

Abstract
We have developed a bacterial two-hybrid system for the detection of interacting proteins that capitalizes on the folding quality control mechanism of the Twin Arginine Transporter (Tat) pathway. The Tat export pathway is responsible for the membrane translocation of folded proteins, including proteins consisting of more than one polypeptide, only one of which contains a signal peptide (“hitchhiker export”). Here, one protein (bait) is expressed as a fusion to a Tat signal peptide, whereas the second protein (prey) is fused to a protein reporter that can confer a phenotype only after export into the bacterial periplasmic space. Since the prey–reporter fusion lacks a signal peptide, it can only be exported as a complex with the bait–signal peptide fusion that is capable of targeting the Tat translocon. Using maltose-binding protein as a reporter, clones expressing interacting proteins can be grown on maltose minimal media or on MacConkey plates. In addition, we introduce the use of the cysteine disulfide oxidase DsbA as a reporter. Export of a signal peptide–prey:bait–DsbA complex into the periplasm allows complementation of dsbA− mutants and restores the formation of active alkaline phosphatase, which in turn can be detected by a chromogenic assay.
Keywords: protein interaction, Tat pathway, protein export, two-hybrid system, folding, fusion protein, maltose-binding protein, DsbA
In recent years, the numbers of genes identified in a broad spectrum of organisms has grown exponentially. A major challenge will be categorizing the corresponding proteins into their functional units within the cell. Several approaches have been used to assign newly identified proteins into cellular networks (Galperin and Koonin 2000; Bork et al. 2004; de Lichtenberg et al. 2005). In vitro, interacting proteins can be detected by mass spectrometry or by chromatographic techniques, typically following genetic fusion with an appropriate affinity tag (Rigaut et al. 1999; Gavin et al. 2002; Butland et al. 2005). In vivo, genetic techniques for the identification of interacting proteins rely mainly on two-hybrid systems and protein complementation assays. A number of such techniques have been developed and used extensively in Escherichia coli (Hu 2001; Karimova et al. 2002, 2005). So far, the detection of protein interactions in bacteria has capitalized on fusions to transcriptional repressors such as λcI, LexA, or AraC, transcriptional activators (involving the recruitment of RNA polymerase or the dimerization of the Vibrio cholera ToxR), complementation of biosynthetic enzymes such as dihydrofolate reductase, or signaling enzymes, e.g., the Bordetella pertussis adenylate cyclase. Ultimately, the binding of bait and prey is manifested as a change in the activity of a reporter enzyme that in turn allows for colony formation on selective plates, or it can be measured quantitatively using chromogenic substrates. A common complication with genetic methods for the discovery of interacting proteins is a high rate of false positives (Fields 2005). False positives can result when one of the two partner proteins alone can trigger a change in the activity of the reporter; for example, through spurious binding to DNA that alters transcriptional activation/repression, or as a result of nonspecific binding events. Misfolding of the prey or bait gives rise to “sticky” proteins having exposed hydrophobic regions that can engage in nonspecific interactions and therefore result in false positives.
Here, we present the development of a bacterial two-hybrid system that capitalizes on the biological folding quality control mechanism that is inherent to the Twin Arginine Translocation (Tat) pathway for protein export. The Tat pathway of bacteria, archaea, and plants is responsible for the translocation of folded proteins across energy-transducing membranes (Berks 1996; Palmer et al. 2004; Lee et al. 2006; Sargent et al. 2006). In that regard, the Tat pathway is fundamentally different from the general secretory (Sec) pathway in which proteins traverse membranes in an unfolded conformation by threading through the SecYEG (Sec61αβγ in eukaryotes) pore. The Tat pathway mediates the export of proteins that must assemble with cofactors within the reducing environment of the cytoplasm. In addition, certain Tat precursors associate with other polypeptides in the cytoplasm that do not contain signal peptides. In a process called “hitchhiker export,” the two polypeptides form a complex that is targeted to the Tat translocation apparatus and then is exported to the periplasm by virtue of the signal peptide in the precursor (Rodrigue et al. 1996). In the absence of the assembly partner or when cofactor assembly has been impaired by mutation, protein export through Tat does not take place (Rodrigue et al. 1996; Halbig et al. 1999). These observations together with in vitro and genetic evidence point to a protein folding quality control mechanism that is intrinsic to Tat translocation (DeLisa et al. 2003). As a result of the folding quality control feature of the Tat pathway, only folded proteins are accepted for translocation across the membrane. This feature of the Tat pathway was recently exploited to develop a screen for protein solubility (Fisher et al. 2006).
We reasoned that the ability of the Tat pathway to discriminate against misfolded polypeptides may be useful for protein interaction analysis. Thus, we developed a two-hybrid system that capitalizes on “hitchhiker export” as well as on the folding quality control features of Tat (Fig. 1A). We have used two reporter systems for the detection of protein:protein interactions via the “Tat two-hybrid assay”: one based on growth on selective media or MacConkey plates, and a second based on an enzymatic assay that relies on the oxidation of alkaline phosphatase in the periplasm, resulting in the formation of active enzyme that is then detected using a chromogenic substrate.
Figure 1.
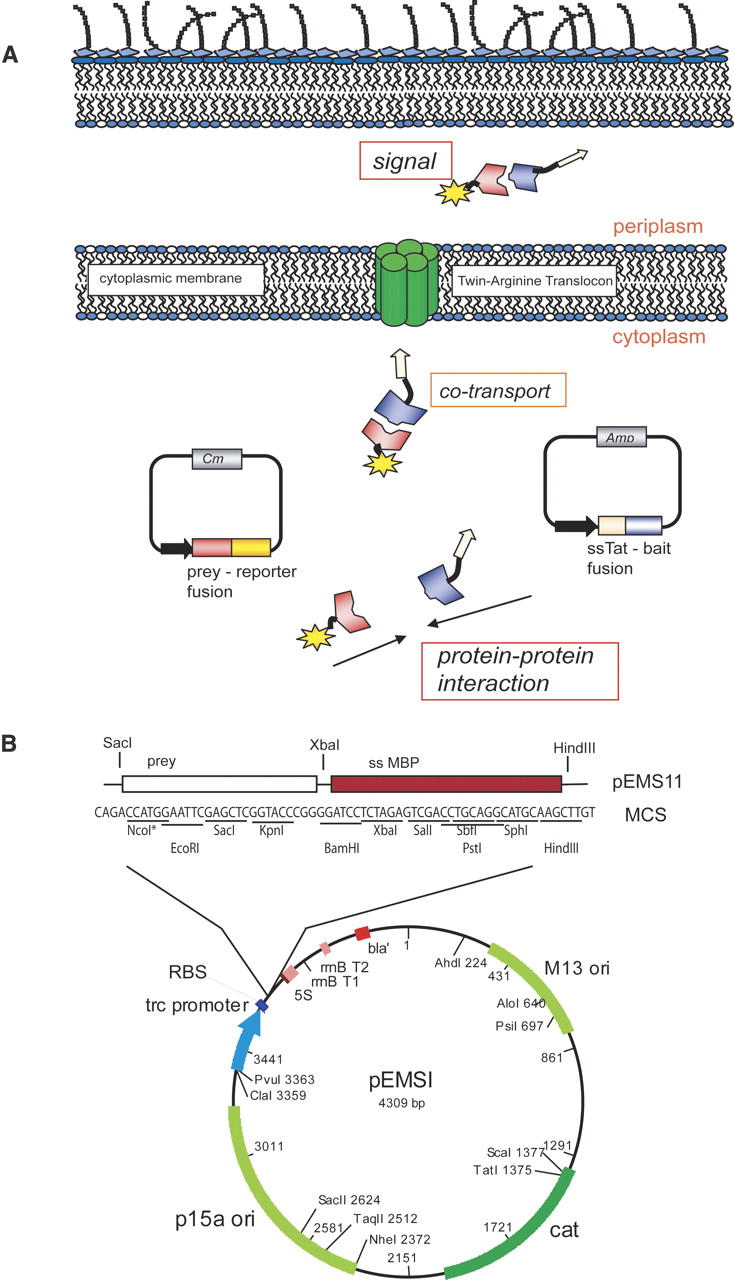
The Tat two-hybrid assay. (A) A pACYC-derivative plasmid expresses the prey fusion to a reporter protein; the fusion can be either C- or N-terminal to the reporter. A compatible pBR322 replicon expresses the bait C-terminal of a Tat signal peptide. Interaction of the bait and prey allows export of the resulting complex through the twin-arginine transporter. In the periplasm, the specific activity of the reporter can be detected. (B) Map of plasmid pEMSI. pEMSI is a derivative of pBAD33 with a trc promoter and the pACYC (p15a) origin of replication. The multiple cloning site (MCS) is indicated. NcoI and EcoRI are not unique in this construct. Plasmid was used for maltose-binding protein assays.
Results
Proteins destined for export via the Tat machinery are synthesized as precursors with an N-terminal signal peptide 30–50 amino acids in length and containing a conserved S/T-RRFLK motif near the N terminus. The Tat pathway was named after the nearly invariant twin arginine dipeptide sequence in the conserved motif of the signal peptide. In Figure 1A, the bait is fused in-frame to the C terminus of a Tat signal peptide. One of the best studied Tat signal peptides is ssTorA of the E. coli trimethylamine N-oxide reductase (TorA), which was selected for the present study. The prey is expressed as a fusion to a reporter that has to be translocated to the periplasm for function. Since the prey–reporter fusion does not contain a signal peptide, it is unable to interact with the Tat translocation pore and therefore remains sequestered within the cytoplasm. However, binding of the prey to the signal peptide–bait fusion results in the formation of a protein complex that can be targeted to the Tat transolocon and is exported into the periplasm.
The maltose-binding protein, MBP, is encoded by the malE gene and has been used extensively as a fusion partner in protein export studies and to aid protein solubility (Boos and Shuman 1998; Kapust and Waugh 1999; Blaudeck et al. 2003). The export of MBP in the periplasm can be detected easily on MacConkey agar plates or by growth on maltose as the sole carbon source. Signal sequenceless ΔssMBP prey fusions were expressed under the control of the ptrc promoter in the low-copy-number plasmid pEMSI (pACYC origin of replication). The signal peptide–bait fusion was similarly transcribed from a ptrc promoter on a compatible plasmid vector containing a pBR322 origin. To avoid cell toxicity due to the high level of expression of the two fusion proteins, we relied on leaky transcription from the ptrc promoter in the absence of inducer. As discussed below, the basal level of transcription from uninduced cells is sufficient to give a high signal to noise ratio.
As model bait and prey proteins, we used the leucine zipper domains of the transcription factor wbfos (rat, amino acids 161–200) and wbjun (rat, amino acids 227–315), respectively (Abate et al. 1990; Crameri and Suter 1993). These two domains represent one of the prototypical systems for interacting proteins (Kouzarides and Ziff 1988) with a KD of 70 nM (Oyama et al. 2006). Plasmids encoding wbFos-ΔssMBP (pEMS11) and ssTorA-wbJun (pEMS12) were transformed into the E. coli malE− strain HS3018 and plated on MacConkey agar plates. Bacteria containing either plasmid alone gave rise to yellow–orange colonies, revealing their inability to utilize maltose as the carbon source. In contrast, cells cotransformed with pEMS11 and pEMS12 acquired the ability to grow on maltose and formed red colonies after growth for 14 h at 37°C (Fig. 2A2). Substitution of the twin arginine dipeptide with a pair of lysines completely abolishes Tat export (DeLisa et al. 2002; Ize et al. 2002): Cells expressing the ssTorA(KK)-wbJun (pEMS16) gave rise to yellow–orange colonies (Fig. 2A1), indicating that the mal+ phenotype shown in Figure 2A2 was dependent on Tat export. Additionally, only cells transformed with both plasmids and encoding a wild-type ssTorA signal sequence formed colonies on minimal media plates with 0.4% maltose as the carbon source (Fig. 2A4). No colony growth was observed for the ssTorA(KK) fusion, whereas the wild-type ssTorA showed growth after 4 d, consistent with the phenotype observed on MacConkey plates (Fig. 2A3).
Figure 2.

Detection of interacting proteins via Tat two-hybrid using MalE or DsbA as reporters. (A) Use of MalE as a reporter. (1) E. coli HS3018 (malE−) cells coexpressing ssTorA(KK)-wbJun fusion with a nonfunctional Tat signal peptide (from pEMS16) and wbFos-MBP (from pEMS11) grown on MacConkey plates with 0.4% maltose; (2) E. coli HS3018 (malE−) coexpressing ssTorA-wbJun (pEMS12) with wbFos-MBP (pEMS11) grown on MacConkey plates with 0.4% maltose; (3) cells as in panel 1 plated on M9 media with 0.4% maltose and incubated for 4 d at 37°C; (4) Cells as in panel 2 grown on maltose minimal media plates as in panel 3. (B) Detection of protein:protein interactions by an enzymatic assay using DsbA as a reporter: Interaction of the prey–ΔssDsbA fusion with ssTorA–bait results in the formation of a complex that is translocated into the periplasm by the Tat pathway via the ssTorA signal peptide. In the periplasm, DsbA catalyzes the oxidative folding of AP, the activity of which can be detected using the chromogenic substrate pNPP. (C) Alkaline phosphatase activity in arbitrary units (au). Cells were grown in MOPS low-phosphate media with or without 0.04% arabinose, harvested after 6 h, and the AP activity was measured as described in the Materials and Methods. (MC1000) E. coli positive control; (MCA) MC1000 ΔdsbA∷kan, negative control; (Im2) E. coli MCA expressing ΔssDsbA-Im2 from pEMS14; (Fos) MCA expressing wbFos-ΔssDsbA from pEMS13; (Im2:E2) coexpression of the ΔssDsbA-immunity protein 2 fusion from pEMS14 together with the ssTorA-endonuclease domain of colicin E2 (H575A) fusion from pEMS15; (Fos:Jun) coexpression of ssTorA-wbJun from pEMS12 with the wbFos-ΔssDsbA fusion (pEMS13); (Jun:Im2) coexpression of ssTorA-Jun from pEMS12 with immunity protein2 fused to ΔssDsbA (pEMS14).
The above results indicate that using MBP as a reporter and growth on selective or indicator plates can be used to detect the interaction of two proteins. We then sought to develop an enzymatic assay for the export of the ssTorA-bait:prey–reporter complex into the periplasm. However, we could not take advantage of the widely used periplasmic reporter alkaline phosphatase (AP) because it has to fold in the cytoplasm before it can be exported via Tat (DeLisa et al. 2003). Cytoplasmic folding of AP can be accomplished only in specialized oxidizing mutant strains (Bessette et al. 1999). In addition, the large size of the folded AP (94 kDa) (Bradshaw et al. 1981) limits the size of the bait and prey proteins in the fusions because it is unlikely that the Tat pore can accommodate signal peptide–bait:prey– complexes >160–180 kDa (Santini et al. 1998). Similarly, β-lactamase, which has been used as a periplasmic reporter enzyme, is not suitable for our purposes because: (a) β-lactamase can fold into an active conformation within the cytoplasm, and therefore the enzymatic activity in total cell lysates is not indicative of export; and (b) a low level of export and resistance to ampicilin are observed in cells expressing signal sequence-less β-lactamase (Bowden et al. 1992).
We elected to investigate the utility of the periplasmic cysteine:disulfide oxidoreductase DsbA as a periplasmic reporter (Fig. 2B). In bacteria, the oxidation of protein thiols to form disulfide bonds normally occurs in the periplasm and is catalyzed by DsbA (Kadokura et al. 2003). DsbA, a thioredoxin superfamily protein, is a highly efficient catalyst of disulfide bond formation. Its activity depends on recycling by the membrane-bound DsbB enzyme, which in turn transfers electrons to the quinones and, ultimately, to the respiratory chain (Nakamoto and Bardwell 2004). The folding of alkaline phosphatase in the periplasm is dependent on oxidation by DsbA. In the E. coli dsbA− mutant MCA (MC1000 ΔdsbA∷kan) grown in low-phosphate MOPS minimum media, AP activity is ∼20-fold lower compared with the isogenic control (Fig. 2C). The level of residual AP activity is partly dependent on the Fe(II) concentration in the growth media (data not shown).
wbFos-ΔssDsbA was expressed from the arabinose-inducible promoter in plasmid pEMS13 to allow differential expression of the bait and prey fusion proteins. Plasmids pEMS12 and pEMS13 were cotransformed into E. coli MCA and grown in low-phosphate media to induce the expression of AP from the chromosomal phoA gene. The synthesis of the wbFos-ΔssDsbA expression from pEMS13 was induced during the logarithmic phase (O.D.600 ∼0.4) with 0.04% arabinose. Cells were harvested ∼6 h after induction and lysed, free thiols were blocked with the sulfhydryl alkylating agent iodoacetamide to prevent oxidation of any reduced AP after lysis (Derman and Beckwith 1991), and the enzymatic activity was determined using the chromogenic substrate pNPP (Fig. 2C). Cells expressing wbFos-ΔssDsbA alone had an AP level slightly above background, whereas coexpression of ssTorA-wbJun from pEMS12 resulted in nearly complete restoration of AP activity to the level observed in cells expressing authentic DsbA from the chromosomal gene. Colicin E2 (E2) and the immunity protein 2 (Im2) constitute a pair of toxin:antitoxin proteins that interact with high affinity (KD = 10−15 M). Im2 binds to the endonuclase domain of Colicin E2 with a 1:1 stoichiometry to inhibit cleavage of DNA prior to export from the cytoplasm. Garinot-Schneider and coworkers (1996) described several mutations that abolish DNase activity. A gene encoding the 86-amino-acid Im2 protein was fused 3′ of the ΔssdsbA gene, giving rise to pEMS14. The C-terminal cloning of Im2 introduced the same restriction sites as those used for the signal peptide–bait plasmid; thus, bait and prey genes can be easily interchanged between the two plasmids encoding the signal peptide and the reporter fusion. The H575A mutation that disables the active site was introduced in ColE2 (amino acids 479–582), and the resulting gene was fused to the C terminus of ssTorA (pEMS15). Cells expressing both ssTorA-E2 in pEMS15 and ΔssDsbA-Im2 in pEMS14 exhibited high AP activity (25-fold above background, Fig. 2C). As expected, expression of ΔssDsbA-Im2 alone gave a very low-level signal. We further examined whether the expression of noninteracting proteins would give us any AP activity. We tested wbFos-ΔssDsbA with the endonuclease domain of colicin E2, ssTorA-wbJun with immunity protein 2, and additionally examined whether the immunity protein could have any dimerization properties using ssTorA-Im2 and Im2-ΔssDsbA. None of these three protein fusion pairs gave signal above background (Fig. 2C; data not shown).
Discussion
We have developed a new genetic assay for the detection of protein:protein interactions by capitalizing on the unique features of the Tat export pathway of E. coli. To the best of our knowledge, this is the first protein interaction assay that takes advantage of protein translocation across an energy-transducing membrane, namely the cytoplasmic membrane of E. coli. Extensive studies have shown that only folded proteins can be translocated via the Tat pathway, including proteins comprising of two or more interacting polypeptides, only one of which contains a signal sequence. Rodrigue et al. (1999) first coined the term “hitchhiker export” after observing that the large subunit HybC of the E. coli hydrogenase 2, which does not contain a signal peptide, was translocated as a complex with HybO, which contains a 37-amino-acid-long Tat signal peptide. More recent studies demonstrated that dimeric FAB antibody fragments can also be exported by a hitchhiker mechanism via a signal peptide on only one of the two polypeptides that comprise the FAB protein (DeLisa et al. 2003). In our approach, one protein is fused to a Tat signal peptide and the second protein to a protein reporter that can confer a phenotype only upon export into the periplasmic space. We demonstrated the detection of interacting proteins via the Tat two-hybrid system using two periplasmic reporters: maltose-binding protein, the export of which can be detected either by selecting for growth on maltose or on MacConkey plates; and DsbA, which catalyzes the formation of active AP in the periplasm. The two reporters allow the detection of protein interactions by growth on selective media, on indicator plates, or by enzymatic assays. Both MBP and DsbA have been used as fusion partners to increase solubility, a feature that can be important for proteins that are susceptible to aggregation. Furthermore, MBP fusions can be easily purified by one-step affinity chromatography on amylose resins, thus expediting subsequent in vitro analyses of protein interactions.
Perhaps the most interesting and useful feature of the Tat two-hybrid assay is that it takes advantage of the requirement that proteins have to be folded in order to be competent for export via the Tat transporter. Neither the detailed mechanism nor the features of the folded polypeptide that are interrogated to determine export competence are understood at the moment. However, Fisher et al. (2006) showed that Tat selects for proteins exhibiting higher solubility. We therefore believe that the Tat two-hybrid assay is likely to be useful for reducing false positives that can arise from the nonspecific interactions or aggregation of noninteracting polypeptides, as well as spurious positives arising from the properties of a single partner (e.g., inadvertent activation/repression of transcription or stress responses that result in a signal).
During the early steps in their biogenesis, proteins destined for Tat export interact with signal-sequence-specific (e.g., TorD) or general chaperones such as DnaK (Graubner et al. 2007; Perez-Rodriguez et al. 2007). There is no evidence that these cytoplasmic chaperones are exported together with their target polypeptide. Consequently, weak interactions, such as those between chaperones and their substrates, are not likely to be detected by the Tat two-hybrid system. This may not be undesirable for studies aimed at finding specific binding partners. The lower limit of protein affinities required to give a positive signal is currently under investigation.
The Tat two-hybrid is expected to give false negatives, i.e., fail to detect interacting proteins, in two instances: first, when the interacting protein partners form a complex that is larger than the size limit that can be accommodated by the Tat pore (<180 kDa). Given that the DsbA reporter has a molecular weight of 21 kDa, this means that the total size of the complex formed by the interacting proteins must be <160 kDa. Second, false negatives may arise when one of the two partner polypeptides exhibits folding kinetics that are substantially slower than the typical transit times for Tat export. When the folding kinetics are slow, the off-pathway reactions leading to aggregation in the cytoplasm predominate and interfere with Tat export (B. Ribnicky, T. Van Blarcom, and G. Georgiou, in prep.).
In conclusion, the Tat two-hybrid assay could improve the accuracy of proteome-wide two-hybrid analyses. In addition, a further useful biotechnological application for the Tat two-hybrid system will be in screening libraries of mutant polypeptides to isolate clones that bind to a target protein with increased affinity or to increase solubility while still binding to its interaction partner.
Material and Methods
Plasmid constructions
Strains and plasmids used in this work are summarized in Table 1 and primers in Supplemental Table S1. Plasmid pEMSI was constructed by removing the araC regulator gene, the pBAD operator, and promoter region of pBAD33 (Guzman et al. 1995) and replacing them with a 220-bp fragment 5′ of the ATG codon in pTrc99A. The latter fragment (with ClaI and HindIII restriction sites) was amplified with the primers EMS11 and EMS12.
Table 1.
Bacterial strains and plasmids

The intermediate plasmid construct pTrc99-ssTorA was generated by amplifying sstorA with primers EMS13 and EMS14 and inserting it into pTrc99 between the NcoI and XbaI sites. wbjun was amplified with primers EMS16 and EMS17 and ligated into the construct pTrc99-ssTorA using the XbaI and HindIII sites, giving rise to pEMS12. The KK version of the TorA signal peptide was generated as above, but using EMS15 as 5′ primer, generating plasmid pEMS16. wbfos was amplified with EMS110 and EMS111, and ΔssmalE with EMS18 and EMS19. wbfos was inserted upstream of ΔssmalE into pEMSI using the SacI, XbaI, and HindIII cut sites for these two genes, resulting in pEMS11. For the construction of the wbFos-ΔssDsbA fusions, wbfos was amplified with the primers EMS112 and EMS111, digested with SacI and XbaI, and inserted into pBAD33. In addition, ΔssdsbA was cloned directly downstream using the primers EMS113 and EMS114, producing pEMS13. The DNase domain of colicin E2 was amplified from the plasmid ColE2-P9 (Mock and Schwartz 1978) with the primers EMS115 and EMS116 containing a codon change of H575A and a 5′ strep2 tag sequence. After digestion with XbaI and HindIII, the DNase domain was inserted downstream of ΔsstorA in pTrc99-ssTorA, generating pEMS15.
Plasmid pEMS14 was generated by PCR amplification of ΔssdsbA with the primers EMS117 and EMS118 using genomic DNA and then insertion of ΔssdsbA in between the SacI and XbaI site of pBAD33, whereas immunity protein 2 was inserted downstream of ΔssdsbA between XbaI and HindIII using primers EMS119 and EMS120 containing an additional Flag tag at the 3′ site. The 5′ position of ΔssdsbA allows an easy exchange of bait and prey since the restriction sites are compatible with the signal peptide construct.
Alkaline phosphate activity assays
Cells were grown overnight in low-phosphate MOPS minimum media (Sambrook et al. 2000) with Peptone P (USBiological) as an amino acid source, subcultured (1/20) into 1 mL of low-phosphate medium and induced with 0.04% arabinose at OD600 0.4. Around 6 h after induction, cultures were diluted with media to OD600 1.0; 20 μL of cell cultures were transferred to a 96-well plate and mixed with 30 μL of lysis buffer, a 2:1 mixture of B-PER (78,248 Pierce Biotechnology, Inc.) with 0.4 M iodoacetamide (Sigma) for 30 min. As substrate for AP, 200 μL of 250 μg/mL p-nitrophenyl phosphate (Sigma) in 0.250 M Tris-HCl pH 8 was added to each well and hydrolysis was measured at A405 on a plate reader (BioTek).
Acknowledgments
We thank Dr. Lluis Masip for helpful discussions, SangTaek Jun for bringing the Im2:ColE2 protein pair to our attention, and Dr. Jamie Link for comments on the manuscript. The research was funded by NIH 1 R01 GM069872 and by the Foundation for Research.
Footnotes
Supplemental material: see www.proteinscience.org
Reprint requests to: George Georgiou, Chemical Engineering/UT Austin, 1 University Station C0400, Austin, TX 78712-0231, USA; e-mail: gg@che.utexas.edu; fax: (512) 471-7963.
Abbreviations: Tat, twin-arginine translocation; Sec, general secretory pathway; TorA, trimethylamine N-oxide reductase; AP, alkaline phosphatase; MBP, maltose-binding protein; Im2, immunity protein 2; E2, endonuclease domain of colicin E2; MCS, multiple cloning site.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.062687207.
References
- Abate C., Luk, D., Gentz, R., Rauscher 3rd, F.J., and Curran, T. 1990. Expression and purification of the leucine zipper and DNA-binding domains of Fos and Jun: Both Fos and Jun contact DNA directly. Proc. Natl. Acad. Sci. 87: 1032–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berks B.C. 1996. A common export pathway for proteins binding complex redox cofactors? Mol. Microbiol. 22: 393–404. [DOI] [PubMed] [Google Scholar]
- Bessette P.H., Aslund, F., Beckwith, J., and Georgiou, G. 1999. Efficient folding of proteins with multiple disulfide bonds in the Escherichia coli cytoplasm. Proc. Natl. Acad. Sci. 96: 13703–13708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaudeck N., Kreutzenbeck, P., Freudl, R., and Sprenger, G.A. 2003. Genetic analysis of pathway specificity during posttranslational protein translocation across the Escherichia coli plasma membrane. J. Bacteriol. 185: 2811–2819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boos W. and Shuman, H. 1998. Maltose/maltodextrin system of Escherichia coli: Transport, metabolism, and regulation. Microbiol. Mol. Biol. Rev. 62: 204–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bork P., Jensen, L.J., von Mering, C., Ramani, A.K., Lee, I., and Marcotte, E.M. 2004. Protein interaction networks from yeast to human. Curr. Opin. Struct. Biol. 14: 292–299. [DOI] [PubMed] [Google Scholar]
- Bowden G.A., Baneyx, F., and Georgiou, G. 1992. Abnormal fractionation of β-lactamase in Escherichia coli: Evidence for an interaction with the inner membrane in the absence of a leader peptide. J. Bacteriol. 174: 3407–3410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradshaw R.A., Cancedda, F., Ericsson, L.H., Neumann, P.A., Piccoli, S.P., Schlesinger, M.J., Shriefer, K., and Walsh, K.A. 1981. Amino acid sequence of Escherichia coli alkaline phosphatase. Proc. Natl. Acad. Sci. 78: 3473–3477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butland G., Peregrin-Alvarez, J.M., Li, J., Yang, W., Yang, X., Canadien, V., Starostine, A., Richards, D., Beattie, B., Krogan, N., et al. 2005. Interaction network containing conserved and essential protein complexes in Escherichia coli. Nature 433: 531–537. [DOI] [PubMed] [Google Scholar]
- Crameri R. and Suter, M. 1993. Display of biologically active proteins on the surface of filamentous phages: A cDNA cloning system for selection of functional gene products linked to the genetic information responsible for their production. Gene 137: 69–75. [DOI] [PubMed] [Google Scholar]
- de Lichtenberg U., Jensen, L.J., Brunak, S., and Bork, P. 2005. Dynamic complex formation during the yeast cell cycle. Science 307: 724–727. [DOI] [PubMed] [Google Scholar]
- DeLisa M.P., Samuelson, P., Palmer, T., and Georgiou, G. 2002. Genetic analysis of the twin arginine translocator secretion pathway in bacteria. J. Biol. Chem. 277: 29825–29831. [DOI] [PubMed] [Google Scholar]
- DeLisa M.P., Tullman, D., and Georgiou, G. 2003. Folding quality control in the export of proteins by the bacterial twin-arginine translocation pathway. Proc. Natl. Acad. Sci. 100: 6115–6120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derman A.I. and Beckwith, J. 1991. Escherichia coli alkaline phosphatase fails to acquire disulfide bonds when retained in the cytoplasm. J. Bacteriol. 173: 7719–7722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields S. 2005. High-throughput two-hybrid analysis. The promise and the peril. FEBS J. 272: 5391–5399. [DOI] [PubMed] [Google Scholar]
- Fisher A.C., Kim, W., and DeLisa, M.P. 2006. Genetic selection for protein solubility enabled by the folding quality control feature of the twin-arginine translocation pathway. Protein Sci. 15: 449–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galperin M.Y. and Koonin, E.V. 2000. Who's your neighbor? New computational approaches for functional genomics. Nat. Biotechnol. 18: 609–613. [DOI] [PubMed] [Google Scholar]
- Garinot-Schneider C., Pommer, A.J., Moore, G.R., Kleanthous, C., and James, R. 1996. Identification of putative active-site residues in the DNase domain of colicin E9 by random mutagenesis. J. Mol. Biol. 260: 731–742. [DOI] [PubMed] [Google Scholar]
- Gavin A.C., Bosche, M., Krause, R., Grandi, P., Marzioch, M., Bauer, A., Schultz, J., Rick, J.M., Michon, A.M., Cruciat, C.M., et al. 2002. Functional organization of the yeast proteome by systematic analysis of protein complexes. Nature 415: 141–147. [DOI] [PubMed] [Google Scholar]
- Graubner W., Schierhorn, A., and Bruser, T. 2007. DnaK plays a pivotal role in Tat targeting of CueO and functions beside SlyD as a general Tat signal binding chaperone. J. Biol. Chem. 282: 7116–7124. [DOI] [PubMed] [Google Scholar]
- Guzman L.M., Belin, D., Carson, M.J., and Beckwith, J. 1995. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J. Bacteriol. 177: 4121–4130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halbig D., Wiegert, T., Blaudeck, N., Freudl, R., and Sprenger, G.A. 1999. The efficient export of NADP-containing glucose-fructose oxidoreductase to the periplasm of Zymomonas mobilis depends both on an intact twin-arginine motif in the signal peptide and on the generation of a structural export signal induced by cofactor binding. Eur. J. Biochem. 263: 543–551. [DOI] [PubMed] [Google Scholar]
- Hu J.C. 2001. Model systems: Studying molecular recognition using bacterial n-hybrid systems. Trends Microbiol. 9: 219–222. [DOI] [PubMed] [Google Scholar]
- Ize B., Gerard, F., Zhang, M., Chanal, A., Voulhoux, R., Palmer, T., Filloux, A., and Wu, L.F. 2002. In vivo dissection of the Tat translocation pathway in Escherichia coli. J. Mol. Biol. 317: 327–335. [DOI] [PubMed] [Google Scholar]
- Kadokura H., Katzen, F., and Beckwith, J. 2003. Protein disulfide bond formation in prokaryotes. Annu. Rev. Biochem. 72: 111–135. [DOI] [PubMed] [Google Scholar]
- Kapust R.B. and Waugh, D.S. 1999. Escherichia coli maltose-binding protein is uncommonly effective at promoting the solubility of polypeptides to which it is fused. Protein Sci. 8: 1668–1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karimova G., Ladant, D., and Ullmann, A. 2002. Two-hybrid systems and their usage in infection biology. Int. J. Med. Microbiol. 292: 17–25. [DOI] [PubMed] [Google Scholar]
- Karimova G., Dautin, N., and Ladant, D. 2005. Interaction network among Escherichia coli membrane proteins involved in cell division as revealed by bacterial two-hybrid analysis. J. Bacteriol. 187: 2233–2243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouzarides T. and Ziff, E. 1988. The role of the leucine zipper in the fos–jun interaction. Nature 336: 646–651. [DOI] [PubMed] [Google Scholar]
- Lee P.A., Tullman-Ercek, D., and Georgiou, G. 2006. The bacterial twin-arginine translocation pathway. Annu. Rev. Microbiol. 60: 373–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mock M. and Schwartz, M. 1978. Mechanism of colicin E3 production in strains harboring wild-type or mutant plasmids. J. Bacteriol. 136: 700–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamoto H. and Bardwell, J.C. 2004. Catalysis of disulfide bond formation and isomerization in the Escherichia coli periplasm. Biochim. Biophys. Acta 1694: 111–119. [DOI] [PubMed] [Google Scholar]
- Oyama R., Takashima, H., Yonezawa, M., Doi, N., Miyamoto-Sato, E., Kinjo, M., and Yanagawa, H. 2006. Protein–protein interaction analysis by C-terminally specific fluorescence labeling and fluorescence cross-correlation spectroscopy. Nucleic Acids Res. 34: e102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer T., Sargent, F., and Berks, B.C. 2004. Light traffic: Photo-crosslinking a novel transport system. Trends Biochem. Sci. 29: 55–57. [DOI] [PubMed] [Google Scholar]
- Perez-Rodriguez R., Fisher, A.C., Perlmutter, J.D., Hicks, M.G., Chanal, A., Santini, C.L., Wu, L.F., Palmer, T., and Delisa, M.P. 2007. An essential role for the DnaK molecular chaperone in stabilizing over-expressed substrate proteins of the bacterial twin-arginine translocation pathway. J. Mol. Biol. 367: 715–730. [DOI] [PubMed] [Google Scholar]
- Rigaut G., Shevchenko, A., Rutz, B., Wilm, M., Mann, M., and Seraphin, B. 1999. A generic protein purification method for protein complex characterization and proteome exploration. Nat. Biotechnol. 17: 1030–1032. [DOI] [PubMed] [Google Scholar]
- Rodrigue A., Boxer, D.H., Mandrand-Berthelot, M.A., and Wu, L.F. 1996. Requirement for nickel of the transmembrane translocation of NiFe-hydrogenase 2 in Escherichia coli. FEBS Lett. 392: 81–86. [DOI] [PubMed] [Google Scholar]
- Rodrigue A., Chanal, A., Beck, K., Muller, M., and Wu, L.F. 1999. Co-translocation of a periplasmic enzyme complex by a hitchhiker mechanism through the bacterial tat pathway. J. Biol. Chem. 274: 13223–13228. [DOI] [PubMed] [Google Scholar]
- Sambrook J.C., Fritsch, E.F., and Maniatis, T. 2000. Molecular cloning: A laboratory manual, 3rd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Santini C.L., Ize, B., Chanal, A., Muller, M., Giordano, G., and Wu, L.F. 1998. A novel sec-independent periplasmic protein translocation pathway in Escherichia coli. EMBO J. 17: 101–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sargent F., Berks, B.C., and Palmer, T. 2006. Pathfinders and trailblazers: A prokaryotic targeting system for transport of folded proteins. FEMS Microbiol. Lett. 254: 198–207. [DOI] [PubMed] [Google Scholar]
- Shuman H.A. 1982. Active transport of maltose in Escherichia coli K 12. Role of the periplasmic maltose-binding protein and evidence for a substrate recognition site in the cytoplasmic membrane. J. Biol. Chem. 257: 5455–5461. [PubMed] [Google Scholar]