

Abstract
A nitroxide side chain (R1) has been substituted at single sites along a helix–turn–helix motif in T4 lysozyme (residues 114–135). Together with previously published data, the new sites reported complete a continuous scan through the motif. Mutants with R1 at sites 115 and 118 were selected for crystallographic analysis to identify the structural origins of the corresponding two-component EPR spectra. At 115, R1 is shown to occupy two rotamers in the room temperature crystal structure, one of which has not been previously reported. The two components in the EPR spectrum apparently arise from differential interactions of the two rotamers with the surrounding structure, the most important of which is a hydrophobic interaction of the nitroxide ring. Interestingly, the crystal structure at 100 K reveals a single rotamer, emphasizing the possibility of rotamer selection in low-temperature crystal structures. Residue 118 is at a solvent-inaccessible site in the protein core, and the structure of 118R1, the first reported for the R1 side chain at a buried site, reveals how the side chain is accommodated in an overpacked core.
Keywords: site-directed spin labeling, EPR spectroscopy, side chain conformation
Site-directed spin labeling (SDSL) has become a powerful tool to study protein structure and dynamics (Hubbell et al. 1996, 1998, 2000; Columbus and Hubbell 2002; Fanucci and Cafiso 2006). In SDSL, a cysteine residue is introduced at a selected site by site-directed mutagenesis and modified with a nitroxide reagent to generate a disulfide-linked nitroxide side chain. The most commonly used spin label side chain is designated R1 (Fig. 1), although other side chains have been used for specific purposes (Mchaourab et al. 1999; Altenbach et al. 2001; Columbus et al. 2001). The EPR spectra of R1, which reflect the motion of the nitroxide, vary greatly as a function of sequence position in folded proteins (Mchaourab et al. 1996). One goal of SDSL is to relate the EPR spectra to local structure and dynamics of the protein using well-characterized proteins of known structure. If this proves to be possible, EPR spectra of spin-labeled proteins can then be used to infer these features in less well-characterized proteins that may not be readily accessible to other methods. This has been largely achieved in the case of simple one-component EPR spectra characteristic of anisotropic motion that arise from R1 at noninteracting sites on the solvent-exposed surface of α-helices (Mchaourab et al. 1996, 1999; Columbus et al. 2001; Columbus and Hubbell 2002), on edge-strands of β-sheets (Lietzow and Hubbell 2004), and, as reported below, in ordered turns. At such sites, where the motion of R1 is weakly ordered, the motion is apparently determined entirely by the internal motion of the side chain and local backbone dynamics (Columbus et al. 2001). Thus, these weakly ordered states contain little direct information on the tertiary fold of the protein, but provide a means for mapping backbone dynamics (Columbus and Hubbell 2004).
Figure 1.

Structures of the nitroxide side chains R1 and R7 showing designation of the various atoms. For R1, the designation of the dihedral angles (χ) is also shown.
Complexity and diversity arise when the nitroxide of R1 is involved in interactions with nearby groups. In such cases, complex multicomponent spectra are found that reflect the secondary and tertiary fold of the protein (Mchaourab et al. 1996). The limit of strong interaction is seen for R1 at sites buried in the interior of the protein, where the EPR spectrum reflects strong immobilization of the nitroxide. In order to reliably interpret such complex EPR spectra of R1 in terms of protein structure and dynamics, it is necessary to determine the structural origins of the spectra and the level of structural perturbation due to R1 substitution in proteins of known structure. The aims of this study are to identify interactions that give rise to complex two-component spectra of R1 at solvent-exposed sites, and to determine the extent of perturbation when the protein accommodates the R1 side chain at a site buried in the hydrophobic core. As in a previous study, the problem is approached using X-ray crystallography of T4L bearing the R1 side chain (Langen et al. 2000). To identify sites suitable for structural studies, R1 was introduced along helix G of T4L (residues 114–123), in the turn connecting helices G and H (residues 124 and 125), and at residues 126 and 127 in helix H. Together with previous SDSL studies of helices G (residue 119) and H (residues 128−135) (Mchaourab et al. 1999), the new data complete a continuous scan of R1 through the helix–turn–helix motif from 114 to 135.
In helix G, residues 115R1 and 119R1 lie on the solvent-exposed surface and have complex two-component spectra, one of which reflects partial immobilization of the nitroxide. The crystal structure of T4L/119R1 at 100 K was previously reported (Langen et al. 2000), and the two components were associated with two rotameric states of the R1 side chain, but the rotamer corresponding to the more immobilized state and the nature of the interactions that give rise to immobilization were not positively identified. Here, the structures of T4L/115R1 at both 100 K and 298 K and the structure of T4L/115R1/R119A at 100 K are reported, and the immobilizing interaction is identified as largely nonspecific and hydrophobic. In addition, the first structure of R1 at a solvent-inaccessible site, T4L/118R1, is reported. In this structure, a short helical segment (helix F, residues 108−113) is rearranged to accommodate the R1 side chain, but the remainder of the structure is essentially identical to the wild-type (WT) protein. Interestingly, helix F is also rearranged upon ligand binding to an engineered cavity in approximately the same location occupied by the nitroxide in T4L/118R1 (Morton and Matthews 1995). Finally, the structure of the 4-Br derivative of R1 (designated R7) (Fig. 1) at site 115 is reported. Such 4-substituted derivatives show increased nitroxide ordering at helix surface sites (Altenbach et al. 2001; Columbus et al. 2001), and the crystal structure of T4L/115R7 provides insight to the structural origin of the increased order.
Results
R1 motion in a helix–turn–helix motif of T4L
Figure 2A shows the location of residues 114−127 at which R1 was introduced in the present study. Residue 114 is in a turn preceding the G-helix, residues 115−123 are in the G-helix proper, residues 124−125 are in the connecting loop between the G- and H-helices, and residues 126−127 are at the N terminus of the H-helix. Also shown for comparison are positions 119 in helix G and 128−135 along helix H and a following loop that were the subject of previous SDSL studies (Mchaourab et al. 1996, 1999). The EPR spectra of R1 at each position are shown in Figure 2B. The previously published spectra for sites 119 and 128−135 are shown for comparative purposes (shaded). Together, these sites complete a helix–turn–helix motif. In each case, the shape of the EPR spectrum reflects the motion of the nitroxide on the nanosecond time scale. One qualitative measure of nitroxide motion is the “scaled mobility” parameter, M s = (δ−1 − δi −1)/(δm −1 − δi −1), where δ is the peak-to-peak width of the central resonance line in the first-derivative spectrum of R1 (Fig. 2B) at the site of interest, δi is the corresponding width for a strongly immobilized R1, and δm is the corresponding width for R1 in a disordered sequence (Hubbell et al. 2000; Columbus and Hubbell 2002). M s varies from 0 to 1, increasing with increasing mobility. Figure 3 shows the plot of M s versus residue number. In helical segments G and H, there is a regular periodic variation of M s, revealing the helices. Loss of periodicity at sites 124 and 125, along with high M s, identifies the turn region that connects helices G and H.
Figure 2.
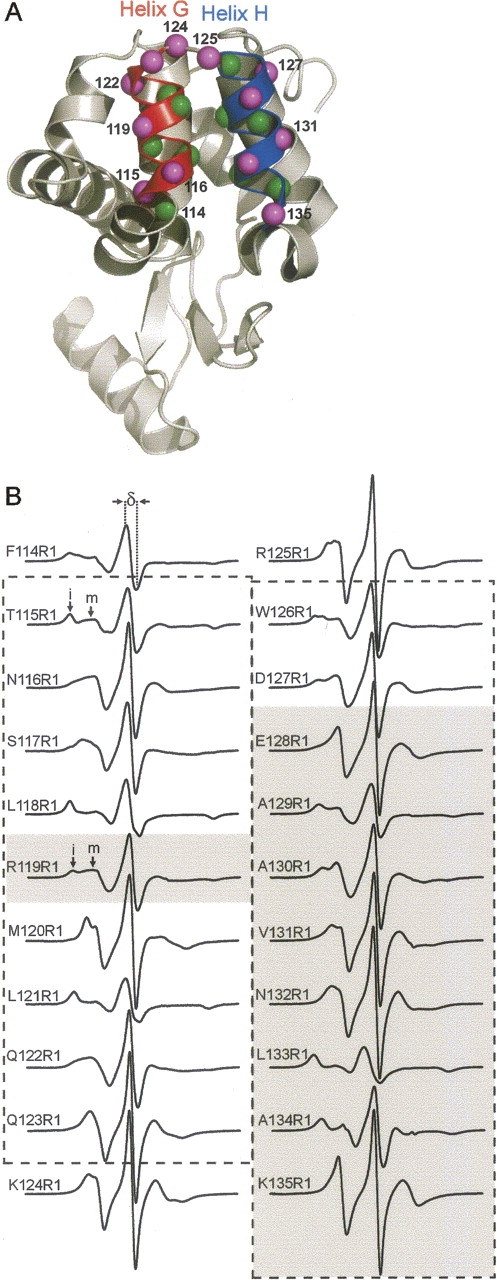
Sites in T4L where R1 was placed along a helix–turn–helix motif and the corresponding EPR spectra. (A) Ribbon diagram of the T4L structure. Spheres at the Cα atoms identify residues where R1 has been introduced. (Magenta) The Cα atoms of the most solvent-exposed sites (fractional solvent accessibility >0.2). (Red) Helix G; (blue) helix H. (B) EPR spectra of R1 at the indicated positions. The dashed box indicates the extent of helix G (left) and helix H (right). The shaded areas indicate previously published spectra. Arrows “i” and “m” indicate immobile and mobile states in the 115R1 and 119R1 spectra, respectively; δ is the width of the central resonance line.
Figure 3.
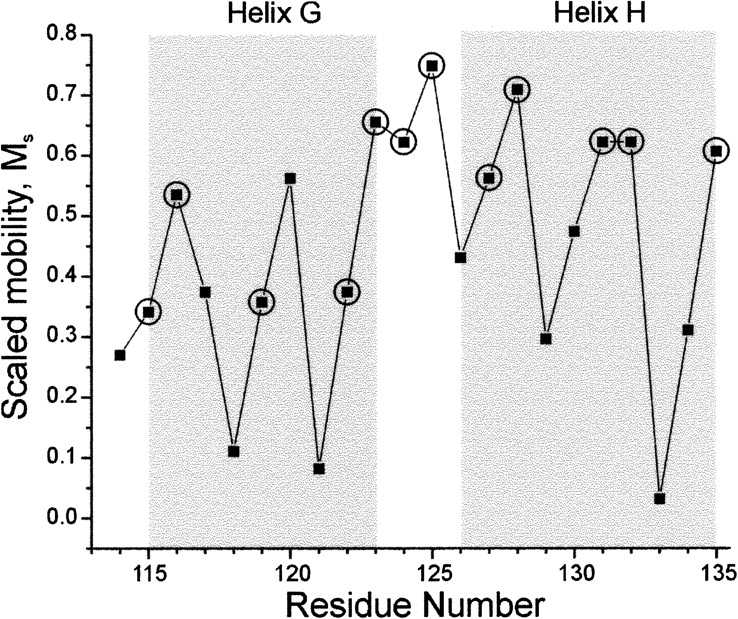
Plot of scaled mobility (M s) versus residue number. Circles identify residues with the highest fractional solvent accessibility (>0.2) corresponding to the magenta spheres in Figure 2A.
In general, M s is correlated with the fractional solvent accessibility of the native residue calculated from the crystal structure (Isas et al. 2002). The circled points in Figure 3 identify the residues of largest fractional solvent accessibility calculated for the native side chain from the wild-type T4L (PDB ID 3LZM) crystal structure (>0.2) (Fig. 2A, magenta spheres). For helix H, there is good correspondence between the largest M s and largest fractional solvent accessibility. The poorer correlation and generally lower M s for the solvent-exposed residues in helix G compared to those in helix H are due largely to immobilization of the nitroxide ring arising from interaction with the local environment; the interaction reduces M s below the level expected for a high solvent accessibility. The presence of both relatively immobilized and mobile nitroxide states is recognized in the spectra of R1 at four of the five most solvent-exposed sites (115, 116, 119, and 122) by spectral intensity in the regions identified as “i” and “m,” respectively, in Figure 2B (the reader is referred to Crane et al. 2006 and Kusnetzow et al. 2006 for a qualitative analysis of EPR spectra). Such multicomponent spectra were identified as arising from “tertiary contact” sites in the original work on the subject (Mchaourab et al. 1996). The immobilized component is well resolved in 115R1 and 119R1 (Fig. 2B, arrows), and is revealed as a broadening of the low field region of the spectra of 116R1 and 122R1. This is in contrast to helix H, where the most solvent-accessible residues 128R1, 131R1, 132R1, and 135R1 have spectra dominated by an R1 population with high mobility. The spectra of 131R1 and 132R1 in particular reveal a single population of R1 in which the nitroxide undergoes anisotropic motion, characteristic of R1 at solvent-exposed sites in ordered helices where the nitroxide has no interactions with neighboring residues (Mchaourab et al. 1999; Columbus et al. 2001).
In the G–H interhelical loop, solvent-exposed 124R1 has a single-component spectrum reflecting rapid anisotropic motion, similar to those of 131R1 and 132R1. This illustrates that this motion can also arise from R1 in an ordered loop, and is not a fingerprint of a helical structure (M. Fleissner and W.L. Hubbell, unpubl.).
Owing to the well-resolved and substantial fraction of immobilized component and the absence of crystal lattice contacts at the site, T4L/115R1 was selected for crystallographic analysis to identify the structural origin of the two states of different mobility.
Native residue L118 in helix G has low solvent accessibility (6%) and is buried in the hydrophobic core of the helix bundle in which it resides. As is discussed below, the EPR spectrum of 118R1 reflects a strongly ordered population (Fig. 2B) and was selected for crystallographic analysis to determine how the R1 side chain is accommodated within the structure at such sites.
Motion of 115R1
Figure 4A shows the EPR spectrum of 115R1 in solution (solid trace) together with a two-component fit (dotted trace; see Materials and Methods). The best fit is obtained with an isotropic motion for the immobilized component with correlation time τ = 20 nsec (β in Fig. 4A) (52% of the population), close to the rotational correlation time of 18 nsec for T4L in 30% sucrose (η = 3 cP) estimated from the Stokes-Einstein equation. Thus, the population of R1 residues giving rise to the β component is effectively immobilized with respect to the protein. The more mobile component (α in Fig. 4A) (48% of the population) has an ordered anisotropic motion with S = 0.48 and τ = 2.2 nsec, similar to those for solvent-exposed sites on ordered helices (Columbus et al. 2001). The spectra corresponding to the individual components determined from the fit are shown in Figure 4B.
Figure 4.

The EPR spectra of 115R1 in solution and in the crystalline state. (A) The 115R1 spectrum in solution (solid trace) superimposed on a two-component MOMD fit (dotted trace; see Materials and Methods). Components α and β correspond to relatively mobile and immobile populations of R1, respectively. (B) The individual spectra that correspond to the α and β components as determined from the fit shown in A. The α and β components are characterized by S = 0.48, τ = 2.2 nsec and S = 0, τ = 20 nsec, respectively. (C) The 115R1 spectrum in the crystalline state at room temperature.
The spectrum of 115R1 in the crystalline state (isotropic suspension of crystallites) at room temperature also has two spectral components (Fig. 4C), but the motion of the individual components is altered by the unusual environment in the crystal lattice; this is not surprising considering the unusual ionic and hydration environment in the crystal. Indeed, the previously published EPR spectra of 119R1 and 80R1 also showed that the spectra of the mobile component in crystals are different from those in solution (Langen et al. 2000). However, the important point for the results to be presented below is that two motional components, one immobilized and the other mobile, are seen in both the solution and crystalline states of 115R1 at room temperature. To ascertain the structural origin of the two components, the crystal structure of 115R1 was determined.
The structure of 115R1 at 100 K
In all structures of R1 determined to date, including those reported here, electron density is resolved for the Cα–Cβ–Sγ–Sδ group (Langen et al. 2000). Thus, dihedral angles χ1 and χ2 can be determined for all cases, and rotamers of R1 are designated with respect to these dihedral angles (Fig. 1). The convention for the dihedral angles follows Lovell et al. (2000), where nominally m (minus) = −60°, p (positive) = +60°, and t (trans) = ±180°. In the various structures, χ1 and χ2 vary considerably from these nominal values, but the simple notation is retained.
The structure of 115R1 at 100 K was determined and refined to 1.4 Å with an R-factor of 15.3% (Tables 1 and 2). Because the EPR spectra revealed two components (at room temperature), it was anticipated that two conformations of the side chain would be resolved. However, the 100 K structure reveals a single conformation of the R1 side chain with the electron density of the entire side chain well-resolved (Fig. 5A). The side-chain torsion angles are given in Table 3. The 115R1 side chain at 100 K is an {m,m} state (χ1 = −81, χ2 = −57), similar to that observed for helical residue 80R1 and one rotamer of 119R1 in T4L (Langen et al. 2000). Interactions that may stabilize this rotamer are shown in Figure 5B. As in the previous structures of the {m,m} rotamer, the Sδ lies close to the H atom modeled on 115Cα (3.2 Å), and it is likely that a CαH···Sδ interaction is a common stabilizing interaction for the {m,m} rotamer of R1 (Langen et al. 2000). The Sγ and Sδ of the disulfide are each ≈3.5 Å from the backbone carbonyl oxygen of Ala 112, and {m,m} in 115R1 may be additionally stabilized by S···O=C interactions (see Discussion). Ordering of the nitroxide ring apparently results from interactions of the Cɛ atom of R1 with the side chain of Lys 83, the oxygen atom of the nitroxide with the side chain of Arg 119, and an α-methyl of the nitroxide ring with the backbone carbonyl of 115 (Fig. 5B). Additional interactions of the nitroxide side chain may be made with the Nζ atom of K83, but electron density is not resolved for this atom (Fig. 5A). Figure 5C shows a space-filling model of R1 and the surrounding side chains, where the close contacts with K83, A112, and R119 are evident.
Table 1.
Summary of data collection statistic

Table 2.
Summary of refinement statistics

Figure 5.
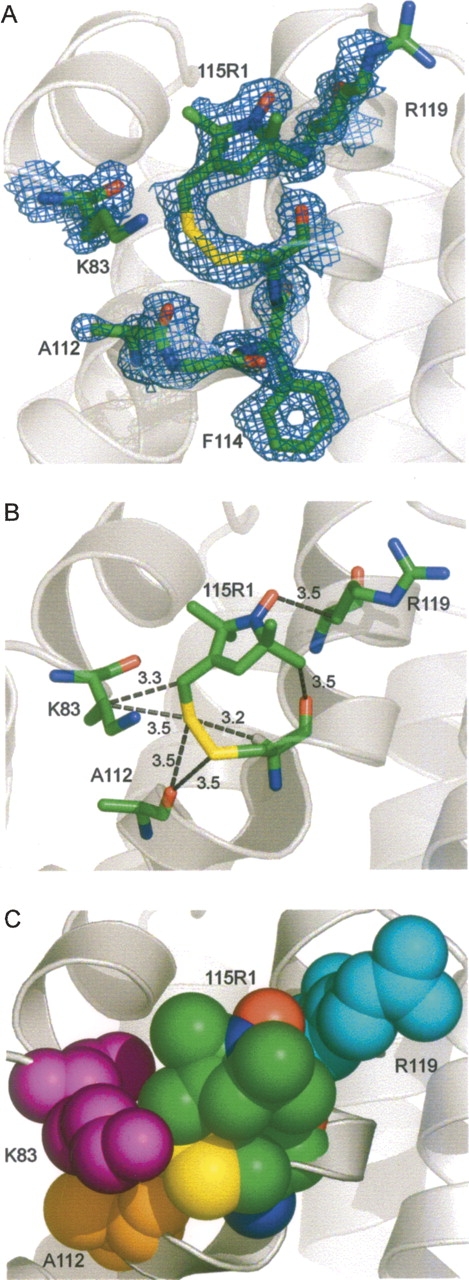
The structure of 115R1 at 100 K. (A) Electron density superimposed on a stick model of R1 and some neighboring side chains. The electron density map was calculated as an unweighted 2F o − F c map contoured at 1.3σ. (B) Distances from R1 to nearest neighbor atoms (≤3.5 Å). The hydrogen on the Cα atom of 115R1 is modeled as a white stick. (C) Space-filling model of 115R1 and neighbors.
Table 3.
Summary of R1 dihedral angles

The structure of 115R1/R119A at 100 K
The structure of Figure 5 indicates that interactions leading to ordering of the nitroxide ring and perhaps stabilization of the single {m,m} rotamer are with the side chain of R119. To explore this possibility, the structure of the 115R1/R119A mutant was solved at 100 K and refined to 1.55 Å with an R-factor of 15.8% (Tables 1 and 2). Figure 6A shows the electron density of the R1 side chain and a few neighboring residues in the 115R1/R119A mutant at 100 K. Again, a single {m,m} rotamer is observed, with a similar value of χ1 and Sδ···HCα distance (3.1 Å), but with a substantially lower value of |χ2| compared with that of 115R1 (33° vs. 57°) (Table 3). The nitroxide ring is now poorly resolved, indicating spatial disorder. In the new (but similar) conformation of 115R1/R119A, the nitroxide ring has moved away from R119, and the disulfide has moved closer to the backbone oxygen of A112, presumably strengthening the interactions with that residue. These results support the suggestion that the interactions of the nitroxide ring with the side chain of Arg 119 identified in Figure 5B are, in fact, key determinants in ordering the ring in 115R1, but are not solely responsible for stabilizing the single {m,m} state. Figure 5C shows a space filling model where the nitroxide ring has been modeled to minimize steric overlaps. The model suggests that the oxygen of the nitroxide may retain a single contact with the Cβ of A119.
Figure 6.
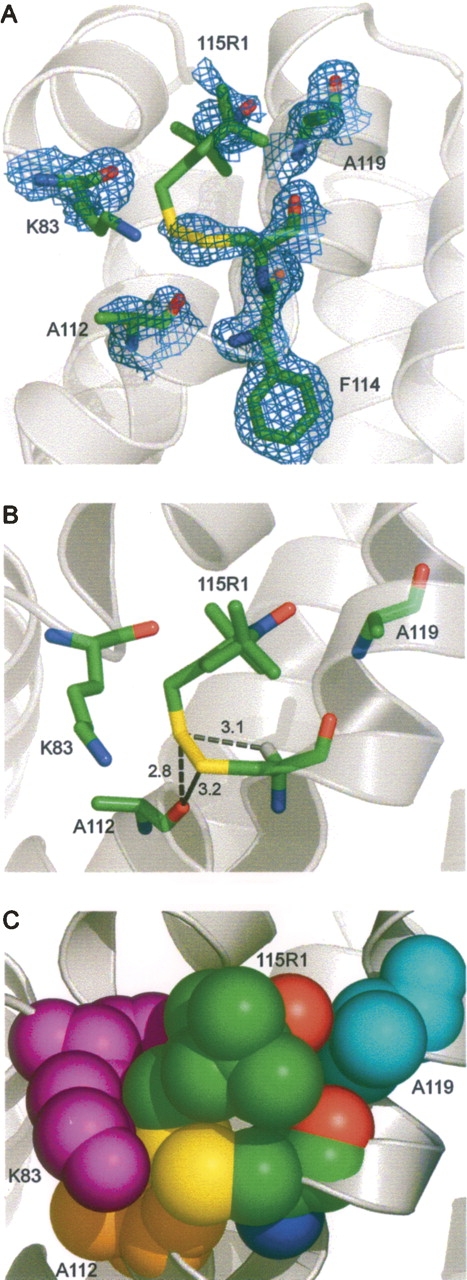
The structure of 115R1/R119A at 100 K. (A) Electron density superimposed on a stick model of R1 and neighboring side chains. The electron density map was calculated as an unweighted 2F o − F c map contoured at 1.3σ. (B) Distances from R1 to nearest neighbor atoms (≤3.5 Å). The hydrogen on the Cα atom of 115R1 is modeled as a white stick. (C) Space-filling model of 115R1 and neighboring groups.
The structure of 115R1 at room temperature
As discussed above, the EPR spectrum of 115R1 in solution and in the crystalline state at room temperature reflects two motional states of the nitroxide. This is not consistent with the structures at 100 K, which have a single well-ordered state of R1, suggesting that low temperature selects a single rotamer. To investigate this possibility, diffraction data of 115R1 were collected at room temperature, and the structure was refined to 1.8 Å with an R-factor of 18.7% (Tables 1 and 2). Figure 7A shows the electron density map where two orientations of the disulfide are now partially resolved that correspond to two rotamers of roughly equal occupancy, namely, {m,m} and {t,m}. Electron densities for the sulfurs are poorly resolved compared to those at 100 K because of thermal motion in the lattice, and electron density for the nitroxide ring is not resolved in either rotamer. The {m,m} rotamer adopts similar χ1 and χ2 angles to those in the low-temperature 115R1/R119A structure (Table 3), and is presumably stabilized by interactions similar to those discussed above and identified in Figure 7B.
Figure 7.

The structure of 115R1 at 298 K. (A) Electron density superimposed on a stick model of R1 and neighboring side chains. The electron density map was calculated as an unweighted 2F o−F c map contoured at 1.1σ. (B) Distances from R1 to nearest neighbor atoms (≤3.5 Å) in the {m,m} rotamer. The hydrogen on the Cα atom of 115R1 is modeled as a white stick. (C) Distances to nearest neighbor atoms in the {t,m} rotamer. (D) Surface representation with a stick model of the 115R1 rotamers. (Red) Nonpolar surfaces.
The {t,m} rotamer has not been observed in previously determined structures of R1 in T4L (Langen et al. 2000). In the {t,m} state, the Sδ atom of the side chain has apparent interactions with the backbone carbonyl oxygen of 115R1 (3.3 Å), the backbone amide nitrogen of N116 (3.4 Å), and the side chain oxygen (or nitrogen) of N116 (3.4 Å) (Fig. 7C).
Although electron density is not resolved for the nitroxide ring, molecular modeling suggests that the nitroxide ring in the {m,m} conformation lies in proximity to a relatively hydrophobic pocket formed by Val 87, Leu 84, Pro 86, Leu 118, Leu 99, and the hydrophobic alky segments of K83 and R119 side chains (Fig. 7D). Because the nitroxide ring is hydrophobic, a favorable interaction with this pocket is likely. Since hydrophobic interactions are nonspecific, multiple orientations of the ring could have comparable interaction energies, accounting for the lack of resolved electron density of the ring. Nevertheless, any hydrophobically adsorbed state could be immobilized on the EPR timescale. The above considerations suggest that the {m,m} state gives rise to the immobilized β component in the EPR spectrum of 115R1 (Fig. 4).
In the {t,m} rotamer, the nitroxide is positioned away from the hydrophobic pocket and projects into the solvent. For this situation, the motion of the nitroxide, and hence the EPR spectrum, is expected to be similar to R1 residues on solvent-exposed surfaces of α-helices. As shown in Figure 4A, the EPR spectrum of the α component reveals a rapid anisotropic motion characteristic of R1 at solvent-exposed helical sites (Mchaourab et al. 1996, 1999; Columbus et al. 2001). Taken together, the above considerations suggest that the α and β components of the 115R1 spectrum arise from the {t,m} and {m,m} rotamers, respectively.
The interactions of 115R1
To test the hydrophobic adsorption model for the origin of the immobilized state of 115R1, the effect of dioxane on the relative populations of the two states was investigated. Dioxane reduces the magnitude of the hydrophobic effect (Nozaki and Tanford 1971), but was found not to cause unfolding of hen egg white lysozyme in concentrations of up to 50% (v/v) (Hamaguchi and Kurono 1963; Izumi and Inoue 1977). If the immobilized component arises from hydrophobic interactions, low concentrations of dioxane would be expected to shift the rotamer equilibrium toward the mobile state.
Figure 8A–C compares the effect of 0%, 10%, and 20% (v/v) dioxane on the EPR spectra of 115R1 (solid traces). As is evident from the changes in relative intensities of the α and β components, the amount of the more mobile component (α) increases with increasing dioxane concentration at the expense of the immobile component. Also shown are superimposed simulated spectra (dotted traces) for a two-component model where the motional parameters for both components are fixed at the values given above for 115R1 in 0% dioxane (Fig. 4A), and the relative populations of the two components are varied. Thus, this is not an attempt at an optimal fit for each case, but rather a comparison of the data with a model wherein two populations simply shift in relative amounts. The reasonable agreement between the simulated and experimental spectra suggests that a shift in populations occurs without a substantial change in motion of either component. This, in turn, implies that the tertiary structure of the protein is unaltered by the dioxane, and only the rotameric equilibrium is affected. The secondary structure of WT* T4L is unperturbed by 20% dioxane in the same buffer used for recording EPR spectra, as assayed by the circular dichroism spectra (Supplemental Fig. S1).
Figure 8.
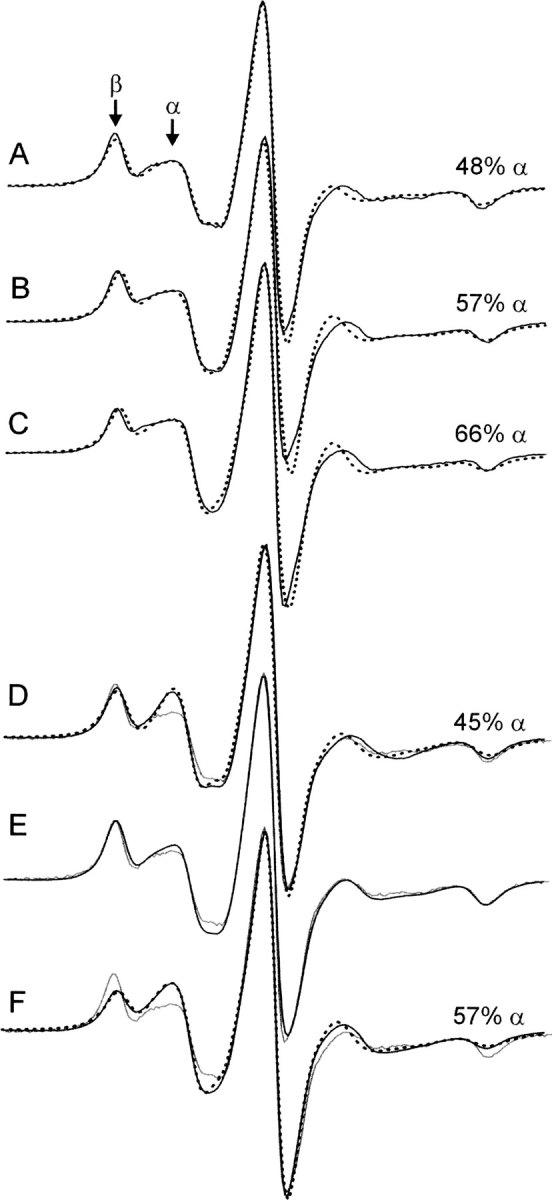
The effect of dioxane and mutation of nearest-neighbor residues on the EPR spectra of 115R1. (A–C) EPR spectra of 115R1 in (A) 0%, (B) 10%, and (C) 20% dioxane (black trace) superimposed on two-component MOMD simulations (dotted traces; see text). The percentage of the α component is indicated for each. (D) The spectrum of 115R1/R119A (black trace) together with a two-component fit (dotted trace) and the spectrum of 115R1 (gray trace). The α and β components for the fit have S = 0.17, τ = 1.9 nsec and S = 0, τ = 20 nsec, respectively. (E) The spectrum of 115R1/R119Q (black trace) superimposed on the 115R1 spectrum (gray trace). (F) The spectrum of 115R1/K83A (black trace) superimposed on a two-component fit (dotted trace) and the spectrum of 115R1 (gray trace). The α and β components for the fit have S = 0.34, τ = 2.1 nsec and S = 0, τ = 17.4 nsec, respectively.
The above results support the proposal that hydrophobic interactions of the nitroxide ring give rise to the immobile state, and the structure suggests that it is likely the {m,m} rotamer that is involved in such interactions. Unlike the {m,m} rotamer and the {t,p} rotamer previously reported (Langen et al. 2000), the {t,m} rotamer projects in a direction that permits direct interaction of the nitroxide ring with either the i + 1 or i + 4 residues in a helix (Fig. 7D). To investigate the possibility of a nitroxide interaction with the bulky arginine residue at 119 (i + 4), the mutant 115R1/R119A was prepared, and the EPR spectrum (black trace) is compared with 115R1 (gray trace) in Figure 8D. In 115R1/R119A, the line shape of the mobile α component is narrowed, suggesting an increase in mobility, leaving the immobilized β component relatively unchanged. A fit to the 115R1/R119A spectrum (dotted trace) supports this conclusion and indicates that the α component has a similar rate but substantially lower order (τ = 1.9 nsec, S = 0.17) compared to that in 115R1 (τ = 2.2 nsec, S = 0.48), and that the β component is essentially unchanged (τ = 20 nsec, S = 0, 55% of population). This result is compatible with an interaction of the nitroxide ring with R119 and lends support to the proposal that the {t,m} state gives rise to the mobile α component of the 115R1 spectrum. To determine whether or not the interaction involves an electrostatic component, 115R1/R119Q was prepared. The glutamine side chain is sufficiently large to make contact with the nitroxide ring but lacks the charge of arginine. The EPR spectrum of 115R1/R119Q shows only a slight increase in mobility of the mobile component (Fig. 8E), suggesting that electrostatic interactions are not central to the apparent nitroxide ring interaction with i + 4. Mutation of the i + 1 asparagine residue to alanine has no effect on the EPR spectrum of 115R1 (C. López, M. Fleissner, and W.L. Hubbell, unpubl.), showing that the putative interaction with N116 (Fig. 7C) is not expressed in the motion of the nitroxide.
The structure of 115R1 suggests that the {m,m} rotamer may be stabilized in part by interactions with K83 (Fig. 7B,D). If so, the mutation K83A may result in a shift of the rotameric equilibrium toward the {t,m} state and, according to the model, an increase in the α population relative to 115R1. The EPR spectrum of 115R1/K83A reveals that this is, indeed, the case (Fig. 8F). Fitting of the spectrum (Fig. 8F, dotted trace) indicates that the population of the α component has increased by ∼10%, but in addition both the α and β components have slight increases in mobility compared to 115R1 (see legend to Fig. 8 for τ and S for each component). Although K83 is not involved in a salt bride, changes in electrostatics in the vicinity of the hydrophobic pocket could give rise to hydration changes that modulate local structure and/or dynamics that are reflected in the side-chain motion.
Collectively, the above results support a model in which the 115R1 side chain is in equilibrium between {m,m} and {t,m} rotamers in slow exchange on the EPR timescale, and in which the rotamers are resolved in the EPR spectrum by differential interactions of the nitroxide ring with the environment. The nitroxide in the {m,m} rotamer is likely immobilized by nonspecific hydrophobic interactions, while that in the {t,m} rotamer is partially ordered by interactions with the i + 4 residue.
The structure of 115R7
It has been shown that a 4-CH3 or 4-Br substituent on the ring of the R1 side chain dramatically decreases the mobility of the nitroxide (Mchaourab et al. 1996; Altenbach et al. 2001) through effects on both order and rate of motion (Columbus et al. 2001). On the basis of modeling, it was suggested that the substituent increases ordering of the side chain because of repulsive interaction with the Sδ atom of the disulfide, which limits the amplitude of motion about χ5 (Fig. 1). Because of the increased order, such side chains have potential applications as sensors for backbone motion with increased dynamic range compared to R1. In addition to repulsive interactions, the 4-substituent must also have an attractive van der Waal's interaction with Sδ at larger internuclear separations that would account for a reduced rate of motion.
The spectrum of 115R7 is dominated by an immobilized component (arrow), consistent with previous observations (Fig. 9A; Altenbach et al. 2001). The 115R7 mutant offers an opportunity to investigate the structural origins of the hindered motion, and the structure of 115R7 was determined at 100 K and refined to 1.4 Å with an R-factor of 15.5% (Tables 1 and 2). Figure 9B shows the electron density contours for the R7 side chain and neighboring residues Arg 119 and Asn 116. Nitroxide ring density is only partially resolved, with strong density for the 4-Br atom. Remarkably, the side chain adopts a {t,m} conformation (χ1 = −179, χ2 = −85) similar to that of the room temperature 115R1 structure, but not observed in the corresponding 100 K structure of 115R1. The rotamer is similarly stabilized by Sδ interactions with the carbonyl oxygen of 115 and the backbone nitrogen and Oδ atom of Asn 116 (Fig. 9C). Unlike the other structures, the nitroxide ring makes contacts with side chains of a symmetry-related molecule in the lattice (Fig. 9D). This makes it impossible to equate the side-chain structure in the crystal with that in solution. Nevertheless, the structure is presented here because it reveals intramolecular interactions of the 4-Br atom that could occur in the solution state. Thus, the 4-Br atom makes close contacts with the Sδ atom in the side chain (3.7 Å) as predicted from modeling, and in addition makes a close contact with the Oδ atom in the i + 1 side chain (Asn 116, 3.3 Å) and contact with the Cδ of the i + 4 side chain (Arg 119, 4.0 Å). If these intramolecular interactions persist in the solution state, they could account for the immobilized state of the nitroxide.
Figure 9.

The EPR spectrum of 115R7 in solution and the structure at 100 K. (A) The EPR spectrum of 115R7 in solution; the arrow identifies the more immobilized component. (B) Electron density superimposed on a stick model of 115R7 and neighboring groups. The electron density map was calculated as an unweighted 2F o−F c map contoured at 1.2σ. (C,D) Distances from R1 to nearest-neighbor atoms (≤3.5 Å).
The structure of 118R1
The R1 side chain has been incorporated at buried (solvent-inaccessible) sites in several proteins. Leu 118 (molar volume 69 Å3) in helix G has a fractional solvent accessibility of 0.06 and is categorized as a buried site. Substitution by R1 (molar volume 195 Å3) overpacks the hydrophobic core by 126 Å3. The thermal unfolding of 118R1 was reversible at pH 3.0 with a melting temperature 7°C lower than the WT* protein, corresponding to a destabilization of ∼2 kcal/mol (Supplemental Fig. S2). To examine the response of the structure to the R1 substitution, the crystal structure of 118R1 was determined and refined to 1.8 Å with an R-factor of 18.5% (Tables 1 and 2).
Figure 10A shows the electron density for the R1. The R1 side chain including the nitroxide ring is completely resolved in a single well-ordered conformation (Table 3). The global structure of T4L in the 118R1 mutant is extremely similar to the WT* structure, except the region of the short one-turn helix F that connects helices E and G (residues 108−113); in the 118R1 structure, helix F adopts a loop configuration (Fig. 10B). As is discussed below, this helix has been previously identified as a highly plastic element in T4L. The 118R1 side chain is well packed in the structure, with one edge exposed to solvent (Fig. 10C).
Figure 10.
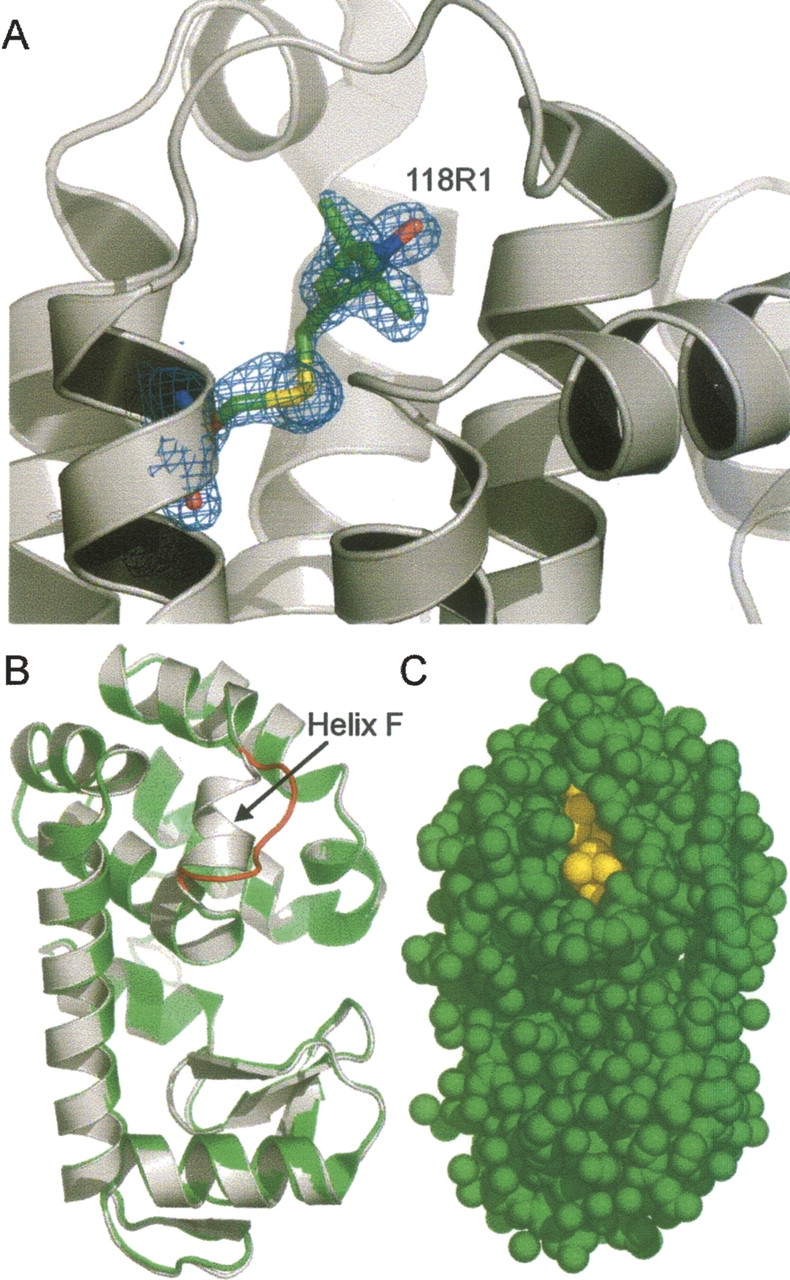
The structure of 118R1 at 100 K. (A) Electron density superimposed on a stick model of 118R1. The electron density map was calculated as an unweighted 2F o−F c map contoured at 1.1σ. (B) Structural alignment of T4L/WT* (white) and T4L/118R1 (green) with the segment corresponding to helix F of WT* shown in red. (C) Space-filling model with 118R1 colored yellow and the remaining structure in green.
Residues on the rearranged loop are less ordered than those in the corresponding helix F of WT*. The average B-factor of the main-chain atoms in the loop of T4L/118R1 is 37 Å2 compared to 18 Å2 for the entire structure, while the corresponding values in the WT* are 23 Å2 and 20 Å2. Figure 11A shows the electron density of residues in the loop in T4L/118R1. The side chains of Glu 108 and Thr 109 have poor density, but the rest of the residues are well defined. The neighboring side chains that are in contact with 118R1 adopt their WT* rotamers with only slight adjustments of the side-chain torsion angles (Fig. 11B). This is consistent with the findings from several other small-to-large substitutions in the protein core (Baldwin et al. 1993, 1996; Lim et al. 1994; Wynn et al. 1995, 1996, 1997; Liu et al. 2000). In these studies, it was also found that changes of side-chain rotamers are only rarely seen when repacking of the protein core is necessary.
Figure 11.
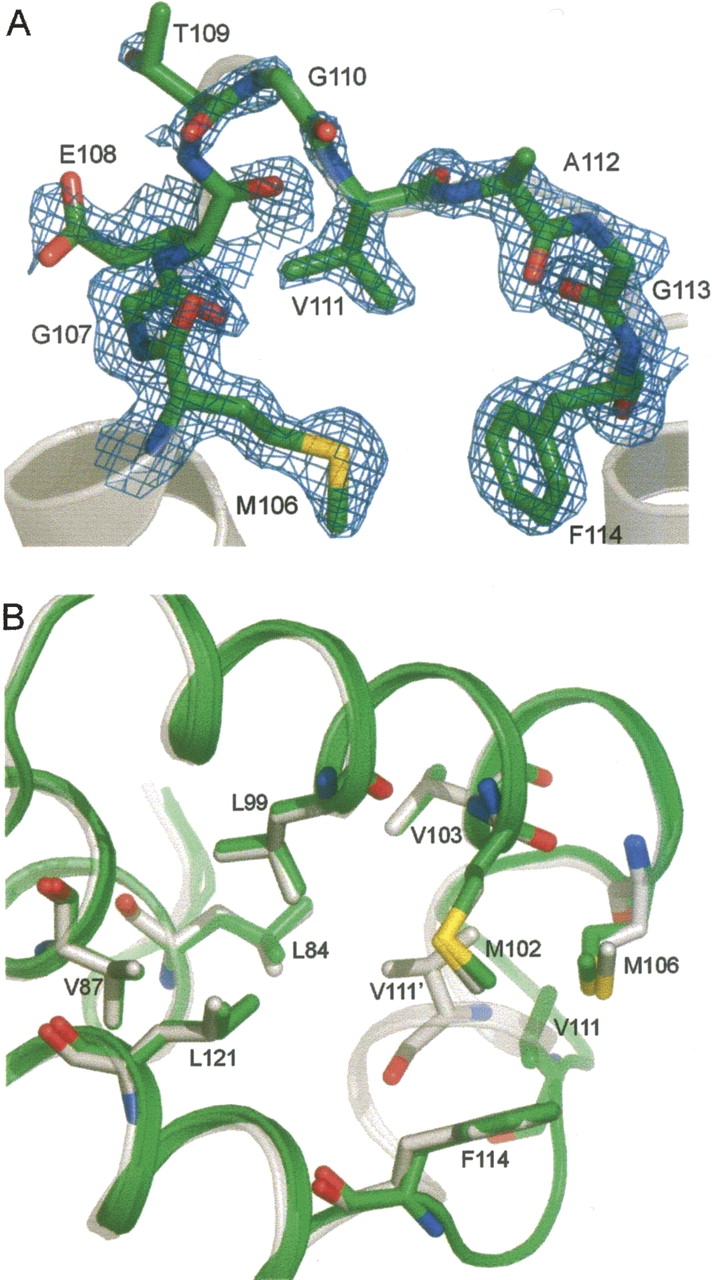
The structure of the neighboring side chains of 118R1. (A) Electron density of residues 106−114. The electron density map was calculated as an unweighted 2F o−F c map contoured at 1.1σ. (B) Structure alignment of residues around 118R1 for T4L/WT* and T4L/118R1. (White) WT*; (green) 118R1.
The core packing in the WT* and T4L/118R1 structures was compared by means of the internal cavities in the proteins. The WT* structure has five internal cavities on the C-terminal domain that contains the labeling site (Fig. 12A). The mutation L118R1 results in a decrease in the volume of cavity II and the disappearance of cavities III, IV, and V. Overall, the mutation to L118R1 resulted in a decrease in the cavity volume of the hydrophobic core of the C-terminal subdomain.
Figure 12.

Cavities in the structure of (A) T4L/WT* and (B) T4L/118R1. (B) The stick model of 118R1 shows the absence of cavities around the nitroxide ring. The cavity numbering is the same as in A, with the new cavity numbered VI.
The crystal structure of 118R1 at 100 K shows the R1 side chain to be completely ordered in the hydrophobic core. If this is the case at room temperature in solution, it is anticipated that the EPR spectrum should correspond to a state immobilized with respect to the protein, having motion determined by the essentially isotropic rotary diffusion of T4L. Figure 13A shows the EPR spectrum of 118R1 in 30% sucrose solution, where T4L has a correlation time for rotational diffusion of ∼18 nsec. The spectrum cannot be well fit assuming a single-component isotropic rotary motion of this correlation time, or assuming a two-component model with one component rotating with the protein. A good fit (Fig. 13A, dashed trace) is only obtained with a dominant component of relatively fast anisotropic motion of high order (τ = 4.6 nsec, S = 0.9, 80%), and a smaller second component with high mobility (τ = 4.3 nsec, S = 0, 20%). The dominant highly ordered component clearly corresponds to the buried state observed in the crystal structure. The low-amplitude motion is fast compared to the protein rotary diffusion, and presumably reflects internal modes of the side chain permitted by thermal fluctuations of the structure. This motion is apparently common for R1 at buried sites; for example, the spectrum of 133R1 (Fig. 2, helix H) is well fit with a single component of similar fast anisotropic motion (τ = 3.9 nsec, S = 0.8) (Fig. 13C, dotted trace).
Figure 13.
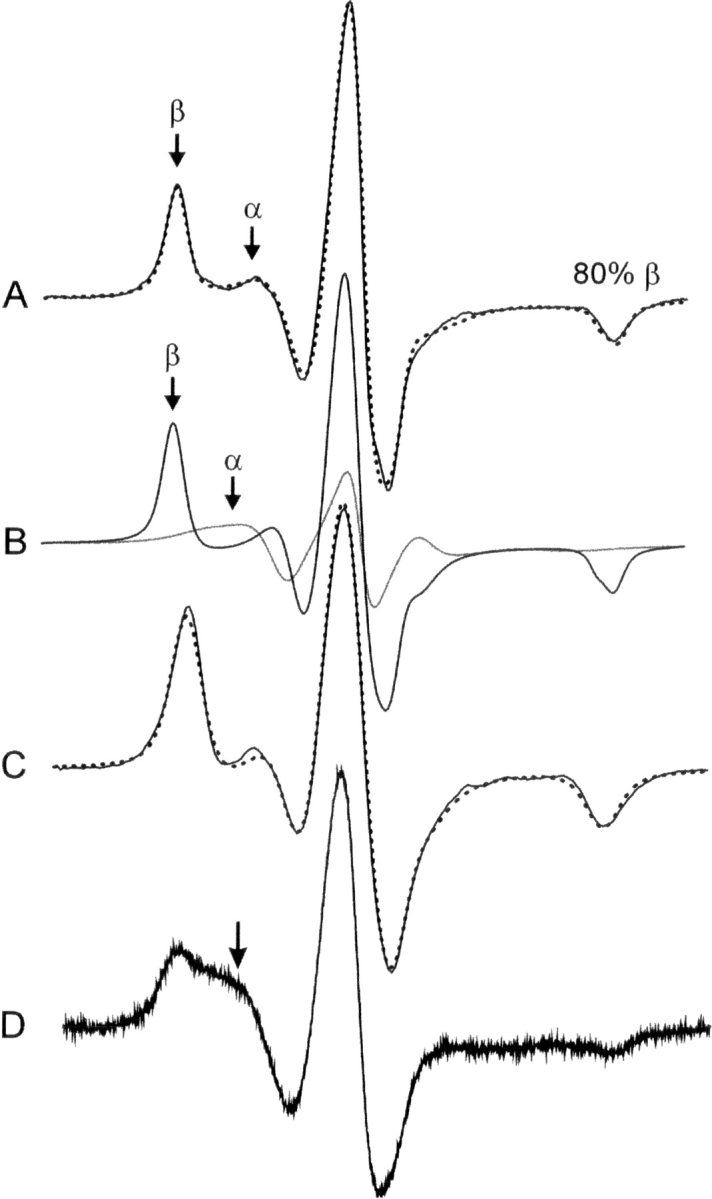
The EPR spectra of 118R1 and 133R1 in solution and 118R1 in the crystalline state. (A) The 118R1 spectrum in solution (solid trace) superimposed on a two-component MOMD fit (dotted trace; see Materials and Methods). Components α and β arise from relatively mobile and immobile populations of R1, respectively. (B) The individual α (gray trace) and β (black trace) components determined from the fit shown in A. The α and β components have S = 0, τ = 4.3 nsec and S = 0.90, τ = 4.6 nsec, respectively. (C) The EPR spectrum of 133R1 in solution (solid trace) superimposed on a single-component MOMD fit (dotted trace) with S = 0.8, τ = 3.9 nsec. (D) The 118R1 spectrum in crystalline state. The arrow indicates a component corresponding to a relatively mobile population of R1.
Interestingly, the EPR spectrum of 118R1 in crystals (isotropic suspension) also reveals two components, one immobilized and the other reflecting a substantial amount of a more mobile component (Fig. 13D, arrow). Thus, the spectrum of 118R1 at room temperature reflects two dynamic modes of R1 in both solution and the crystal. Presumably, a single immobilized state is selected at 100 K in the crystal. The origin of the component of high mobility in 118R1 is uncertain, but may reflect a partly unfolded state of the F-helix.
Discussion
Quantitative application of SDSL to determining protein structure and dynamics relies heavily on knowledge of the rotameric states and interactions of the spin-labeled side chains used. The present report is part of an ongoing study to develop a rotameric library for the commonly used R1 side chain, and to identify the origin of multiple dynamic modes of R1 revealed in the EPR spectra.
115R1 structure: Low-temperature selection of rotamer
The structure of 115R1 at room temperature revealed two conformations of the R1 side chain with respect to χ1 and χ2 (Fig. 7A), while at 100 K, only one conformation is present with well-resolved density for the entire side chain (Fig. 5A). Apparently, favorable enthalpic contributions dominate at 100 K and select a single rotamer that makes the maximum number of contacts, whereas at room temperature entropic contributions to the free energy become significant and allow population of a second state with more degrees of freedom. The crystal structure of 115R1/R119A also shows a single rotamer with similar χ1 and χ2 as 115R1, but with disorder in the distal part of the side chain. This suggests that interactions with R119 are involved in low-temperature ordering of the nitroxide ring, but that this is not the sole source of the low-temperature stabilization of the {m,m} rotamer. Rather, the ensemble of interactions in the {m,m} state is involved.
Variable-temperature crystallography has been used to study the effects of temperature on the structure of sperm whale myoglobin (Hartmann et al. 1982; Frauenfelder et al. 1987), ribonuclease A (Tilton et al. 1992), hen egg white lysozyme (Young et al. 1994), and others (Juers and Matthews 2001). Crystal packing, protein volume, and the role of bulk solvent have been extensively analyzed. Juers and Matthews (2004) found that the intermolecular contacts between proteins in the crystal lattice of β-galactosidase were dominated by long polar side chains, which tend to be somewhat disordered at room temperature but can form extensive hydrogen-bonded networks on cooling. It is now commonplace to collect diffraction data at cryogenic temperature (∼100 K) for improved quality. Juers and Matthews (2004) pointed out that caution must be taken when interpreting protein–protein interactions based on low-temperature crystal structures, and the cautionary note apparently extends to interpreting room temperature experimental data related to side-chain structure as well. As is discussed below, low temperature also apparently selects a specific structure in the 118R1 mutant. However, it should be emphasized that this effect does not preclude useful interpretation of cryogenic data obtained for spin-labeled proteins; room temperature EPR spectra will reveal when it occurs (as for 115R1), and the conformation of the side chain at low temperature closely corresponds to one rotamer in the room temperature structure.
Interactions of 115R1: Interactions that stabilize the rotamers and potential origins of multiple components in the EPR spectra
Compelling evidence is now accumulating to indicate that disulfide interactions with the backbone are key elements in stabilizing the rotamers of R1 at solvent-exposed helical sites. The first such evidence was that removing the Sδ atom from the R1 side chain (to produce a thioether-linked side chain) results in increased nitroxide mobility, although the number of bonds between the nitroxide ring and the backbone is fewer (Mchaourab et al. 1996). The crystal structures of 80R1 and 119R1 offered an explanation for this effect, namely, the existence of an {m,m} rotamer in which the Sδ atom of the disulfide lies sufficiently close to the Cα hydrogen atom to suggest an attractive Sδ···HCα interaction (Langen et al. 2000). The appearance of an {m,m} rotamer in the 115R1 structure at both cryogenic and room temperature adds support to the importance of this putative interaction, as does the absence of an {m,t} rotamer in any of the solvent-exposed structures obtained so far; in this rotamer, Sδ is moved away from the backbone. Finally, the anisotropic motion of R1 at solvent-exposed sites on α-helices is in complete accord with effective immobilization of the disulfide group that would result from the proposed interaction (Columbus et al. 2001).
Other evidence for Sδ···HCα interactions exists in the literature. Van Wart and Scheraga (1977) first noted a potential Sδ···HCα interaction in aliphatic disulfides. Recent surveys of the protein database reveal a preponderance of the {m,m} configuration for the half cysteines of the disulfide in which Sδ is close to the HCα (Dani et al. 2003; Bhattacharyya et al. 2004). Considering the paucity of the {m,m} configuration for other aliphatic side chains (see below), this is suggestive of an attractive interaction.
The new {t,m} rotamer observed for 115R1 at room temperature also appears to be stabilized by interactions of the Sδ of the disulfide with main-chain atoms, in this case the C=O of 115R1 and the backbone N of the i + 1 residue. S···O=C and S···N interactions have been identified in several structures (Rosenfield et al. 1977; Burling and Goldstein 1993; Nagao et al. 1998; Pal and Chakrabarti 2001; Bhattacharyya et al. 2004), including the {t,p} states of 65R1 and 119R1 in T4L (Langen et al. 2000), and appear to be important interactions stabilizing these rotamers of R1. The {t,m} rotamer has been rarely observed in the rotamer libraries of natural amino acids (Schrauber et al. 1993; Dunbrack Jr. and Cohen 1997; Lovell et al. 2000), a situation that can be understood if the disulfide interaction of the R1 side chain is key in the {t,m} stabilization. In addition to these specific noncovalent interactions of the Sδ atom, it is possible that desolvation plays a role in partially sequestering the relatively hydrophobic disulfide away from water near the helical backbone.
The interactions with main-chain atoms discussed above are those that are believed to stabilize the rotamers with respect to the χ1, χ2 dihedral angles. However, additional interactions of the nitroxide ring itself are required to differentially modulate the motion of the nitroxide in different rotamers and give rise to multiple components of the EPR spectrum. In the case of 115R1, the structure and the effect of dioxane support a model in which the interaction giving rise to immobilization is a hydrophobic one in the {m,m} rotamer. In this rotamer at room temperature, modeling suggests that the nitroxide ring projects away from the i + 4 arginine at 119, toward an adjacent hydrophobic pocket (Fig. 7D). Hydrophobic interaction of the nonpolar nitroxide is expected to be quite general, and the use of organic solvents like dioxane may provide a test for the interaction.
In the room temperature {t,m} rotamer, modeling suggests that the ring projects toward the i + 1 and i + 4 residues (Fig. 7D). In this position, interaction with the bulky R119 is possible, and that such an interaction exists is indicated by the change in the order parameter of 115R1 upon the substitution of R119A (Fig. 8D). In an earlier study, it was concluded that nearest-neighbor residues i ± 3 and i ± 4 at solvent-exposed helix surface sites would not interact with the R1 residue (Mchaourab et al. 1996, 1999), but this conclusion must now be modified in view of the existence of the {t,m} rotamer; it is likely that i + 1 and/or i + 4 interactions will be observed for R1 in this configuration, and such intrahelical interactions may account for the appearance of multiple dynamic modes in some EPR spectra.
Previous studies documented the dramatic effect of numerous 4-substituents on reducing the mobility of the nitroxide ring in R1 derivatives (Columbus et al. 2001). It was suggested that the substituents hindered internal motion of the ring by interaction with the Sδ sulfur of the disulfide. The structure of the 4-Br derivative 115R7 provides some support for this proposal in that the Br atom lies within van der Waals interaction distance of Sδ, but in the crystal also has additional potential intra- and inter-residue interactions, including some with a symmetry-related molecule.
The 118R1 structure and motion of the nitroxide
The structure of 118R1 is the first reported for the R1 side chain at a buried site in a protein fold, and illustrates a novel example of how and to what extent a protein changes its structure to release strain in small-to-large side-chain mutations. One of the major findings from the previous studies of the small-to-large mutations within the hydrophobic core is that relaxation of strain is achieved by a combination of small shifts in the backbone (usually only a few tenths of an Angstrom unit) and small adjustments in the side-chain rotamer angles (Baldwin et al. 1993, 1996; Lim et al. 1994; Wynn et al. 1995, 1996, 1997; Liu et al. 2000). Changes of side-chain rotamers are only rarely seen when repacking of the protein core is needed to accommodate larger side chains. The case of 118R1 is distinct in that the single rotamer of the R1 side chain (Fig. 10A) is accommodated by a backbone rearrangement in the short (1.5 turn) helix F that connects helices E and G of a helix bundle that constitutes the major feature of the C-terminal domain of T4L (Fig. 10B). The remainder of the structure is essentially identical to the WT* (Fig. 10B), although the disappearance of some cavities present in the WT* and the appearance of new ones in the T4L/118R1 structure distant from the mutation site (Fig. 12) reveal subtle changes in the global fold. The rotamers of the surrounding native side chains in the bundle are not altered (Fig. 11B), and the structure is well packed with a total cavity volume less than that of the native structure (Fig. 12).
Interestingly, the short helix F has relatively high B-factors in the native structure, and has been shown to be a “plastic” element distorted by binding of ligands to an engineered cavity mutant (L99A) in T4L (Morton and Matthews 1995); the ligand-binding site is in the same general vicinity as the nitroxide ring of 118R1. A rearrangement of helix F similar to that in T4L/118R1 was, in fact, observed in a structure of the cavity mutant containing bound isobutylbenzene (Morton and Matthews 1995). Moreover, helix F has been found to be in conformational exchange in the cavity mutant, presumably transiently moving out of the way to allow access of the ligand to the cavity (Skrynnikov et al. 2002).
The EPR spectrum of 118R1 in solution has a powder-like line shape that would qualitatively be interpreted as a strongly immobilized state, consistent with a buried location in the structure. However, a fit of the spectrum requires two components, the dominant of which has a relatively fast motion (4.6 nsec) of high order (S = 0.9), consistent with a low-amplitude rocking motion about the χ5 dihedral (15°) (Fig. 13A). The protein is well packed around the nitroxide ring (Figs. 10C and 12B), and the motion presumably represents thermal fluctuations of the protein core. Similar motion is also observed for 133R1, another buried site (Fig. 13C), and may be a general feature of R1 in protein interiors.
The origin of the second component in the 118R1 spectrum is a matter of speculation. The dynamic parameters (τ = 4.3 nsec, S = 0) reflect a relatively unconstrained motion. One possibility is fluctuation of the flexible sequence 106−113 between ordered and dynamically disordered structures on a microsecond-to-millisecond timescale. Remarkably, the spectrum of 118R1 in the crystalline state at room temperature also reveals two (or more) dynamic modes of R1 (Fig. 13D), but with a substantial enhancement of the more mobile component compared to that in the solution state. In the context of the above model, the environment in the crystal favors a more unfolded state at room temperature, with selection of the well-ordered state at 100 K.
Rotamers of nitroxide side chains
Native amino acids are rotameric; that is, the side-chain conformation can be represented by a limited number of χ angle combinations. The χ1, χ2 rotamer preferences for unbranched aliphatic side chains that have δ atoms in helices are given in Figure 14A along with those for cysteine for comparison. Previously published data (Langen et al. 2000) and that of the present study show that the R1 side chain is also rotameric in this sense with some interesting qualifications. In total, 10 partial structures of the R1 side chain, and one of R7, have now been determined at helical sites in T4L that allow assignment of the rotamers with respect to the χ1 and χ2 dihedral angles. Five of the rotamers are {m,m}, two {t,m}, two {t,p}, one {m,t}, and one is highly distorted that does not fall within the boundaries of the usual rotamer definitions (118R1) (Fig. 14B).
Figure 14.
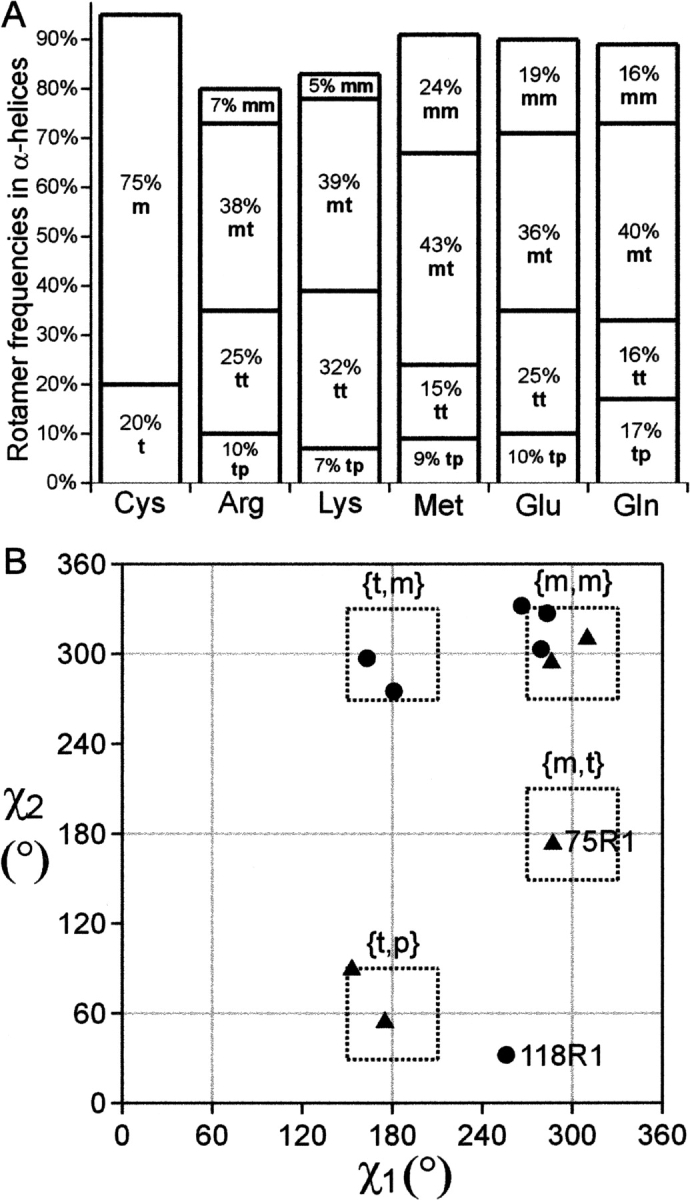
The rotamer preferences of native and R1 side chains. (A) The rotamer populations of cysteine and some unbranched aliphatic side chains that contain δ atoms. The data were taken from Lovell et al. (2000). (B) Rotamer preference of R1 based on the structures in this work (Table 3) and those of Langen et al. (2000) [65R1, 75R1, 80R1, and 119R1 (two rotamers)]. The rotamer of 115R7 is included. The dotted boxes are drawn at ±30° from the nominal rotamer designations [m (−60° = 300°), p (+60°), t (180°)].
As can be seen in Figure 14, the {m,m} rotamer is a minority for the native side chains, but is dominant for the R1 side chain (and 4-Br derivative). As discussed above, it is considered likely that the predominance of the {m,m} state for R1 is determined by the Sδ···HCα interaction and possibly Sδ desolvation that is absent in the native residues, except for methionine, which also has an Sδ atom. Although methionine has the highest proportion of {m,m} rotamers of the native residues, the fact that it is not dominant suggests that unique properties of the disulfide are involved in stabilizing the {m,m} state. As noted above, half cysteines also have a preponderance of the {m,m} configuration.
Conversely, the {m,t} rotamer is preferred in native side chains, but represented by only one of 11 structures of R1, namely, 75R1 (Langen et al. 2000). In this rotamer, the disulfide is moved away from interaction with the backbone, and the rarity of this rotamer in the limited number of structures of R1 solved so far is likely a result of the correspondingly higher energy. Residue 75R1 is located at a crystal contact site, completely buried in the interface, and packing constraints may determine the {m,t} rotamer. The 118R1 rotamer is highly distorted and not classifiable in one of the traditional rotameric states (Fig. 14B). Like 75R1, the conformation is likely determined by packing interactions in the highly constrained interior of the protein. Although too few structures of R1 are available for a final conclusion, it appears at this point that R1 is “rotameric” at solvent-exposed sites on helical surfaces, being either {m,m}, {t,p}, or {t,m}, but that it may adopt a range of χ1-, χ2-values in response to packing constraints at buried sites. Apparently, the strength of the backbone interactions with Sδ is sufficient to restrict internal motions of the side chain so that the nitroxide motion is not completely averaged by rotational motions about the five bonds connecting it to the backbone, but is sufficiently weak that it may rearrange in response to packing interactions and be accepted at buried sites.
Materials and Methods
Construction, expression, purification, and spin labeling of T4L mutants
The template T4L gene used for mutagenesis was that coding for the cysteine-free “pseudo-wild-type” protein containing the substitutions C54T and C97A (Matsumura and Matthews 1989; Nicholson et al. 1991) kindly provided by F.W. Dahlquist (University of California, Santa Barbara); this is referred to as WT*. Cysteine mutants at positions 119 and 128−135 have been previously reported (Mchaourab et al. 1996, 1999). Other mutants were constructed using the overlap extension PCR method as described by Ho et al. (1989). Genes for all mutants were verified by sequencing.
T4L proteins were expressed and purified as previously described (Mchaourab et al. 1996; Columbus et al. 2001). Protein purity was at least 95% judged by SDS-PAGE. Spin labeling of single cysteine substitution mutants in labeling buffer (50 mM MOPS, 25 mM NaCl at pH 6.8) was carried out at 4°C overnight with a 10-fold molar excess of either 1-oxyl-3-methanesulfonythiomethyl-2,5-dihydro-2,2,5,5-tetramethyl-1H-pyrrole (to generate side chain R1) or 1-oxyl-3-bromo-4-methanesulfonythiomethyl-2,5-dihydro-2,2,5,5-tetramethyl-1H-pyrrole (to generate side chain R7). Excess spin label was removed using a HiTrap (Amersham) desalting column, and the labeled proteins were concentrated using YM-10 Microcon filter concentrators (Millipore). The spin-labeling reagents were synthesized as reported (Hankovszky et al. 1980; Berliner et al. 1982; Kálai et al. 1998).
Crystallization of T4L containing nitroxide side chains
Crystallization of spin-labeled T4L proteins was carried out under the nonreducing conditions described previously using the hanging-drop vapor diffusion method with a mixture of sodium and potassium phosphate as the precipitant (Jacobson et al. 1992). Crystallization trials were set up at room temperature (∼21°) by mixing 2 μL of protein solution (10−20 mg/mL in 50 mM MOPS, 25 mM NaCl at pH 6.8) with an equal volume of well buffer (2.0 M Na/KPO4, 0.24 M NaCl, 40 mM 2-hydroxyethyl disulfide) of four different pH values (6.2, 6.6, 6.9, and 7.2). Drops of the protein solution were suspended over 1 mL of the same buffer. Crystals of 115R1 and 115R1/R119A appeared within 1 wk, and crystals of 115R7 and 118R1 appeared in 4–6 wk.
Data collection, structure determination, and refinement
For data collection, crystals were flash-frozen in a cryostream of N2 gas at 100 K using mineral oil as a cryoprotectant. Diffraction data for 115R1, 115R7, and 118R1 were collected at 100 K at Brookhaven National Laboratory using beamline X8C of the National Synchrotron Light Source. Diffraction data of mutant 115R1/R119A were collected at 100 K using a Rigaku FRD generator with an R-AXIS IV++ detector. For room temperature data of 115R1, the crystal was mounted in a siliconized glass capillary using well buffer to maintain vapor pressure, and the data were collected using a Rigaku FRD generator with an R-AXIS IV++ detector. Data reduction and scaling were done with the programs DENZO and SCALEPACK (Otwinowski and Minor 1996). The data processing statistics are listed in Table 1. The crystals of all mutants grew in space group P3221 and were isomorphous with WT T4L crystals with similar cell dimensions.
Structures were determined by the molecular replacement method using the program EPMR (Kissinger et al. 1999). The WT T4L coordinates (PDB ID 3LZM) were used as a search model with residues 54, 97, and other mutation sites replaced by glycine. The initial R-factors before any refinement ranged from 34% to 39%. The structure of 118R1 was refined using the program CNS (Brunger et al. 1998). The structures of 115R1, 115R1/R119A, and 115R7 were refined using the program SHELXL (Sheldrick and Schneider 1997). The refinement was monitored using the free R-factor. A subset (5%) of the diffraction data was chosen randomly and omitted from refinement for calculation of the free R-factor. The model was rebuilt manually during refinement using the program O (Jones et al. 1991). The spin label side chains were manually built when the R-factor dropped to ∼20%. The final structures were validated with the programs ProCheck (Laskowski et al. 1993), SFCHECK (Vaguine et al. 1999), Errat (Colovos and Yeates 1993), and WHAT_CHECK (Hooft et al. 1996). The final refinement statistics are reported in Table 2. The final coordinates and structure factors have been deposited in the Protein Data Bank under entries 2IGC, 2OU8, 2OU9, 2NTG, and 2NTH (Table 2).
Cavity volumes were calculated using GRASP (Nicholls et al. 1991) with a 1.2 Å probe. Accessible surface areas were calculated using the algorithm of Lee and Richards (1971) as implemented in the program NACCESS (Hubbard and Thorton 1996), using a probe size of 1.4 Å. The fractional solvent accessibility for the side chain of a given residue X was calculated from the ratio of the ASA in the protein to that of the same residue in the extended Ala-X-Ala tripeptide, as recommended by Hubbard et al. (1991).
EPR spectroscopy and spectral simulations
EPR spectra were recorded on a Varian E-109 X-band spectrometer fitted with a loop-gap resonator at room temperature using a microwave power of 2 mW and modulation amplitude optimized to the natural linewidth of each individual spectrum. Solution spectra were obtained with spin-labeled T4L mutant proteins (100−500 μM). All protein samples also contained 30% (w/w) sucrose in order to increase solution viscosity and thereby minimize the contribution of protein rotation to the EPR spectral line shape (Mchaourab et al. 1996, 1999). To obtain polycrystalline spectra of the labeled proteins, several crystals were crushed in the mother liquor; the spectrum was found to be independent of the orientation of the sample in the resonator.
Experimental EPR spectra of 115R1 were fit to a two-component model using the program NLSL developed by Freed and coworkers (Schneider and Freed 1989; Budil et al. 1996). The crystal structure of 115R1 indicates that the nitroxides corresponding to both components are solvent exposed, and values for the magnetic A and g tensors appropriate for a generic aqueous environment were selected as starting values (Columbus et al. 2001). A final set of values based on global fits to the spectra from the various mutants and conditions explored is (1) for the immobile component Axx = 5.9 ± 0.5, Ayy = 5.6 ± 0.2, Azz = 37.3 ± 0.2; gxx = 2.0083 ± 0.0001, gyy = 2.0060 ± 0.0001, and gzz = 2.0022; (2) for the mobile component, Axx = 6.3 ± 0.3, Ayy = 6.9 ± 0.1, Azz = 36.9; gxx = 2.0072; gyy = 2.0053, and gzz = 2.0022. The g-values are all given relative to gzz = 2.0022. These values were used in fits of all 115R1 spectra; slight improvements were obtained by small variations that did not exceed those indicated by ± values.
Axially symmetry motion of the nitroxide is assumed for simplicity. In terms of the elements of the rotational diffusion tensor, Rz ≡ R par, Rx = Ry ≡ R perp. In the modified spherical tensor representation, the motion is described by the geometric mean rotational diffusion constant 〈R〉 = (R par R perp 2)1/3 and the rotational anisotropy parameter N = R par/R perp (Budil et al. 1996). The effective correlation time is computed as τ = 1/6〈R〉. To constrain the number of fitting parameters, the immobilized state is taken to have a simple isotropic motion (N = 1), as would be the case if R1 motion is determined by rotational diffusion of the roughly spherical T4L molecule. The results presented below justify this assumption. The more mobile state is described by the MOMD model (Budil et al. 1996) with a z-axis anisotropic motion characteristic of R1 at noninteracting surface sites (Columbus et al. 2001). Spatial ordering of the anisotropic motion is accounted for by an order parameter, S, computed from the single C 20 coefficient of the ordering potential varied in the simulations (Budil et al. 1996). To further constrain the number of fitting parameters, the symmetry axis of the diffusion tensor is tilted by 36° relative to the z-molecular axis of the nitroxide, as found for R1 on helix surface sites (Columbus et al. 2001).
Least-squares fitting of the two-component spectra thus involved variation of only 〈R〉, N, and C 20 as dynamic parameters to describe the motion of the mobile component, and only 〈R〉 for the more immobile component (N = 1). For the mobile component, N = 0.4 was found to give good global fits to the various 115R1 spectra, and was fixed at that value to further reduce the number of fitting parameters. The program NLSL does not allow for independent variation of the populations in two-component fits, and the relative populations are an output of the fitting.
The spectrum of buried site 118R1 also has two components, although it is strongly dominated by an immobilized state. For fitting of this spectrum, starting values of the A and g tensors for the immobilized state were obtained from fitting the spectrum obtained in the absence of motion in frozen solution (203 K). Values of Axx and Ayy are not well determined by this procedure and were allowed to vary in the final fits. The final values used were Axx = 6.5, Ayy = 5.9, Azz = 35.3, gxx = 2.0075, gyy = 2.0057, and gzz = 2.0022. The mobile component was assumed to be solvent exposed, and appropriate values were Axx = 5.9, Ayy = 6.3, Azz = 37.0, gxx = 2.0080, gyy = 2.0060, and gzz = 2.0022. Residue 133R1 is also a buried site but whose spectrum represents a single dynamic component. Values of A and g were again obtained by fitting the spectrum obtained at 203 K and were Axx = 5.9, Ayy = 5.4, Azz = 35.3, gxx = 2.0088, gyy = 2.0060, and gzz = 2.0022. Fitting procedures for the two-component spectrum of 118R1 followed those given above for 115R1.
Electronic supplemental material
Supplemental Figure S1 shows far-UV circular dichroism spectra of WT* T4L in the presence and absence of 20% dioxane. Supplemental Figure S2 shows the thermal melting of 118R1 and WT* T4L. These figures are included in a Microsoft Word file named: guo_supplementary_material.doc.
Acknowledgments
Research reported was supported by NIH Grant EY 05200, the Jules Stein Professor Endowment, and grant OTKA T048334 from the Hungarian National Research Fund. The authors thank Mark Fleissner for helpful discussions.
Footnotes
Supplemental material: see www.proteinscience.org
Reprint requests to: Wayne L. Hubbell, Jules Stein Eye Institute, UCLA School of Medicine, Los Angeles, CA 90095-7008, USA; e-mail: hubbellw@jsei.ucla.edu; fax: (310) 794-2114.
Abbreviations: SDSL, site-directed spin labeling; EPR, electron paramagnetic resonance; T4L, T4 lysozyme; WT*, pseudo-wild-type T4L; MOMD, microscopic order and macroscopic disorder.
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.062739107.
References
- Altenbach C., Oh, K.J., Trabanino, R.J., Hideg, K., and Hubbell, W.L. 2001. Estimation of inter-residue distances in spin labeled proteins at physiological temperatures: Experimental strategies and practical limitations. Biochemistry 40: 15471–15482. [DOI] [PubMed] [Google Scholar]
- Baldwin E.P., Hajiseyedjavadi, O., Baase, W.A., and Matthews, B.W. 1993. The role of backbone flexibility in the accommodation of variants that repack the core of T4 lysozyme. Science 262: 1715–1718. [DOI] [PubMed] [Google Scholar]
- Baldwin E., Xu, J., Hajiseyedjavadi, O., Baase, W.A., and Matthews, B.W. 1996. Thermodynamic and structural compensation in “size-switch” core repacking variants of bacteriophage T4 lysozyme. J. Mol. Biol. 259: 542–559. [DOI] [PubMed] [Google Scholar]
- Berliner L.J., Grunwald, J., Hankovszky, H.O., and Hideg, K. 1982. A novel reversible thiol-specific spin label: Papain active site labeling and inhibition. Anal. Biochem. 119: 450–455. [DOI] [PubMed] [Google Scholar]
- Bhattacharyya R., Pal, D., and Chakrabarti, P. 2004. Disulfide bonds, their stereospecific environment and conservation in protein structures. Protein Eng. Des. Sel. 17: 795–808. [DOI] [PubMed] [Google Scholar]
- Brunger A.T., Adams, P.D., Clore, G.M., DeLano, W.L., Gros, P., Grosse-Kunstleve, R.W., Jiang, J.S., Kuszewski, J., Nilges, M., Pannu, N.S., et al. 1998. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 54: 905–921. [DOI] [PubMed] [Google Scholar]
- Budil D.E., Lee, S., Saxena, S., and Freed, J.H. 1996. Nonlinear-least-squares analysis of slow-motion EPR spectra in one and two dimensions using a modified Levenberg-Marquardt algorithm. J. Magn. Reson. 120: 155–189. [Google Scholar]
- Burling F.T. and Goldstein, B.M. 1993. A database study of nonbonded intramolecular sulfur nucleophile contacts. Acta Crystallogr. B 49: 738–744. [DOI] [PubMed] [Google Scholar]
- Colovos C. and Yeates, T.O. 1993. Verification of protein structures: Patterns of nonbonded atomic interactions. Protein Sci. 2: 1511–1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Columbus L. and Hubbell, W.L. 2002. A new spin on protein dynamics. Trends Biochem. Sci. 27: 288–295. [DOI] [PubMed] [Google Scholar]
- Columbus L. and Hubbell, W.L. 2004. Mapping backbone dynamics in solution with site-directed spin labeling: GCN4–58 bZip free and bound to DNA. Biochemistry 43: 7273–7287. [DOI] [PubMed] [Google Scholar]
- Columbus L., Kálai, T., Jeko, J., Hideg, K., and Hubbell, W.L. 2001. Molecular motion of spin labeled side chains in α-helices: Analysis by variation of side chain structure. Biochemistry 40: 3828–3846. [DOI] [PubMed] [Google Scholar]
- Crane J.M., Suo, Y.Y., Lilly, A.A., Mao, C.F., Hubbell, W.L., and Randall, L.L. 2006. Sites of interaction of a precursor polypeptide on the export chaperone SecB mapped by site-directed spin labeling. J. Mol. Biol. 363: 63–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dani V.S., Ramakrishnan, C., and Varadarajan, R. 2003. MODIP revisited: Re-evaluation and refinement of an automated procedure for modeling of disulfide bonds in proteins. Protein Eng. 16: 187–193. [DOI] [PubMed] [Google Scholar]
- Dunbrack R.L. and Cohen, F.E. 1997. Bayesian statistical analysis of protein side-chain rotamer preferences. Protein Sci. 6: 1661–1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fanucci G.E. and Cafiso, D.S. 2006. Recent advances and applications of site-directed spin labeling. Curr. Opin. Struct. Biol. 16: 644–653. [DOI] [PubMed] [Google Scholar]
- Frauenfelder H., Hartmann, H., Karplus, M., Kuntz, I.D., Kuriyan, J., Parak, F., Petsko, G.A., Ringe, D., Tilton, R.F., Connolly, M.L., et al. 1987. Thermal expansion of a protein. Biochemistry 26: 254–261. [DOI] [PubMed] [Google Scholar]
- Hamaguchi K. and Kurono, A. 1963. Structure of muramidase (lysozyme). III. Effect of 2-chloroethanol, ethanol and dioxane on the stability of muramidase. J. Biochem. 54: 497–505. [DOI] [PubMed] [Google Scholar]
- Hankovszky H.O., Hideg, K., and Lex, L. 1980. Nitroxyls. VII. Synthesis and reactions of highly reactive 1-oxyl-2,2,5,5-tetramethyl-2,5-dihydropyrrole-3-ylmethyl sulfonates. Synthesis (Mass.) 1980: 914–916. [Google Scholar]
- Hartmann H., Parak, F., Steigemann, W., Petsko, G.A., Ponzi, D.R., and Frauenfelder, H. 1982. Conformational substates in a protein—Structure and dynamics of metmyoglobin at 80-K. Proc. Natl. Acad. Sci. 79: 4967–4971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho S.N., Hunt, H.D., Horton, R.M., Pullen, J.K., and Pease, L.R. 1989. Site-directed mutagenesis by overlap extension using the polymerase chain-reaction. Gene 77: 51–59. [DOI] [PubMed] [Google Scholar]
- Hooft R.W., Vriend, G., Sander, C., and Abola, E.E. 1996. Errors in protein structures. Nature 381: 272. [DOI] [PubMed] [Google Scholar]
- Hubbard S.J. and Thornton, J.M. 1996. NACCESS computer program, 2.1.1 ed. University College, London.
- Hubbard S.J., Campbell, S.F., and Thornton, J.M. 1991. Molecular recognition: Conformational analysis of limited proteolytic sites and serine proteinase protein inhibitors. J. Mol. Biol. 220: 507–530. [DOI] [PubMed] [Google Scholar]
- Hubbell W.L., McHaourab, H.S., Altenbach, C., and Lietzow, M.A. 1996. Watching proteins move using site-directed spin labeling. Structure 4: 779–783. [DOI] [PubMed] [Google Scholar]
- Hubbell W.L., Gross, A., Langen, R., and Lietzow, M.A. 1998. Recent advances in site-directed spin labeling of proteins. Curr. Opin. Struct. Biol. 8: 649–656. [DOI] [PubMed] [Google Scholar]
- Hubbell W.L., Cafiso, D.S., and Altenbach, C. 2000. Identifying conformational changes with site-directed spin labeling. Nat. Struct. Biol. 7: 735–739. [DOI] [PubMed] [Google Scholar]
- Isas J.M., Langen, R., Haigler, H.T., and Hubbell, W.L. 2002. Structure and dynamics of a helical hairpin and loop region in annexin 12: A site-directed spin labeling study. Biochemistry 41: 1464–1473. [DOI] [PubMed] [Google Scholar]
- Izumi T. and Inoue, H. 1977. Effect of dioxane on the conformation of lysozyme at low pH.I. Studies by circular dichroism and viscosity measurements. J. Biochem. 81: 1769–1779. [DOI] [PubMed] [Google Scholar]
- Jacobson R.H., Matsumura, M., Faber, H.R., and Matthews, B.W. 1992. Structure of a stabilizing disulfide bridge mutant that closes the active-site cleft of T4 lysozyme. Protein Sci. 1: 46–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones T.A., Zou, J.Y., Cowan, S.W., and Kjeldgaard, M. 1991. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A 47: 110–119. [DOI] [PubMed] [Google Scholar]
- Juers D.H. and Matthews, B.W. 2001. Reversible lattice repacking illustrates the temperature dependence of macromolecular interactions. J. Mol. Biol. 311: 851–862. [DOI] [PubMed] [Google Scholar]
- Juers D.H. and Matthews, B.W. 2004. Cryo-cooling in macromolecular crystallography: Advantages, disadvantages and optimization. Q. Rev. Biophys. 37: 105–119. [DOI] [PubMed] [Google Scholar]
- Kálai T., Balog, M., Jeko, J., and Hideg, K. 1998. 3-Substituted 4-bromo-2,2,5,5-tetramethyl-2,5-dihydro-1H-pyrrol-1-yloxyl radicals as versatile synthons for synthesis of new paramagnetic heterocycles. Synthesis (Mass.) 1476–1482.
- Kissinger C.R., Gehlhaar, D.K., and Fogel, D.B. 1999. Rapid automated molecular replacement by evolutionary search. Acta Crystallogr. D Biol. Crystallogr. 55: 484–491. [DOI] [PubMed] [Google Scholar]
- Kusnetzow A.K., Altenbach, C., and Hubbell, W.L. 2006. Conformational states and dynamics of rhodopsin in micelles and bilayers. Biochemistry 45: 5538–5550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langen R., Oh, K.J., Cascio, D., and Hubbell, W.L. 2000. Crystal structures of spin labeled T4 lysozyme mutants: Implications for the interpretation of EPR spectra in terms of structure. Biochemistry 39: 8396–8405. [DOI] [PubMed] [Google Scholar]
- Laskowski R.A., MacArthur, M.W., Moss, D.S., and Thornton, J.M. 1993. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26: 283–291. [Google Scholar]
- Lee B. and Richards, F.M. 1971. The interpretation of protein structures: Estimation of static accessibility. J. Mol. Biol. 55: 379–400. [DOI] [PubMed] [Google Scholar]
- Lietzow M.A. and Hubbell, W.L. 2004. Motion of spin label side chains in cellular retinol-binding protein: Correlation with structure and nearest-neighbor interactions in an antiparallel β-sheet. Biochemistry 43: 3137–3151. [DOI] [PubMed] [Google Scholar]
- Lim W.A., Hodel, A., Sauer, R.T., and Richards, F.M. 1994. The crystal structure of a mutant protein with altered but improved hydrophobic core packing. Proc. Natl. Acad. Sci. 91: 423–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu R.J., Baase, W.A., and Matthews, B.W. 2000. The introduction of strain and its effects on the structure and stability of T4 lysozyme. J. Mol. Biol. 295: 127–145. [DOI] [PubMed] [Google Scholar]
- Lovell S.C., Word, J.M., Richardson, J.S., and Richardson, D.C. 2000. The penultimate rotamer library. Proteins 40: 389–408. [PubMed] [Google Scholar]
- Matsumura M. and Matthews, B.W. 1989. Control of enzyme activity by an engineered disulfide bond. Science 243: 792–794. [DOI] [PubMed] [Google Scholar]
- Mchaourab H.S., Lietzow, M.A., Hideg, K., and Hubbell, W.L. 1996. Motion of spin-labeled side chains in T4 lysozyme: Correlation with protein structure and dynamics. Biochemistry 35: 7692–7704. [DOI] [PubMed] [Google Scholar]
- Mchaourab H.S., Kalai, T., Hideg, K., and Hubbell, W.L. 1999. Motion of spin-labeled side chains in T4 lysozyme: Effect of side chain structure. Biochemistry 38: 2947–2955. [DOI] [PubMed] [Google Scholar]
- Morton A. and Matthews, B.W. 1995. Specificity of ligand binding in a buried nonpolar cavity of T4 lysozyme: Linkage of dynamics and structural plasticity. Biochemistry 34: 8576–8588. [DOI] [PubMed] [Google Scholar]
- Nagao Y., Hirata, T., Goto, S., Sano, S., Kakehi, A., Iizuka, K., and Shiro, M. 1998. Intramolecular nonbonded S···O interaction recognized in (acylimino)thiadiazoline derivatives as angiotensin II receptor antagonists and related compounds. J. Am. Chem. Soc. 120: 3104–3110. [Google Scholar]
- Nicholls A., Sharp, K.A., and Honig, B. 1991. Protein folding and association: Insights from the interfacial and thermodynamic properties of hydrocarbons. Proteins 11: 281–296. [DOI] [PubMed] [Google Scholar]
- Nicholson H., Anderson, D.E., Dao-pin, S., and Matthews, B.W. 1991. Analysis of the interaction between charged side chains and the α-helix dipole using designed thermostable mutants of phage T4 lysozyme. Biochemistry 30: 9816–9828. [DOI] [PubMed] [Google Scholar]
- Nozaki Y. and Tanford, C. 1971. The solubility of amino acids and two glycine peptides in aqueous ethanol and dioxane solutions. Establishment of a hydrophobicity scale. J. Biol. Chem. 246: 2211–2217. [PubMed] [Google Scholar]
- Otwinowski Z. and Minor, W. 1996. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276: 307–326. [DOI] [PubMed] [Google Scholar]
- Pal D. and Chakrabarti, P. 2001. Non-hydrogen bond interactions involving the methionine sulfur atom. J. Biomol. Struct. Dyn. 19: 115–128. [DOI] [PubMed] [Google Scholar]
- Rosenfield R.E., Parthasarathy, R., and Dunitz, J.D. 1977. Directional preferences of nonbonded atomic contacts with divalent sulfur. 1. Electrophiles and nucleophiles. J. Am. Chem. Soc. 99: 4860–4862. [Google Scholar]
- Schneider D.J. and Freed, J.H. 1989. Calculating slow motional magnetic resonance spectra. In Spin labeling: Theory and applications (ed. J. Reuben), pp. 1–76. Plenum Press, New York.
- Schrauber H., Eisenhaber, F., and Argos, P. 1993. Rotamers: To be or not to be? An analysis of amino acid side-chain conformations in globular proteins. J. Mol. Biol. 230: 592–612. [DOI] [PubMed] [Google Scholar]
- Sheldrick G.M. and Schneider, T.R. 1997. SHELXL: High resolution refinement. Methods Enzymol. 277: 319–343. [PubMed] [Google Scholar]
- Skrynnikov N.R., Dahlquist, F.W., and Kay, L.E. 2002. Reconstructing NMR spectra of “invisible” excited protein states using HSQC and HMQC experiments. J. Am. Chem. Soc. 124: 12352–12360. [DOI] [PubMed] [Google Scholar]
- Tilton R.F., Dewan, J.C., and Petsko, G.A. 1992. Effects of temperature on protein structure and dynamics: X-ray crystallographic studies of the protein ribonuclease-A at nine different temperatures from 98 to 320 K. Biochemistry 31: 2469–2481. [DOI] [PubMed] [Google Scholar]
- Vaguine A.A., Richelle, J., and Wodak, S.J. 1999. SFCHECK: A unified set of procedures for evaluating the quality of macromolecular structure-factor data and their agreement with the atomic model. Acta Crystallogr. D Biol. Crystallogr. 55: 191–205. [DOI] [PubMed] [Google Scholar]
- Van Wart H.E. and Scheraga, H.A. 1977. Stable conformations of aliphatic disulfides: Influence of 1,4 interactions involving sulfur atoms. Proc. Natl. Acad. Sci. 74: 13–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wynn R., Anderson, C.L., Richards, F.M., and Fox, R.O. 1995. Interactions in nonnative and truncated forms of staphylococcal nuclease as indicated by mutational free energy changes. Protein Sci. 4: 1815–1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wynn R., Harkins, P.C., Richards, F.M., and Fox, R.O. 1996. Mobile unnatural amino acid side chains in the core of staphylococcal nuclease. Protein Sci. 5: 1026–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wynn R., Harkins, P.C., Richards, F.M., and Fox, R.O. 1997. Comparison of straight chain and cyclic unnatural amino acids embedded in the core of staphylococcal nuclease. Protein Sci. 6: 1621–1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young A.C.M., Tilton, R.F., and Dewan, J.C. 1994. Thermal expansion of hen egg-white lysozyme. Comparison of the 1.9 Å resolution structures of the tetragonal form of the enzyme at 100 K and 298 K. J. Mol. Biol. 235: 302–317. [DOI] [PubMed] [Google Scholar]