). Selected residues of the protease (yellow) and inhibitors (green) are shown. In B, aprotinin induces the active conformation of the oxyanion hole, which is occupied by the P1 C=O. In C, the oxyanion hole is disrupted owing to a flip of the peptide bond at Gly133–Thr132, which creates a 310 helical conformation (the C=O of Thr132 is marked with an arrow in B and C). The P1 C=O and P1′ side chain of aprotinin (shown as black stick in C) would clash with Thr132 in this conformation. Selected hydrogen bonds are shown with dashed lines.
Enzyme–substrate main-chain interactions are strongly conserved in chymotrypsin-like proteases (Perona and Craik 1995). The main chain of aprotinin residues 13–19 (PCKARII) forms antiparallel β-sheet interactions with strands E2B and B1 of WNV NS2B-NS3 (Fig. 2A; Table 2), while the side chains occupy the presumed subsites S3–S1 and S1′–S4′, exactly as observed in the trypsin–aprotinin complex. The main-chain conformations of residues occupying the S3 and S2 sites (P3 and P2) are closely superposable with those of the peptidic inhibitor-bound complex (see Supplemental Fig. 1). A second loop of aprotinin (residues 36–39 GGCR) also interacts with WNV protease. The buried surface area in the WNV complex (700 Å2) is very similar to that in the trypsin complex (650 Å2). Moreover, there are only two significant differences in hydrogen bonding to the aprotinin main chain: (1) An additional bond between the Ala16 (P2 position) C=O and Thr132 Oγ1, which is not possible in trypsin and which we propose to be relevant to substrate specificity at the P1′ position (see below); and (2) the Pro13 (P3 position) main chain C=O binds to the side chain of Tyr161 in WNVpro rather than to the main chain of Gly153 in trypsin; there is, however, no substrate preference at this position for either enzyme.
Table 2.
H-bonds between aprotinin and WNV NS2B-NS3pro or bovine trypsin

Evidence for induced fit in the oxyanion hole
Aprotinin binding induces a catalytically competent conformation of the “oxyanion hole” (Fig. 2B). This hole, lined by main-chain nitrogens (from Gly133 to Ser135 in WNV) is improperly formed in the substrate-free and peptidic inhibitor-bound structures (Fig. 2C); in those structures the peptide bond between Thr132 and Gly133 is flipped. Interestingly, the flipped bond creates a helical (310) conformation for residues 131–135 that is stabilized by two hydrogen bonds that are absent in the productive conformation. Thus, it is possible that the nonproductive conformation of the oxyanion hole is energetically favored in the absence of substrate, only acquiring the productive conformation in the presence of substrate with an appropriate P1′ residue. That is, it would suggest an “induced fit” mechanism (Koshland 1958), which might contribute to substrate specificity or enzyme turnover (see below).
Substrate specificity of the Flaviviruses
The aprotinin-bound WNV structure mimics a classic Michaelis–Menten complex, allowing for a more authentic and complete view of the enzyme–substrate precleavage complex than was available from the peptidic inhibitor-bound structure, which occupied only the S1–S4 sites. It is now straightforward to model an authentic substrate into the S2 through S2′ specificity pockets of the WNV protease. WNV and related flaviviral proteases have a requirement for basic residues at both the P1 and P2 positions of their substrates and Gly, Ser, or Thr at the P1′ position. In our model, the P2 side chain (Lys/Arg) interacts both directly with Asn84 of NS2B and indirectly with a negatively charged surface created by the invading hairpin of NS2B, including Asp80 and Asp82, as previously proposed (Otlewski et al. 2001). In contrast, trypsin, with no P2 preference, has no such negatively charged patch or binding pocket in this region. NS2B binding also helps to define the S1 pocket in WNV protease, by changing the conformation of NS3 residues 116–132, such that Asp129 is introduced into the base of the pocket, where it can salt-bridge with the P1 Lys/Arg (Fig. 2A). Accordingly, in WNV or DV proteases, mutation of Asp129 to Glu (which may be too long) or Ala (which cannot salt-bridge) abrogates catalysis (Chappell et al. 2005). In trypsin, Asp189 plays an analogous role (Perona and Craik 1995; Hedstrom 2002), consistent with its similar P1 specificity.
The S1′ pocket in WNV NS2B-NS3pro comprises a cavity between strand B1 and the helical turn (residues 50–53) following strand C1 (Fig. 3A,C) and is lined on one side by the catalytic histidine and on the other by an invariant glycine (Gly37 in WNV). The pocket is well formed but only large enough to accommodate small P1′ side chains such as the consensus residues Gly, Ser, or Thr. Ser and Thr have the potential to H-bond to the main-chain C=O of the adjacent Ala36, rationalizing the P1′ preference for these residues, while glycine may leave enough space for a water molecule. The inability of the Ala side chain to H-bond may explain its rare appearance in flaviviral cleavage sequences. In trypsin, no such pocket is formed (Fig. 3B), since a disulfide bridge occupies this site, and, as a result, trypsin has no specificity at the S1′ position.
Figure 3.
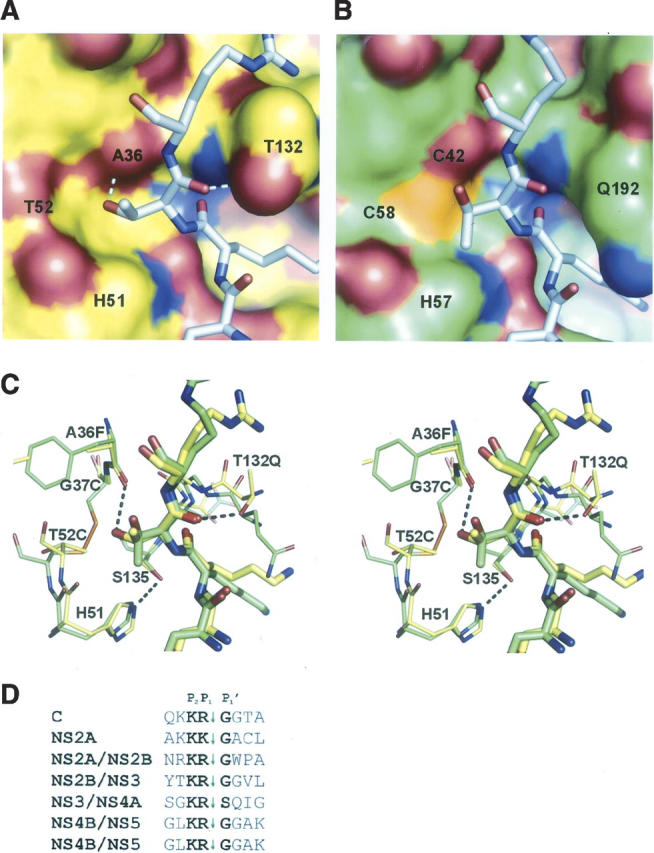
Substrate specificity at the S1′ pocket. Close-up of the surfaces of aprotinin-bound (A) WNV NS2B-NS3pro and (B) bovine trypsin, showing the pronounced pocket in the WNV protease that is absent in trypsin (where a disulfide bridge occludes the pocket). Ala of aprotinin has been replaced in silico by Thr, a consensus residue for flaviviral substrates. Potential H-bonds to NS3 residues Ala36 and Thr132 are shown as dashed lines in A, and the oxyanion hole (blue) occupied by the P1 C=O is evident at the center of both figures. WNV NS2B-NS3pro actually prefers glycine, perhaps owing to the additional H-bond to Thr132, which restrains the P1–P1′ dihedral angles. (C) Stereo stick comparison of A and B with WNV complex in yellow and trypsin in green. Selected H-bonds are shown as dashed lines. (D) WNV NS2B-NS3pro cleavage sequences, showing consensus at the P2, P1, and P1′ positions.
WNV protease, unusually, has a strong preference for Gly at the P1′ position, although Ser is tolerated in one case (Fig. 3D); this preference may correlate with the presence of a Thr residue at position 132 of NS3 rather than the Pro residue found in DV. The Thr side chain makes a H-bond with the main-chain C=O of the P1′ residue (Fig. 3A,C), which is flanked on one side by a short H-bond between the P2′ main-chain nitrogen and the C=O of Ala36 and on the other side by the P1 C=O that occupies the oxyanion hole. We propose that the additional H-bond in WNV protease places stringent restraints on the substrate dihedral angles, allowing for limited “wiggle room” for the P1′ side chain, which is positioned close to the catalytic His; this may explain the preference for glycine. Although the P2′ main chain makes important contacts in the enzyme-inhibitor complex, its side chain points out into solution, consistent with the lack of specificity at this position.
Two conformations of WNV NS2A-NS3pro
The major difference between the inhibitor-bound and -free WNV NS2A-NS3pro structures lies in the conformation of the NS2B cofactor (Fig. 4A). In the inhibitor-free structure, the β1 strand of NS2B is formed as in the aprotinin complex, augmenting the β-barrel of the N-terminal lobe of NS3pro. However, beyond strand β1, the last NS2B residue that adopts the same conformation as in the aprotinin complex is Trp62, which is buried in a pocket on the C-terminal lobe of NS3. Trp62 appears to act as an “anchor” for NS2B: accordingly, mutation of either Trp62 (Niyomrattanakit et al. 2004) or of the NS3 residue, Gln96 (Matusan et al. 2001), which lines the base of the Trp62 acceptor pocket, abrogates catalytic activity. Following Trp62, the NS2B chain adopts a new conformation: A new turn-and-a-half of helix (“α1”) is followed by an abrupt reversal of chain direction at G70 (conserved as Gly or Ala in flaviviruses) followed by a four-residue β-strand (which we call β1′) that augments the central β-sheet of the NS3 C-terminal domain by making main-chain interactions with β-strand B2A. The chain then continues in this reverse direction, toward the N terminus of NS2B, i.e., opposite from that in the inhibitor-bound structure, and is ordered at its C terminus by virtue of a crystal contact (see below). These structures show for the first time that the same flavivirus protease can adopt two distinct cofactor-bound conformations. Despite these major changes in the interaction with NS2B, the NS3 coordinates are closely superposable (RMSD = 0.8 Å for 148 out of 151 Cα atoms).
Figure 4.

Two conformations of the NS2B cofactor. (A) Superposition of WNV NS2B-NS3 proteases: substrate-free (green and blue) and aprotinin-bound (gray and red). The secondary elements of NS2B unique to the substrate-free (α1 and β1′) and the substrate-bound (β2 and β3) structures, as well as the alternative C termini are indicated. The point of departure (Trp62) for the two NS2B elements is labeled and shown as a stick model. The elements β2′ and β3′ in the substrate-free structure are stabilized by crystal contacts (see Supplemental Fig. 3). Active site residues are shown as black circles. (B) Superposition of substrate-free DV (gray and red) and WNV (green and blue) NS2B-NS3pro. The collapse of the E2B–F2B loop observed in DV is indicated by the arrow.
Evidence for two conformations common to flaviviral NS2B-NS3 proteases
The inhibitor-free DV NS2B-NS3pro structure (Erbel et al. 2006) is overall very similar to the inhibitor-free WNV structure (RMSD = 0.9 Å for 133 out of 149 Cα atoms). Remarkably, the NS2B chain departs the body of NS3 at the same point and adopts an almost identical turn–helix–strand conformation (Fig. 4B). The sequence of this region is well conserved (consensus WExxAEITGSS) among flaviviruses, raising the strong possibility that the two conformations are a conserved feature of flaviviruses. The two inhibitor-free proteases do differ significantly, however, in the region close to the active site, although this may be a consequence of an extensive lattice contact in the WNV protease crystals. Thus, in DV protease, the E2B–F2 loop collapses onto the B2B–C2 loop, both occluding the hydrophobic cavity that accepts the NS2B β2–β3 hairpin in the substrate-bound structure and also distorting the S1 pocket, leading to the expulsion of Asp129, which salt-bridges to the P1 Lys/Arg in the productive conformation; that is, both the S1 and S2 pockets are disrupted (see Supplemental Fig. 2). However, in the WNV protease, despite the absence of the NS2B terminal hairpin, the catalytic site is otherwise well formed (except that the oxyanion hole adopts the helical nonproductive conformation); furthermore, the surrounding loops adopt conformations very similar to those in the aprotinin-bound structure. The sequence of this region is well conserved among the flaviviruses, and the more “productive” conformation observed for WNV may be explained by an extensive crystal contact comprising a symmetric dimer in which the hydrophobic A1–B1 loops from each molecule insert between the B2B–C2 and E2B–F2 loops of the other molecule, mimicking binding of the β2–β3 hairpin from NS2B (Supplemental Fig. 3). The dimer is presumably a crystallization artifact, since the protein is monomeric in solution. The WNV structure therefore probably reflects a propensity for the productive conformation that happens to be stabilized by particular crystal contacts.
Evolution of the Flaviviridae protease fold
Among the Flaviviridae family, protease structures are now known for the flaviviruses, DV and WNV, and the hepacivirus, HCV. There are no structures for the pestiviruses, such as Bovine virus diarrhea, although they appear to be more akin to the hepaciviruses, utilizing a downstream NS4A cofactor (Xu et al. 1997). The structure of HCV NS3pro-NS4A is distinct from those of the flaviviral NS2B-NS3pro (Fig. 5A). This is particularly true of the C-terminal lobe, which adopts a chymotrypsin-like fold in HCV, but is radically different in the flaviviruses. Although there is no evidence that functional flaviviral NS3 protein exists in infected cells in the absence of its cofactor, nevertheless high resolution crystal structures of cofactor-free DV NS3 have been obtained (Murthy et al. 1999, 2000). Moreover, they reveal an astonishingly close structural similarity (RMSD = 1.4 Å for 157 residues) with cofactor-bound HCV (Fig. 5B,C), both in their three-dimensional organization and in the order and alignment of their secondary structural elements, including N- and C-terminal helices that are not present in WNV and DV NS2B-NS3pro. The only major difference, other than a longer C terminus in HCV, is an eight-residue inserted loop in cofactor-free DV that is conserved in the flaviviruses (see below). Thus, the flaviviral NS3 protein appears to have a strong propensity to adopt an HCV-like fold in the absence of NS2B. Perhaps this conformation is utilized as an intermediate in the assembly of the NS2B-NS3 complex. Notwithstanding, the existence of this essentially identical fold strongly suggests that the flaviviral fold is an evolutionary adaptation of an HCV-type fold in which the cofactor plays a key role.
Figure 5.
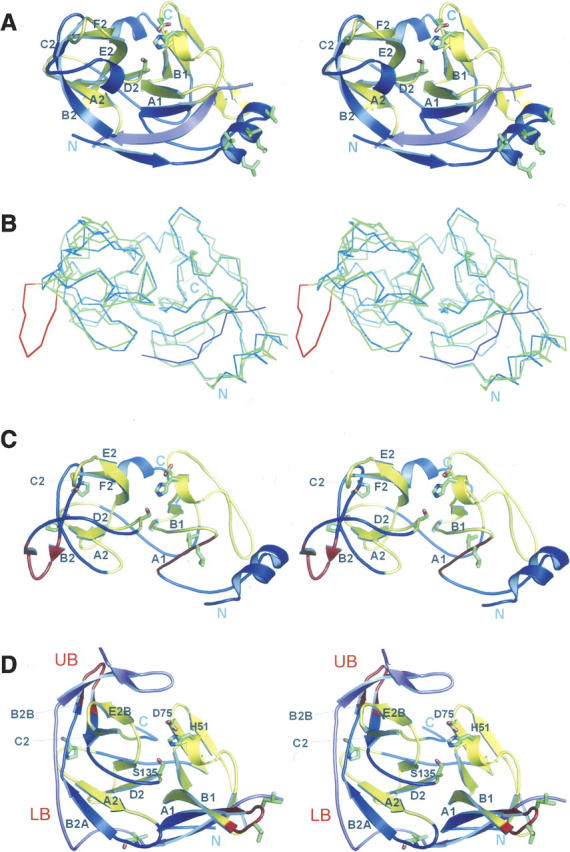
Stereo comparisons of flaviviral and HCV protease domains. (A) HCV NS3pro-NS4A protease (PDB entry 1JXP). NS4A is in purple; NS3 is in yellow, except for regions analogous to the mobile regions in flaviviruses (in blue). Hydrophobic side chains on the NS3 N-terminal helix implicated in membrane attachment are in green. (B) Cα superposition of HCV NS3pro-NS4A (blue/magenta) and cofactor-free DV NS3pro (green; PDB entry 1DF9). The RMSD between Cα atoms is 1.44 Å for 157 residues. The seven-residue insertion unique to flaviviruses (residues 117–124 in WNV) is highlighted in red. (C,D) Cofactor-free DV NS3pro (C) and aprotinin-bound WNV NS2B-NS3pro (D) colored to illustrate conformational differences. Regions of NS3 that are structurally similar are in yellow; those that acquire alternate conformations upon cofactor binding are in blue, except for residues 26–32 and 117–123, which are shown in red to highlight their major relocations. Cofactor binding leads to a separation of the C-terminal barrel into an open “lower barrel” (LB) and a closed “upper barrel” (UB). Catalytic residues, selected side chains that may be important for the conformational changes (Pro/Val106 and Pro113), and the hydrophobic hairpin at the A1–B1 turn of NS3 exposed by NS2B binding are shown as green sticks. NS2B is in purple. Selected secondary structure elements are labeled. β-strands of DV NS3 that do not form regular β-sheet H-bonds are shown as loops.
A detailed comparison between the WNV NS2A-NS3pro and HCV NS3pro-NS4A (and cofactor-free DV NS3) domains reveals that within the N-terminal β-barrel there are substantial reorganization and changes in strand register upon cofactor binding for the flaviviruses that do not occur in HCV. Thus, in HCV (Figs. 1C, 5A), the first β-strand (A1) is formed by residues 27–32 (WNV numbering), while in WNV NS2B-NS3pro (Figs. 1C, 5D) the homologous residues adopt a β-turn between strands A1 and B1, and strand A1 is formed instead by residues from the preceding loop. These changes lead to the exposure of two hydrophobic residues conserved among flaviviruses (L30 and L31) on the surface of the protein. Intriguingly, these are spatially analogous to the hydrophobic N-terminal helix, α0, of the HCV protease, which has been implicated in membrane association (Yan et al. 1998) and is lost in the flaviviruses (see also Supplemental Fig. 4). The change in register of the N-terminal residues in WNV NS2B-NS3pro also appears to disrupt the N-terminal helix, freeing the first ∼20 residues from contacts with the domain. The additional slack created could account for the ability of flaviviral proteases to autoproteolyze the NS2B-NS3 linkage (Chambers et al. 1993; Wu et al. 2003), which may offer an evolutionary advantage; thus, in HCV NS3pro-NS4A, these residues remain ordered, and another protease is required to cleave the analogous linkage (Grakoui et al. 1993). The new location of the ordered N terminus also displaces the interdomain linker (residues 82–92) toward the C terminus, which is helical in HCV, but which becomes disordered in both WNV and DV.
In HCV NS3pro-NS4A, the C-terminal lobe has a chymotrypsin-like β-barrel conformation. The NS4A cofactor of HCV does not extend into the C-terminal lobe of NS3pro and does not affect its structure. In contrast, in both the WNV and DV NS2B-NS3pro structures, the longer NS2B cofactor invades the C-terminal lobe, inducing a remarkable reorganization of the secondary and tertiary structures. Thus, the entire segment between β-strands A2 and D2 (cf. Fig. 5C,D) undergoes extensive rearrangements of strands, loops, and β-hairpins, such that the formerly closed single barrel is split into two barrels: an open β-barrel (“lower barrel”), with a smaller closed β-barrel (“upper barrel”) above it. The reorganization includes: (1) A lengthening of the A2–B2 loop and change in register of the B2 strand (which we now call “B2A”) and (2) a large-scale flip and change in register of a β-hairpin comprising B2B and C2, leading to a change in its location from the bottom of the lower barrel to the top of the upper barrel. The tip of this new hairpin originates from the seven-residue insertion (residues 117–124 in WNV) that is a hallmark of the flaviviruses but is absent in HCV, as noted above. This insertion enables the conformational changes that create the new β-barrel, including the new B2B–C2 hairpin that cradles the productive conformation of the NS2B β2–β3 hairpin. In the absence of such a loop, such a conformational change is not possible in HCV. Two prolines (Pro102 and Pro113) that are invariant in flaviviruses (but not in HCV) are located in strands in the cofactor-free DV NS3 structure but relocate to loops (typically their preferred location) in the cofactor-bound structures, which may help to promote the conformational switch upon NS2B binding. Consistent with the conformational sensitivity of this region, in a random screen for temperature-sensitive replication-restricted mutants in the DV type IV polyproprotein, two out of the eight were found within NS3pro (Blaney et al. 2003); moreover, both of these (L128F and S158P) map to the upper β-barrel.
Discussion
Our complex of WNV NS2B-NS3pro with the substrate-mimetic inhibitor aprotinin allows, for the first time, a comprehensive analysis of the substrate specificity of WNV and flaviviruses in general. As previously shown, the preference for basic residues at the S1 and S2 sites follows from the trypsin-like organization of the S1 site together with a negatively charged surface introduced by the invading hairpin of NS2B at the S2 site. However, our structure now reveals the interactions at the S1′ and S2′ sites and offers a rationale for the preference for serine and threonine at the P1′ site for most flaviviruses, as well as the particular preference for glycine in the case of WNV protease.
The aprotinin-bound structure further shows that the oxyanion hole is only correctly formed in the presence of an authentic substrate (or a very close mimic). Such a mechanism has been proposed in the case of the highly specific serine proteases of the complement system (Jing et al. 2000), where the inactive conformation of the oxyanion hole is similarly stabilized by other interactions in the absence of substrate. Whether such a mechanism actually provides greater specificity is unclear, as Fersht's (1977) classic analysis suggests that specificity is not altered by induced fit in the case where chemistry is the rate-limiting step. Given the apparent stability of the inactive conformation of the oxyanion hole, the induced fit mechanism might instead provide for a more rapid substrate release. Experimental data in this regard are limited (Pasternak et al. 2001), and the flaviviral proteases may provide a useful system for testing this fundamental concept in enzymology.
We found that WNV NS2B-NS3 can adopt two distinct conformations that differ in the placement of the NS2B cofactor: In the first, NS2B wraps around NS3, completing the structure of the active site; in the second, the NS2B chain changes direction shortly after invading the C-terminal lobe, and we assume this to be an inactive conformation, given the deleterious effects of mutations in the β2–β3 loop on catalytic activity (Niyomrattanakit et al. 2004). The remarkable similarity between the substrate-free conformations of the NS2B cofactors in WNV and DV NS2B-NS3pro (Erbel et al. 2006) raises the distinct possibility that flaviviral proteases possess a second conformational state in vivo that lacks proteolytic activity. A mechanism to “turn off” the protease activity once its activity is no longer required could plausibly offer an advantage at subsequent stages in viral pathogenesis, when the “nonstructural” proteins become actively involved in viral RNA replication, and the helicase activity of NS3 is now required while further proteolytic activity could be counterproductive.
The astonishing similarity between the structures of HCV NS3pro-NS4A and cofactor-free DV NS3pro strongly suggests that the flaviviral NS2B-NS3pro fold has evolved from the HCV fold and has been embellished in ways that allow for additional levels of regulation. The reorganization of the C-terminal lobe of NS3 allows the longer NS2B segment to form β-sheet interactions that hold it in the active site, as previously shown (Erbel et al. 2006). Our analysis reveals that the reorganization is facilitated by a seven-residue insertion common to flaviviruses but absent in HCV, and also perhaps by strategically placed proline residues that promote strand-to-coil transitions. The change in strand register also frees the N terminus, allowing the NS2B-NS3 linkage to be self-cleaved, in cis, rather than being dependent on other proteases as in HCV, which may offer a further biological advantage. However, one consequence of this reorganization is the loss of the N-terminal helix, which in HCV is implicated in membrane attachment (Yan et al. 1998). It is tempting to speculate that the change in strand register upon NS2B binding that exposes a hydrophobic hairpin in the N-terminal domain serves to compensate for this loss (see Supplemental Fig. 4). Finally, we note that the β2–β3 loop of NS2B, once placed at the active site by a few key hydrophobic interactions, lacks further structural constraints, which could allow for rapid evolution of substrate specificity in response to host adaptation, a concept proposed by Harrison (1996) in a nonpathogenic context.
Materials and Methods
Protein cloning, expression, and purification
Construction of the recombinant NS2B-NS3, which contained residues 49–96 of NS2B, a nine-residue GGGGSGGGG linker, and the NS3pro sequence (residues 2–184) was described previously (Shiryaev et al. 2006). The construct was recloned into the pET101/D-TOPO vector (Invitrogen) and C-terminally tagged with a His-6 tag. The resulting construct was additionally modified, by using a QuikChange mutagenesis kit (Stratagene), to substitute the C-terminal Gly–Gly residues for Arg–Arg in the linker sequence, and the active site His51 for Ala to obtain the catalytically inert H51A mutant. In the second construct used for cocrystallization with aprotinin, Lys96 of NS2B was replaced with Ala, which significantly reduced self-proteolysis at this site. Competent Escherichia coli BL21 (DE3) Codon Plus cells (Stratagene) were transformed with recombinant pET101 vectors. Transformed cells were grown in Luria–Bertani broth containing 100 mg/L ampicillin and 34 mg/L chloramphenicol at 37°C. Cultures were induced with 0.6 mM isopropyl b-D-thiogalactoside, and growth was continued for an additional 12–20 h at 18°C. The cells (5 g) were then collected by centrifugation (6000g, 15 min), resuspended in 50 mM Tris buffer (pH 7.5), containing 300 mM NaCl, 2 mM β-mercaptoethanol, and 5 μM leupeptin, and disrupted by two passages through a French press. After a 1 h centrifugation at 40,000g, the supernatant fraction (35 mL) was purified on a 5-mL Ni-affinity column (GE/Amersham). The impurities were removed by washing the column with 20 mM Tris buffer (pH 7.5) containing 500 mM NaCl and 30 mM potassium phosphate The NS2B-NS3 constructs were eluted on a 20–300-mM gradient of imidazole. Concentration of the samples with a 5-kDa-cutoff concentrator was followed by gel filtration on a high-resolution Superdex S200 or S75 column for the removal of minor impurities and for buffer exchange into 20 mM HEPES (pH 7.0), 200 mM LiCl, and 2 mM β-mercaptoethanol. A typical yield of the purified NS2B-NS3pro protein was 30 mg/L of E. coli culture. To obtain the selenomethionine derivative of NS2B-NS3, methionine biosynthesis was inhibited in E. coli cells, and the newly synthesized NS2B-NS3 was biosynthetically labeled with SeMet (Doublie 1997). Following purification, DTT was added to a final concentration of 10 mM.
Crystallization and structure solution
Owing to autoproteolysis, we were unable to crystallize the isolated catalytically active NS2B-NS3pro protein. The inert H51A mutant, however, crystallized under a broad range of conditions. Hexagonal prisms with typical dimensions of 20 × 20 × 200 μm3 were obtained by vapor diffusion in hanging drops at room temperature for 2 wk. 2.5 μL of the H51A mutant protein (10 mg/mL in 10 mM HEPES at pH 7.4, 100 mM LiCl, and either 2 mM β-mercaptoethanol or 10 mM dithiothreitol [DTT]) were mixed with 2.5 μL of well solution containing 100 mM Tris (pH 7.5), 200 mM LiCl, and 30% (w/v) PEG 1500 (Hampton Research). Crystals were flash frozen without further cryoprotectant in liquid nitrogen. Multiwavelength anomalous dispersion (MAD) data from the selenomethionine-substituted crystals were collected at the Brookhaven National Synchrotron Light Source (NSLS) beamline X26C through the mail-in service. Three hundred sixty frames with an oscillation width of 0.5° were collected for each wavelength of the MAD experiment. The data were processed using the HKL2000 package (Otwinowski and Minor 1997). The native data set was collected at the Berkeley Advanced Light Source (ALS) beamline 12.3.1. The set comprises 100 frames with an oscillation width of 1.0° (Table 1). Eight of the 10 selenium atoms were located by SHELXD (Schneider and Sheldrick 2002), verified by SOLVE (Terwilliger 2003), and refined by AUTOSHARP (Bricogne et al. 2003). Automatic model building with RESOLVE (Terwilliger 2003) assigned 20 of 211 residues. The remainder of the model was built manually using XFIT (McRee 1999) and COOT (Emsley and Cowtan 2004). Initial refinement was carried out against native data to 2.2 Å resolution using CNS version 1.0 (Brünger et al. 1998). Rigid body refinement was followed by 400 cycles of simulated annealing at 2000 K, 100 cycles of individual coordinates refinement, and 30 cycles of individual B-factor refinement. Further refinement was performed with REFMAC (Murshudov et al. 1997) from the CCP4 version 5.0 (Collaborative Computational Project No. 4 1994) using data from 50 to 1.8 Å resolution. Solvent was added automatically in 20 cycles of ARP/WARP (Perrakis et al. 2001).
The protease–aprotinin complex was prepared by mixing the catalytically active protein at 0.5 mg/mL with bovine lung aprotinin (Sigma) at twice the molar concentration in 150 mM NaCl and 50 mM Tris (pH 8). Concentrating the complex to 10 mg/mL using a 30-kDa cutoff removed the excess aprotinin. The complex was then mixed with the same buffer containing 15% (w/v) PEG 6000 (Hampton Research). Crystals (orthorhombic prisms) with typical dimensions of 20 × 20 × 100 μm3 were obtained by vapor diffusion in hanging drops after 2 wk at room temperature. A native data set was collected at ALS beamline 12.3.1 and processed as described above. The structure of the WNV NS2B-NS3pro–aprotinin complex was determined by molecular replacement using PHASER (Read 2001), using structures of the inhibitor-free protein and bovine aprotinin (PDB code 1F5R) as search models. Refinement proceeded, as above, to a resolution of 2.3 Å (Table 1). Both models were validated with PROCHECK (Laskowski et al. 1993). The aprotinin-bound WNV NS2B-NS3pro model comprises residues E49–N89 of NS2B and K14–P176 of NS3pro; disordered residues are 90–96 of NS2B, 9 residues of the linker, 13 residues at the N terminus and 10 residues at the C terminus of NS3pro, and 6 residues of the His-tag. The inhibitor-free model comprises residues E50–A93 of NS2B and P10-G178 of NS3pro; disordered residues are 94–96 in the cofactor, 9 residues of the linker, 9 residues at the N terminus and 8 residues at the C terminus of NS3pro, and 6 residues of the His-tag. The disordered regions are very similar to those in the peptide-bound structure (Erbel et al. 2006). One exception is residues 28–32 at the tip of the A1–B1 hairpin, which are ordered in our structures but disordered in the peptide-bound structure. Coordinates and structure factors for the inhibitor-free WNV NS2B-NS3pro (H51A mutant) and the NS2B-NS3pro–aprotinin complex have been deposited with the PDB (codes 2GGV and 2IJO).
Acknowledgments
We are especially grateful to Laurie Bankston for critical reading of the manuscript. We are grateful for access to and user support from the ALS and NSLS national synchrotron facilities supported by the DOE and NIH. We thank Annie Heroux and Kenneth Frankel for assistance in data collection, Boris Ratnikov for kinetic characterization, Andrey Bobkov for calorimetric analysis, and Maurizio Pellecchia for NMR analysis. This work was supported by grants to R.C.L. (DAMD17-03-2-0038 and NIH P01 AI055789) and A.Y.S. (NIH U54 RR020843).
Footnotes
Supplemental material: see www.proteinscience.org
Reprint requests to: Robert C. Liddington, Infectious and Inflammatory Disease Center, Burnham Institute for Medical Research, 10901 North Torrey Pines Road, La Jolla, CA 92037, USA; e-mail: rlidding@burnham.org; fax: (858) 713-9925.
Abbreviations: NS, nonstructural; NS3pro, NS3 protease domain; WNV, West Nile Virus; DV, Dengue Virus; HCV, Hepatitis C Virus; JEV, Japanese Encephalitis Virus; KV, Kunjin Virus; YFV, Yellow Fever Virus; TM, transmembrane (helix); RSMD, root mean square deviation; NSLS, Brookhaven National Synchrotron Light Source; ALS, Berkeley Advanced Light Source; MAD, multiwavelength anomalous dispersion; DTT, dithiothreitol.
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.072753207.
References
- Blaney J.E., Manipon, G.G., Murphy, B.R., and Whitehead, S.S. 2003. Temperature sensitive mutations in the genes encoding the NS1, NS2A, NS3, and NS5 nonstructural proteins of dengue virus type 4 restrict replication in the brains of mice. Arch. Virol. 148: 999–1006. [DOI] [PubMed] [Google Scholar]
- Bricogne G., Vonrhein, C., Flensburg, C., Schiltz, M., and Paciorek, W. 2003. Generation, representation and flow of phase information in structure determination: Recent developments in and around SHARP 2.0. Acta Crystallogr. D Biol. Crystallogr. 59: 2023–2030. [DOI] [PubMed] [Google Scholar]
- Brünger A.T., Adams, P.D., Clore, G.M., DeLano, W.L., Gros, P., Grosse-Kunstleve, R.W., Jiang, J.S., Kuszewski, J., Nilges, M., Pannu, N.S., et al. 1998. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 54: 905–921. [DOI] [PubMed] [Google Scholar]
- Burke D.S. and Monath, T.P. 2001. Flaviviruses. In Fields Virology (eds. D.M. Knipe and P.M. Howley), pp. 1043–1125. Lippincott Williams & Wikins, Philadelphia.
- Chambers T.J., Weir, R.C., Grakoui, A., McCourt, D.W., Bazan, J.F., Fletterick, R.J., and Rice, C.M. 1990. Evidence that the N-terminal domain of nonstructural protein NS3 from yellow fever virus is a serine protease responsible for site-specific cleavages in the viral polyprotein. Proc. Natl. Acad. Sci. 87: 8898–8902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers T.J., Nestorowicz, A., Amberg, S.M., and Rice, C.M. 1993. Mutagenesis of the yellow fever virus NS2B protein: Effects on proteolytic processing, NS2B-NS3 complex formation, and viral replication. J. Virol. 67: 6797–6807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chappell K.J., Nall, T.A., Stoermer, M.J., Fang, N.X., Tyndall, J.D., Fairlie, D.P., and Young, P.R. 2005. Site-directed mutagenesis and kinetic studies of the West Nile Virus NS3 protease identify key enzyme–substrate interactions. J. Biol. Chem. 280: 2896–2903. [DOI] [PubMed] [Google Scholar]
- Clum S., Ebner, K.E., and Padmanabhan, R. 1997. Cotranslational membrane insertion of the serine proteinase precursor NS2B-NS3(Pro) of dengue virus type 2 is required for efficient in vitro processing and is mediated through the hydrophobic regions of NS2B. J. Biol. Chem. 272: 30715–30723. [DOI] [PubMed] [Google Scholar]
- Collaborative Computational Project No. 4 1994. The CCP4 suite: Programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50: 760–763. [DOI] [PubMed] [Google Scholar]
- Doublie S. 1997. Preparation of selenomethionyl proteins for phase determination. Methods Enzymol. 276: 523–530. [PubMed] [Google Scholar]
- Emsley P. and Cowtan, K. 2004. Coot: Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60: 2126–2132. [DOI] [PubMed] [Google Scholar]
- Erbel P., Schiering, N., D'Arcy, A., Renatus, M., Kroemer, M., Lim, S.P., Yin, Z., Keller, T.H., Vasudevan, S.G., and Hommel, U. 2006. Structural basis for the activation of flaviviral NS3 proteases from dengue and West Nile virus. Nat. Struct. Mol. Biol. 13: 372–373. [DOI] [PubMed] [Google Scholar]
- Falgout B., Pethel, M., Zhang, Y.M., and Lai, C.J. 1991. Both nonstructural proteins NS2B and NS3 are required for the proteolytic processing of dengue virus nonstructural proteins. J. Virol. 65: 2467–2475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fersht A. 1977. Enzyme structure and mechanism. W.H. Freeman and Company, Reading and San Francisco.
- Grakoui A., McCourt, D.W., Wychowski, C., Feinstone, S.M., and Rice, C.M. 1993. A second hepatitis C virus-encoded proteinase. Proc. Natl. Acad. Sci. 90: 10583–10587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison S.C. 1996. Peptide-surface association: The case of PDZ and PTB domains. Cell 86: 341–343. [DOI] [PubMed] [Google Scholar]
- Hedstrom L. 2002. Serine protease mechanism and specificity. Chem. Rev. 102: 4501–4524. [DOI] [PubMed] [Google Scholar]
- Jing H., Xu, Y., Carson, M., Moore, D., Macon, K.J., Volanakis, J.E., and Narayana, S.V. 2000. New structural motifs on the chymotrypsin fold and their potential roles in complement factor B. EMBO J. 19: 164–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J.L., Morgenstern, K.A., Lin, C., Fox, T., Dwyer, M.D., Landro, J.A., Chambers, S.P., Markland, W., Lepre, C.A., O'Malley, E.T., et al. 1996. Crystal structure of the hepatitis C virus NS3 protease domain complexed with a synthetic NS4A cofactor peptide. Cell 87: 343–355. [DOI] [PubMed] [Google Scholar]
- Koshland D.E. 1958. Application of a theory of enzyme specificity to protein synthesis. Proc. Natl. Acad. Sci. 44: 98–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski R.A., MacArthur, M.W., Moss, D.S., and Thornton, J.M. 1993. PROCHECK: A program to check the sterochemical quality of protein structures. J. Appl. Crystallogr. 26: 283–291. [Google Scholar]
- Li J., Lim, S.P., Beer, D., Patel, V., Wen, D., Tumanut, C., Tully, D.C., Williams, J.A., Jiricek, J., Priestle, J.P., et al. 2005. Functional profiling of recombinant NS3 proteases from all four serotypes of dengue virus using tetrapeptide and octapeptide substrate libraries. J. Biol. Chem. 280: 28766–28774. [DOI] [PubMed] [Google Scholar]
- Lin C., Thomson, J.A., and Rice, C.M. 1995. A central region in the hepatitis C virus NS4A protein allows formation of an active NS3-NS4A serine proteinase complex in vivo and in vitro. J. Virol. 69: 4373–4380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matusan A.E., Kelley, P.G., Pryor, M.J., Whisstock, J.C., Davidson, A.D., and Wright, P.J. 2001. Mutagenesis of the dengue virus type 2 NS3 proteinase and the production of growth-restricted virus. J. Gen. Virol. 82: 1647–1656. [DOI] [PubMed] [Google Scholar]
- McRee D.E. 1999. XtalView/Xfit—A versatile program for manipulating atomic coordinates and electron density. J. Struct. Biol. 125: 156–165. [DOI] [PubMed] [Google Scholar]
- Murshudov G.N., Vagin, A.A., and Dodson, E.J. 1997. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53: 240–255. [DOI] [PubMed] [Google Scholar]
- Murthy H.M., Clum, S., and Padmanabhan, R. 1999. Dengue virus NS3 serine protease. Crystal structure and insights into interaction of the active site with substrates by molecular modeling and structural analysis of mutational effects. J. Biol. Chem. 274: 5573–5580. [DOI] [PubMed] [Google Scholar]
- Murthy H.M., Judge, K., DeLucas, L., and Padmanabhan, R. 2000. Crystal structure of Dengue virus NS3 protease in complex with a Bowman-Birk inhibitor: Implications for flaviviral polyprotein processing and drug design. J. Mol. Biol. 301: 759–767. [DOI] [PubMed] [Google Scholar]
- Nall T.A., Chappell, K.J., Stoermer, M.J., Fang, N.X., Tyndall, J.D., Young, P.R., and Fairlie, D.P. 2004. Enzymatic characterization and homology model of a catalytically active recombinant West Nile virus NS3 protease. J. Biol. Chem. 279: 48535–48542. [DOI] [PubMed] [Google Scholar]
- Niyomrattanakit P., Winoyanuwattikun, P., Chanprapaph, S., Angsuthanasombat, C., Panyim, S., and Katzenmeier, G. 2004. Identification of residues in the dengue virus type 2 NS2B cofactor that are critical for NS3 protease activation. J. Virol. 78: 13708–13716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otlewski J., Jaskolski, M., Buczek, O., Cierpicki, T., Czapinska, H., Krowarsch, D., Smalas, A.O., Stachowiak, D., Szpineta, A., and Dadlez, M. 2001. Structure–function relationship of serine protease–protein inhibitor interaction. Acta Biochim. Pol. 48: 419–428. [PubMed] [Google Scholar]
- Otwinowski Z. and Minor, W. 1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276: 307–326. [DOI] [PubMed] [Google Scholar]
- Pasternak A., Ringe, D., and Hedstrom, L. 1999. Comparison of anionic and cationic trypsinogens: The anionic activation domain is more flexible in solution and differs in its mode of BPTI binding in the crystal structure. Protein Sci. 8: 253–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasternak A., White, A., Jeffery, C.J., Medina, N., Cahoon, M., Ringe, D., and Hedstrom, L. 2001. The energetic cost of induced fit catalysis: Crystal structures of trypsinogen mutants with enhanced activity and inhibitor affinity. Protein Sci. 10: 1331–1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perona J.J. and Craik, C.S. 1995. Structural basis of substrate specificity in the serine proteases. Protein Sci. 4: 337–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrakis A., Harkiolaki, M., Wilson, K.S., and Lamzin, V.S. 2001. ARP/wARP and molecular replacement. Acta Crystallogr. D Biol. Crystallogr. 57: 1523–1526. [DOI] [PubMed] [Google Scholar]
- Read R.J. 2001. Pushing the boundaries of molecular replacement with maximum likelihood. Acta Crystallogr. D Biol. Crystallogr. 57: 1373–1382. [DOI] [PubMed] [Google Scholar]
- Rice C.M. 1996. Flaviviridae: The viruses and their replication. Lippincott-Raven, Philadelphia/New York.
- Schneider T.R. and Sheldrick, G.M. 2002. Substructure solution with SHELXD. Acta Crystallogr. D Biol. Crystallogr. 58: 1772–1779. [DOI] [PubMed] [Google Scholar]
- Shiryaev S.A., Ratnikov, B.I., Chekanov, A.V., Sikora, S., Rozanov, D.V., Godzik, A., Wang, J., Smith, J.W., Huang, Z., Lindberg, I., et al. 2006. Cleavage targets and the D-arginine-based inhibitors of the West Nile virus NS3 processing proteinase. Biochem. J. 393: 503–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speight G., Coia, G., Parker, M.D., and Westaway, E.G. 1988. Gene mapping and positive identification of the non-structural proteins NS2A, NS2B, NS3, NS4B and NS5 of the flavivirus Kunjin and their cleavage sites. J. Gen. Virol. 69: 23–34. [DOI] [PubMed] [Google Scholar]
- Terwilliger T.C. 2003. SOLVE and RESOLVE: Automated structure solution and density modification. Methods Enzymol. 374: 22–37. [DOI] [PubMed] [Google Scholar]
- Wu C.F., Wang, S.H., Sun, C.M., Hu, S.T., and Syu, W.J. 2003. Activation of dengue protease autocleavage at the NS2B-NS3 junction by recombinant NS3 and GST-NS2B fusion proteins. J. Virol. Methods 114: 45–54. [DOI] [PubMed] [Google Scholar]
- Xu J., Mendez, E., Caron, P.R., Lin, C., Murcko, M.A., Collett, M.S., and Rice, C.M. 1997. Bovine viral diarrhea virus NS3 serine proteinase: Polyprotein cleavage sites, cofactor requirements, and molecular model of an enzyme essential for pestivirus replication. J. Virol. 71: 5312–5322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Y., Li, Y., Munshi, S., Sardana, V., Cole, J.L., Sardana, M., Steinkuehler, C., Tomei, L., De Francesco, R., Kuo, L.C., et al. 1998. Complex of NS3 protease and NS4A peptide of BK strain hepatitis C virus: A 2.2 A resolution structure in a hexagonal crystal form. Protein Sci. 7: 837–847. [DOI] [PMC free article] [PubMed] [Google Scholar]