

Abstract
Calmodulin (CaM) is an EF-hand protein composed of two calcium (Ca2+)-binding EF-hand motifs in its N-domain (EF-1 and EF-2) and two in its C-domain (EF-3 and EF-4). In this study, we examined the structure, dynamics, and Ca2+-binding properties of a fragment of CaM containing only EF-2 and EF-3 and the intervening linker sequence (CaM2/3). Based on NMR spectroscopic analyses, Ca2+-free CaM2/3 is predominantly unfolded, but upon binding Ca2+, adopts a monomeric structure composed of two EF-hand motifs bridged by a short antiparallel β-sheet. Despite having an “even–odd” pairing of EF-hands, the tertiary structure of CaM2/3 is similar to both the “odd–even” paired N- and C-domains of Ca2+-ligated CaM, with the conformationally flexible linker sequence adopting the role of an inter-EF-hand loop. However, unlike either CaM domain, CaM2/3 exhibits stepwise Ca2+ binding with a K d1 = 30 ± 5 μM to EF-3, and a K d2 > 1000 μM to EF-2. Binding of the first equivalent of Ca2+ induces the cooperative folding of CaM2/3. In the case of native CaM, stacking interactions between four conserved aromatic residues help to hold the first and fourth helices of each EF-hand domain together, while the loop between EF-hands covalently tethers the second and third helices. In contrast, these aromatic residues lie along the second and third helices of CaM2/3, and thus are positioned adjacent to the loop between its “even–odd” paired EF-hands. This nonnative hydrophobic core packing may contribute to the weak Ca2+ affinity exhibited by EF-2 in the context of CaM2/3.
Keywords: calmodulin, calcium-binding protein, EF-hand, NMR spectroscopy, protein structure and dynamics
Calcium (Ca2+) binding proteins are often composed of EF-hand motifs associated into folded EF-hand domains. An EF-hand domain, which is typically composed of two EF-hands connected by a flexible loop, is stabilized by an antiparallel β-sheet and by the packing of hydrophobic side chains from neighboring helices. A key feature of this hydrophobic packing is the presence of four stacked aromatic residues found on the first and the fourth helices of the domain (Babu et al. 1988). As a prototypical EF-hand protein, calmodulin (CaM) contains four Ca2+ binding EF-hand motifs: EF-1 and EF-2 associate to form its N-domain, and EF-3 and EF-4 associate to form its C-domain. The two domains are tethered through a flexible intervening sequence, known as the linker (Babu et al. 1988).
The arrangement of EF-hands in CaM is thought to be a consequence of its evolution from a progenitor EF-hand by gene duplication. In this theory, a progenitor EF-hand gave rise to a progenitor EF-hand domain. This in turn duplicated into a two-domain progenitor EF-hand protein, which finally diverged into a family of Ca2+-binding proteins, including CaM and troponin C (TnC) (Nakayama et al. 1992). Accordingly, the “odd–even” paired EF-1 and EF-2 have evolved to optimize their association to form the N-domain of CaM, as have EF-3 and EF-4 to form the C-domain. This optimization includes the ability to cooperatively bind Ca2+ with an overall affinity necessary to respond to changes in the physiological concentrations of this cation and thereby regulate numerous signaling pathways.
Although EF-hand sequences have evolved to specifically pair into domains, there is still remarkable plasticity in their potential modes of association. This is evidenced by the peptide models of single EF-hands that form heterodimers, as well as those that self-associate as homodimers. Examples of homodimers include single EF-hand peptides of CaM (EF-3) (Reid 1987a), TnC (EF-3 and EF-4) (Kay et al. 1991; Shaw et al. 1992), and chimeras of parvalbumin and CaM (Franchini and Reid 1999). Heterodimers include EF-1:EF-2 of CaM or calbindin D9k and EF-3:EF-4 of CaM or TnC (Finn et al. 1992; Shaw and Sykes 1996; Shuman et al. 2006). The tertiary structures of the two TnC homodimers are very similar to the C-domain of native TnC and, by extension, all EF-hand domains (Kay et al. 1991; Shaw et al. 1992). Despite this structural similarity, Ca2+ binding and cooperativity are compromised in the homodimeric EF-hand models (Reid 1987b; Kay et al. 1991; Shaw et al. 1991). Heterodimers of EF-hand peptides have improved Ca2+ affinities over those of homodimers, yet are still significantly lower than in their respective native proteins (Finn et al. 1992; Shaw and Sykes 1996).
To investigate further how EF-hand association affects the structure, Ca2+ affinity, and cooperativity of CaM, we examined a previously uncharacterized fragment of this protein spanning residues 46–113 (CaM2/3) (Fig. 1). This fragment encompasses EF-2 and EF-3, as well as the intervening sequence that normally serves as the flexible linker between its N- and C-domains. Although predominantly unstructured in its apo form, Ca2+ binding induces the folding and nonnative intramolecular association of EF-2 and EF-3. Using NMR spectroscopy, we demonstrate that, despite an “even–odd” arrangement of EF-hands, CaM2/3 adopts a tertiary structure very similar to both the N- and C-domains of Ca2+-ligated CaM. However, in contrast to the high affinity and positive cooperativity for Ca2+ binding exhibited by these two domains, either in isolation or in native CaM (Linse et al. 1991), NMR spectroscopic and CD titration studies show that CaM2/3 binds Ca2+ in a stepwise manner, with a K d1 of 30 ± 5 μM to EF-3, and a K d2 of >1000 μM to EF-2. A similar behavior of one moderate-affinity and one low-affinity Ca2+-binding site was found for the homodimer EF-hand peptides from TnC (Kay et al. 1991; Shaw et al. 1992).
Figure 1.

The sequence and NMR-derived secondary structure of CaM2/3. (A) The amino acid sequence of CaM2/3 (residues 46–113) is highlighted in light gray within the sequence of full-length human CaM. The sequences of EF-3 and EF-2 are also aligned with those of EF-1 and EF-4, respectively, using the 12-residue Ca2+-binding segments (red type). Conserved aromatic residues important for forming part of the hydrophobic core of each domain are highlighted in green. Residues corresponding to the linker between the N- and C-domains of native CaM are double underlined. Residues corresponding to the loops between EF-hands in each domain are single underlined. (B) The amino acid sequence of CaM2/3 with the positions of (h) helices (cylinder) and (b) β-strands (arrows) identified by PROMOTIF (Hutchinson and Thornton 1996) within the ensemble of NMR-derived structures for CaM2/3.
In the N-domain (or C-domain) of native CaM, and almost all other related EF-hand proteins, conserved aromatic residues on the first helix of EF-1 (or EF-3) and the last helix of EF-2 (or EF-4) partake in aromatic stacking interactions (Fig. 1; Babu et al. 1988). Along with the loop between the second helix of EF-1 (or EF-3) and the first helix of EF-2 (or EF-4), these aromatic residues help hold the “odd–even” paired EF-hands together in a cooperatively folded structural arrangement. We propose that the absence of either the cluster of aromatic residues or the covalent inter-EF-hand loop can reduce high-affinity, cooperative Ca2+ binding. In the case of CaM2/3, with an “even–odd” arrangement of EF-hands, the aromatic residues are on its middle two helices and thus adjacent to the loop formed by the intervening linker sequence. The nonnative positioning of these aromatic residues with respect to the loop may contribute to the resulting sequential binding of Ca2+ to one moderate- and one low-affinity site.
Results
CaM2/3 structure is stabilized by Ca2+ binding
CD and NMR spectroscopy were used to monitor the behavior of CaM2/3 as a function of Ca2+ concentration. The CD spectra of Ca2+-free CaM2/3 show minima near 208 and 222 nm, indicative of at least some transient helical content (Fig. 2, inset). However, the 15N-HSQC spectrum of fully Ca2+-free CaM2/3 shows poorly resolved amide resonances with 1H chemical shifts clustered near 8.5 ppm (Fig. 3A). Therefore, Ca2+-free CaM2/3 does not adopt a stable, folded structure, but rather exhibits features characteristic of a molten globule state (Redfield 2004). However, upon the incremental addition of Ca2+, a progressive disappearance of the Ca2+-free peaks and the concomitant appearance of a new set of well-dispersed amide peaks were observed in the 15N-HSQC spectra. These new dispersed peaks correspond to folded Ca2+-ligated CaM2/3 (Fig. 3B,C). In parallel, a significant decrease in ellipticity was observed by CD upon addition of Ca2+ (Fig. 2, inset). Together, these results demonstrate that Ca2+ binding induces the cooperative folding of CaM2/3 in the slow exchange regime on the NMR chemical shift timescale.
Figure 2.

Ca2+ titration of CaM2/3 monitored by CD spectropolarimetry. The mean ellipticity at 222 nm with standard deviations from four replicates (triangles), is plotted against total Ca2+ concentration and the fit line generated with the CaLigator software (Andre and Linse 2002). The plotted ellipticity data were not corrected for dilution. The total CaM2/3 concentration was 40 μM, and the apparent, stepwise Ca2+ dissociation constants are (mean and standard error) K d1 = 30 ± 5 μM and K d2 > 1000 μM (25°C, pH 7.4). Binding of the first Ca2+ accounts for ∼90% of the total induced CD change. (Inset) The far-UV CD spectra of Ca2+-free (dashed line) and fully Ca2+-ligated (solid line) CaM2/3.
Figure 3.
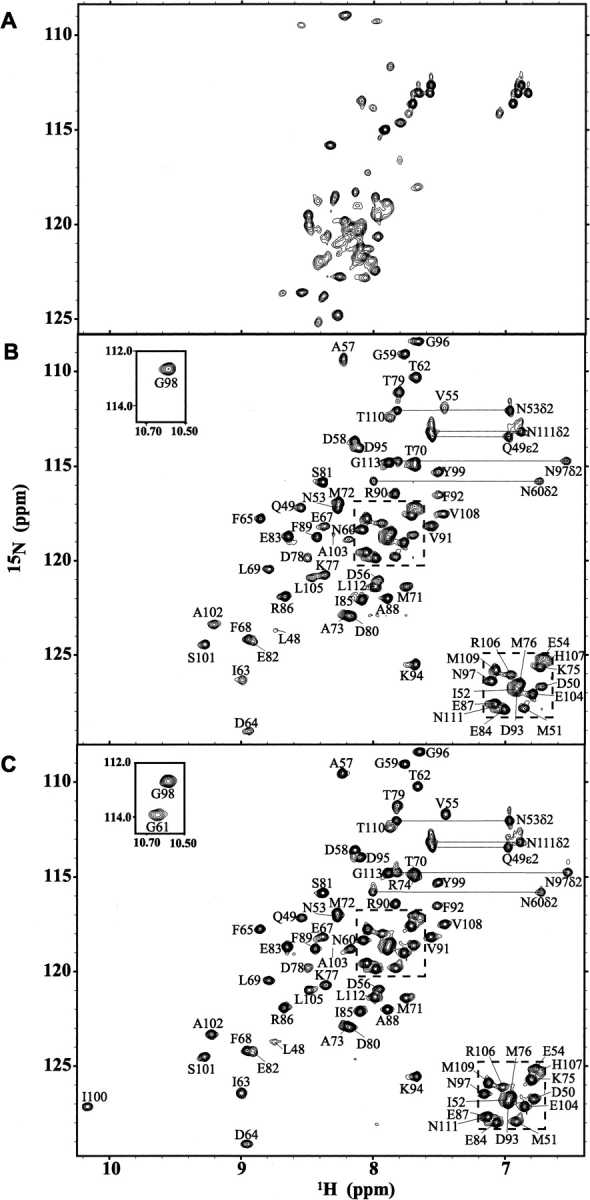
The 15N-HSQC spectra of 15N-CaM2/3. (A) The unassigned spectrum of Ca2+-free CaM2/3 at 25°C and pH 7.4. The poor 1HN dispersion indicates a lack of a predominant tertiary structure. The 15N HSQC spectra of 0.37 mM 15N-CaM2/3 in the presence of (B) 0.74 mM Ca2+ and (C) 1.8 mM Ca2+. Assigned peaks are numbered as in native CaM, with horizontal lines connecting resonances from 15NH2 groups. The insets with dashed lines correspond to the crowded regions near the center of the spectra. The insets bordered with a solid line show the downfield-shifted signals from G98 and G61. The peak for A57 is aliased from 130.9 ppm, and signals corresponding to residues A46 and E47 were not detected. Consistent with the K d values from CD-monitored titrations, the spectrum of CaM2/3 with equimolar Ca2+ was very similar to that of B, whereas signals from the amides of G61 and I100 were only observed in the presence of significantly higher concentrations of Ca2+ (C). Note that while 1.8 mM Ca2+ produces ∼65% occupancy of EF-2, the 15N-HSQC spectrum of CaM2/3 does not significantly change in the presence of 10 mM Ca2+, as used for subsequent structural studies to ensure >90% occupancy.
Most of the 15N-HSQC resonances corresponding to folded CaM2/3 were observed in the presence of ∼2:1 molar ratio of Ca2+:protein (Fig. 3B); however, a large excess of Ca2+ was required to obtain the spectra for fully Ca2+-loaded CaM2/3 (Fig. 3C). This is best illustrated by consideration of the amide hydrogen (1HN) resonances of G61 and G98. The downfield shifts of these nuclei, attributed to hydrogen bonding with the carboxyl side chains of D56 and D93, respectively, are diagnostic of a Ca2+-ligated EF-hand (Ikura et al. 1985; Kay et al. 1991). The signal from G98, corresponding to EF-3 in CaM2/3, is observed upon the initial addition of Ca2+ (Fig. 3B), whereas that from G61 is not detected until ∼5 molar equivalents of Ca2+ (1.8 mM) are present (Fig. 3C). This demonstrates that binding of Ca2+ to EF-3 is linked to the cooperative folding of CaM2/3 to an intermediate, albeit “native-like” structure. Complete folding requires Ca2+ binding to the lower-affinity EF-2. Furthermore, with one Ca2+ bound, the signals from EF-2 residues adjacent to G61, including D56, D58, N60, T62, I63, and D64, were relatively weak. Upon addition of excess Ca2+, these signals sharpened, but did not change in chemical shift. This behavior is suggestive of peak broadening due to localized conformational exchange, on the millisecond-to-microsecond timescale, for these residues about an “EF-hand-like” intermediate state. Ca2+ binding stabilizes the conformation of these residues, giving rise to sharper 15N-HSQC peaks.
Ca2+-binding affinity of CaM2/3
The Ca2+ affinity of CaM2/3 was measured using CD spectropolarimetry (Fig. 2). The titration data were fit with the program CaLigator (Andre and Linse 2002), yielding apparent dissociation constants of K d1 = 30 ± 5 μM and K d2 > 1000 μM. These widely divergent dissociation constants are indicative of sequential, two-step Ca2+ binding by CaM2/3 (Li et al. 1995). In conjunction with the NMR spectra of Figure 3, K d1 corresponds to the coupled folding of CaM2/3 and Ca2+ binding to EF-3, whereas K d2 reflects the subsequent weak binding of Ca2+ to EF-2.
NMR-derived structural ensemble of Ca2+-saturated CaM2/3
NMR spectroscopy was used to determine the tertiary structural ensemble of CaM2/3 (Fig. 4). The restraint data set and final structural statistics are summarized in Table 1. Note that 15N relaxation measurements (below), as well as gel filtration, light scattering, and isotope-filtered NOE experiments (data not shown) verify that CaM2/3 is monomeric under the experimental conditions of this study.
Figure 4.

The NMR-derived structural ensemble of Ca2+-ligated CaM2/3. (A) The lowest-energy structure (left) and 20-member structural ensemble of CaM2/3 superimposed onto each other using the backbone heavy atoms (Cα, C′, and N) within the helical regions. Following the nomenclature for native CaM, the helices are labeled C through F (residues 48–55, 65–73, 82–92, 102–109). (B) The hydrophobic side chains (green) within the lowest energy structure (left) and structural ensemble of CaM2/3, superimposed on the backbone of the lowest energy structure. In panels A and B, the helices are identified in red, the Ca2+ binding segments blue, the β-sheet cyan, the flexible linker region yellow, the disordered regions at the N- and C-termini (residues 46–47 and 110–113) magenta, and the Ca2+ ions black. (C) The surface representation of the lowest energy structure of CaM2/3. The hydrophobic residues are shaded green, aspartate and glutamate residues red, arginine and lysine residues blue, and the remaining residues gray. Residues R74, M76, R106, and M109 are labeled on the surfaces to assist in orientation.
Table 1.
NMR restraints and structural statistics for Ca2+-saturated CaM2/3

CaM2/3 is composed of four helices (using the nomenclature for native CaM: helix C, residues 48–55; D, 65–73; E, 82–92; and F, 102–109) and one small antiparallel β-sheet centered on I63 and I100 (Fig. 4A). The downfield 1HN and 15N chemical shifts of I63 and I100 provide evidence for this β-sheet structure. Based on previous NMR spectroscopic studies of EF-hand domains, these shifts can be attributed to hydrogen bonds between I63 1HN and I100 C=O as well as I100 1HN and I63 C=O (Ikura et al. 1985; Biekofsky et al. 1998). Additional evidence for this antiparallel β-sheet is found in NOE interactions detected between I63 1HN and I100 1HN, Y99 1Hα and F65 1HN, and S101 1Hβ and T62 1Hγ. This secondary structure of CaM2/3, which is nearly identical to that for the corresponding sequence in native CaM (Chattopadhyaya et al. 1992; Chou et al. 2001b), confirms that residues 48–72 and 82–109 form EF-hand motifs in CaM2/3. These motifs associate through the β-strand pairing and hydrophobic side-chain packing, thereby stabilizing the globular tertiary structure of CaM2/3 (Fig. 4B).
CaM2/3 has a well-defined hydrophobic core formed in particular by the packing of aromatic residues (Fig. 4B,C). This packing is defined by NOE interactions detected between F65 1Hα and F89 1Hɛ, F89 1Hβ and F65 1Hɛ, and F89 1HN and F92 1Hδ. The average distances between the aromatic ring centroids of residues F65 to F89, F89 to F68, and F68 to F92 are 4.1 ± 0.1 Å, 5.4 ± 0.2 Å, and 5.9 ± 0.4 Å, respectively. These distances are typical of aromatic residues involved in stacking interactions (McGaughey et al. 1998). They are also consistent with the average inter-centroid distances of 5.5 ± 0.2 Å, 5.5 ± 0.1 Å, and 5.8 ± 0.2 Å between the equivalent aromatic residues in both the N- and C-domains of several crystal structures of Ca2+-ligated CaM [1CLL.pdb, 1PWR.pdb, 1EXR.pdb, and 3CLN.pdb; residues: F65 to F16, F16 to F68, F68 to F19, and F(Y)138 to F89, F89 to F141, F141 to F92] (Babu et al. 1988; Chattopadhyaya et al. 1992; Wilson and Brunger 2000; Fallon and Quiocho 2003). The EF-hands of CaM2/3 are joined by a loop comprised of residues 74–81 (yellow regions in Fig. 4). Thus, in contrast to native CaM, where this sequence is a flexible interdomain linker, these residues now play an intradomain role in CaM2/3 corresponding to that of the loops between EF-1 and EF-2 and between EF-3 and EF-4 in the N- and C-domains of CaM, respectively (Fig. 1). As evidenced by high RMSDs within the structural ensemble, as well as by 15N relaxation measurements (below), this loop region is somewhat conformationally mobile. The flexibility of this region may accommodate the nonnative association of EF-2 and EF-3 in CaM2/3.
Structural evidence for Ca2+ binding by CaM2/3
The CaM2/3 ensemble includes two Ca2+ ions chelated by EF-2 and EF-3 (Fig. 4, black spheres) in a mode similar to that observed in native Ca2+-ligated CaM. Although not directly observable by NMR spectroscopic methods, experimental evidence for Ca2+ chelation at these EF-hands includes the following. First, in the presence of saturating Ca2+, the 1HN resonances of G61 and G98 are shifted dramatically downfield to 10.63 and 10.58 ppm, respectively (Fig. 3C). These chemical shifts, which are diagnostic of Ca2+-ligated EF-hand motifs, reflect the formation of hydrogen bonds to the carboxylates of D56 and D93 in native CaM (Ikura et al. 1985; Kay et al. 1991). Second, the large difference in chemical shifts (0.32–0.96 ppm) between the 1Hβ/1Hβ′ (or 1Hγ/1Hγ′ in the case of glutamate) of residues D58, N60, D64, E67, D93, D95, N97, and S101 is also indicative of their involvement in chelating Ca2+ (Shaw et al. 1992). Third, the downfield 1HN and 15N chemical shifts for I63 and I100 result from both β-sheet formation and Ca2+ chelation (Fig. 3B,C; Biekofsky et al. 1998). Fourth, key NOE interactions were identified between chelating residues D56, T62, and G59 in EF-2, as well as between D93 and G96, K94 and E104, and D95 and D98 in EF-3. The corresponding NOE-based distance restraints positioned the chelating residues close enough to justify the added Ca2+ restraints. Fifth, the conformational arrangement of the EF-hand motifs in CaM2/3 is similar to those observed in crystal structures of Ca2+-ligated CaM, yet distinct from those found in Ca2+-free CaM (Figs. 4 and 5; Chattopadhyaya et al. 1992; Kuboniwa et al. 1995). This is seen in terms of interhelical angles of CaM2/3 compared to both the Ca2+-free and ligated forms of CaM (Table 2), as well as by the lower backbone RMSDs of EF-2 and EF-3 of CaM2/3 superimposed on Ca2+-ligated CaM versus Ca2+-free CaM (Fig. 5; Table 3).
Figure 5.
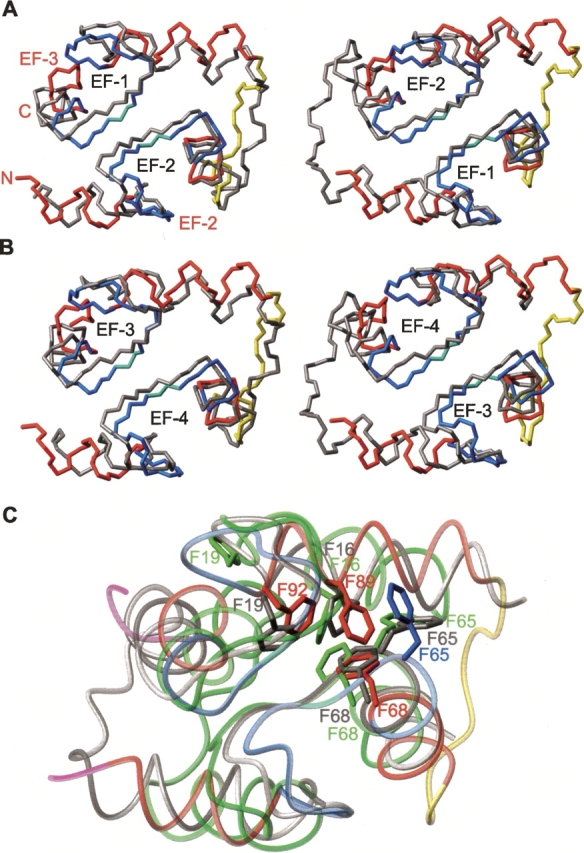
CaM2/3 superimposed onto the domains of CaM. (A,B) The backbone of the N- or C-domains of Ca2+-ligated CaM (gray) (1CLL.pdb) superimposed on the lowest-energy structure of CaM2/3 (colored according to Fig. 4). In this figure, the view of CaM2/3 is held approximately constant, and that of CaM varied, with the EF-hands of CaM black (the EF-hands as well as the N and C termini of CaM2/3 are red in A). (A, left) The superimposition of the N-domain EF-1 and EF-2 of Ca2+-ligated CaM on EF-2 and EF-3 of CaM2/3, respectively. (Right) The opposite superimposition (i.e., EF-2 and EF-1 of CaM on EF-2 and EF-3 of CaM2/3, respectively). (B, left) The superimposition of the C-domain EF-3 and EF-4 of Ca2+-ligated CaM on EF-2 and EF-3 of CaM2/3, respectively. (Right) The opposite superimposition. (C) The superimposition from A, (right) along with the crystal structure of the N-domain of Ca2+-free CaM (1QX5.pdb; green) displayed as transparent Cα tubes. The aromatic residues involved in stacking interactions are shown in gray for Ca2+-ligated CaM, green for Ca2+-free CaM, and according to secondary structure as in Figure 4 for CaM2/3. All superimpositions were made using backbone atoms Cα, C′, and N over the Ca2+-binding segments and eight residues of the flanking helical regions. See Table 3 for the corresponding RMSDs. The disordered regions at the N and C termini (residues 45–47 and 110–113) of CaM2/3 and the Ca2+ ions are removed for clarity.
Table 2.
Interhelical angles for CaM2/3 and native CaM

Table 3.
Structural alignment of the EF-hands and EF-hand domains of Ca2+-ligated CaM2/3 versus Ca2+-ligated and Ca2+-free CaM

Structural similarity of CaM2/3 to the N- and C-domains of native CaM
The overall structure of Ca2+-ligated CaM2/3 is remarkably similar to both the N- and C-domains of native Ca2+-ligated CaM (Fig. 5). As summarized in Table 3, the individual EF-hands of CaM2/3 superimpose well onto each of the individual EF-hands of Ca2+-ligated CaM. In addition, both EF-hands of CaM2/3 fit well onto either domain of Ca2+-ligated CaM. This is true regardless of which EF-hands are superimposed. For example, the superimposition of EF-2 and EF-3 from CaM2/3 onto EF-3 and EF-4, respectively, of the C-domain of CaM has a similar RMSD to that found for the reverse superimposition (i.e., onto EF-4 and EF-3, respectively). There is also substantial overlap between the relative positions of stacked aromatic residues in the core of CaM2/3 and those in both the N- and C-domains of Ca2+-ligated CaM (Fig. 5C). Furthermore, CaM2/3 has a cleft with exposed hydrophobic residues (Fig. 4B,C) that is similar to the hydrophobic target-binding pockets observed in both domains of CaM. The presence of this cleft is consistent with the observation that CaM2/3 also interacts with CaM targets (data not shown).
CaM2/3 backbone dynamics from amide 15N relaxation
The global and local dynamic properties of Ca2+-ligated CaM2/3 were probed by 15N T 1, T 2, and heteronuclear 1H-{15N}NOE measurements (Fig. 6A–C). Excluding residues exhibiting anomalous T 1/T 2 or heteronuclear 1H-{15N}NOE ratios indicative of conformational exchange or a high degree of internal mobility, the relaxation data yielded a fully anisotropic rotational diffusion tensor of Dxx = 2.5 ± 0.2 × 107 sec−1, Dyy = 3.4 ± 0.2 × 107 sec−1, and Dzz = 3.9 ± 0.2 × 107 sec−1 for CaM2/3. These values, indicative of an anisotropic shape, correspond to an effective correlation time of 5.1 ± 0.2 nsec for the global tumbling of CaM2/3. This is close to that of ∼5 nsec expected for a monomeric 68-residue protein (Daragan and Mayo 1997), and similar to the correlation time reported for the isolated C-domain of CaM (Malmendal et al. 1999).
Figure 6.
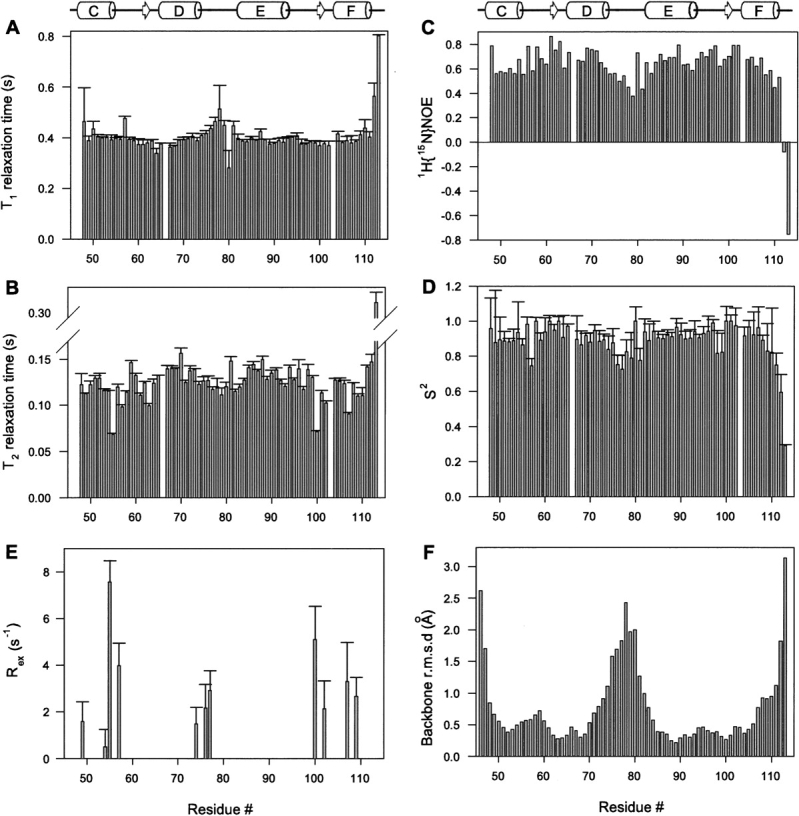
Backbone 15N relaxation data for Ca2+-saturated CaM2/3. (A) The longitudinal (T 1) and (B) transverse (T 2) 15N relaxation times (and standard errors), and (C) the heteronuclear 1H-{15N}NOE ratios measured for CaM2/3 at 25°C, along with (D) the fit anisotropic model free order parameters S 2 and (E) the conformational exchange broadening (R ex) terms. (F) The per-residue RMSDs of backbone heavy atoms (Cα, C′, and N) after superimposition of residues 48–71 and 82–106 for the CaM2/3 structural ensemble. Missing data correspond to P66 and residues with overlapping or very weak signals. The reduced S 2 values and 1H-{15N}NOE ratios, and the elevated RMSDs for residues near the N and C termini of CaM2/3, as well as the loop between helices D and E (corresponding to the linker region in native CaM) indicate that these regions are more conformationally dynamic, and less well structurally defined, than the remainder of the protein. A schematic of the secondary structure of CaM2/3 is also shown.
With consideration of anisotropic diffusion, the internal dynamic properties of Ca2+-ligated CaM2/3 can be described by the Lipari-Szabo model free formalism in terms of a generalized order parameter, S 2, that decreases from 1 to 0 with increasing mobility of the NH bond vector (Fig. 6D). With the exception of its disordered N- and C-terminal residues, CaM2/3 exhibits relatively uniform average S 2 values of 0.91 ± 0.07. This is consistent with a well-folded, globular structure. However, upon closer inspection, a decrease in both S 2 (average 0.80 ± 0.6) and 1H-{15N}NOE values is observed for residues 74–81, indicative of enhanced mobility on the subnanosecond timescale for the loop between EF-2 and EF-3. These residues also exhibit high RMSDs in the CaM2/3 structural ensemble (Figs. 4 and 6F). Thus, reduced structural precision, which is directly attributable to a limited number of measurable restraints, may arise in part from local conformational dynamics. In native CaM, these residues form the flexible linker between the N- and C-terminal domains. In CaM2/3, the same residues now form a flexible loop, similar to the loops between the EF-hands in the N- and C-terminal domains of native CaM. The fast-timescale backbone flexibility of the linker in CaM2/3 is comparable to that of the loops between EF-1 and EF-2, as well as EF-3 and EF-4, in native CaM (i.e., average S 2 values of 0.83 ± 0.06 and 0.79 ± 0.1 were reported for the N- and C-domains, respectively, of CaM complexed with smMLCK) (Lee and Wand 2001).
The T 2 lifetimes for some residues in or near helices C and F, as well as the loop region between EF-2 and EF-3, are anomalously short, suggestive of conformational exchange broadening (R ex) (Fig. 6E). This may result in part from Ca2+ association/dissociation, as signals from several amides in EF-2 are severely broadened in 15N-HSQC spectra acquired at subsaturating concentrations of Ca2+. Alternatively, these terms may reflect motions of Ca2+-saturated CaM2/3 on the millisecond-to-microsecond timescale. Given that helices C and F are distal from the loop between EF-2 and EF-3, it is tempting to speculate that such motions could reflect a hinge-like opening of CaM2/3. However, detailed main-chain and side-chain dynamic measurements are needed to fully test this hypothesis.
Discussion
Coupling of Ca2+ binding and cooperative folding in CaM2/3
In the absence of Ca2+, CaM2/3 shows no discernable tertiary structure reflected by the random coil amide 1HN chemical shifts in its 15N-HSQC spectrum (Fig. 3A). However, CD spectropolarimetry reveals that Ca2+-free CaM2/3 contains at least transiently formed helices (Fig. 2, inset). This behavior, indicative of a molten globule state, indicates that unlike the EF-hand domains of Ca2+-free CaM, unligated EF-2 and EF-3 in CaM2/3 cannot fold into a thermodynamically stable structure.
In contrast, upon the binding of one Ca2+ by EF-3 (K d1 = 30 ± 5 μM), CaM2/3 folds cooperatively to an intermediate, albeit nearly complete, globular structure. In this state, however, the 15N-HSQC signal of G61 is not observed, and those of neighboring residues are weak. The exchange broadening of the Ca2+-binding segment of EF-2 likely arises from millisecond-to-microsecond timescale motions between conformations with distinct amide chemical shifts. This conformational exchange, which could reflect the association/dissociation of Ca2+ with EF-2 at subsaturating levels, propagates to the amide of I100. Although I100 is in EF-3, its amide hydrogen-bonds to I63 of EF-2, forming the short antiparallel β-sheet in CaM2/3. Note that G61 and I100 have the most downfield-shifted 1HN resonances, and thus their amide signals are most severely broadened by this conformational exchange. The binding of a second Ca2+ to CaM2/3 (K d2 > 1000 μM) stabilizes EF-2. Notably, Ca2+ ligation at EF-2 leads to a sharpening of the 15N-HSQC peaks from amides neighboring G61, but does not alter their chemical shifts and induces only a small decrease in CD signal at 208 and 222 nm. Such behavior indicates that, although conformationally dynamic, Ca2+-free EF-2 is likely, on average, native-like when a single Ca2+ is ligated to EF-3. Thus, Ca2+ binding by EF-3 and the folding of EF-2 and EF-3 into an EF-hand domain are tightly coupled in a thermodynamic and structural sense. This is similar to the Ca2+-induced homodimerization and folding of the peptide model of the EF-3 of TnC to form a Ca2+–(TnC3)2 complex, which can subsequently bind a second Ca2+ with low affinity (Shaw et al. 1991).
Sequential Ca2+ binding by CaM2/3
CaM exhibits positive Ca2+-binding cooperativity that manifests as an increase in Ca2+ affinity at a second EF-hand due to previous Ca2+ binding at its partner EF-hand (Linse et al. 1991). This is observed quantitatively by the dissociation constants measured for its N-domain TR1C (K d1 = 15.8 μM and K d2 = 4.0 μM) and C-domain TR2C (K d1 = 7.9 μM and K d2 = 0.2 μM) (Linse et al. 1991). In contrast, CaM2/3 binds Ca2+ in a sequential or stepwise manner, first via EF-3 and then EF-2. This sequential binding is reflected by the wide difference between the two dissociation constants describing the CD-monitored titration of CaM2/3. Unfortunately, owing to the >30-fold difference between the K d1 and K d2 dissociation constants of CaM2/3, it is not possible to ascertain if cooperativity exists between its constituent EF-hands. Such a determination would require the measurement of the Ca2+ affinity of the high-affinity Ca2+-binding EF-3 while EF-2 is occupied, and that of the low-affinity EF-2 while EF-3 is Ca2+ free (Linse and Forsen 1995); structurally, this would require the analysis of the low-population (or “hypothetical”) folded states of CaM2/3 with or without Ca2+ bound at EF-2. Nevertheless, since the sequence of EF-2 can accommodate tight Ca2+ binding in CaM, it is clear that its nonnative structural context in CaM2/3 significantly decreases its effective affinity for Ca2+.
The association of EF-2 and EF-3 in CaM2/3
In the absence of their native EF-hand partners, EF-2 and EF-3 of CaM2/3 fold upon Ca2+ binding into a typical EF-hand domain. The covalent attachment of EF-2 and EF-3 via the linker may be a contributing factor in promoting their association because of the high effective local concentration of each EF-hand with respect to the other. However, it is also likely that any flexible sequence of similar length could play the same role. The covalent attachment of EF-2 and EF-3 might also structurally disfavor the formation of any potential intermolecular dimers or oligomers. Parenthetically, the structure of CaM2/3 also provides additional evidence for the adaptability of the flexible interdomain linker of CaM. In the case of CaM2/3, this sequence now plays the same role that the loops between EF-hands (linking helix B to C and helix F to G) play in CaM. This also raises the possibility that the linker in CaM evolved out of a loop between EF-hands in a progenitor EF-hand protein.
The pairing of EF-2 with EF-3 in CaM2/3 is similar to the association of EF-1 with EF-2 and of EF-3 with EF-4 in the N- and C-domains of Ca2+-ligated CaM. To the first approximation, this reflects the sequence similarity of EF-hand motifs and their symmetric arrangement within an EF-hand domain. However, in native CaM, EF-2 is the C-terminal EF-hand of the N-domain, while EF-3 is the N-terminal EF-hand of the C-domain. Thus, counting from the N to the C terminus, each domain of CaM has an “odd”- then “even”-numbered EF-hand. In contrast, EF-2 is the first (N-terminal) EF-hand in CaM2/3, preceding EF-3 (C-terminal), producing an “even” then “odd” pairing. The fact that “odd” EF-hands (EF-1 and EF-3) are more closely related in sequence to one another than to “even” EF-hands (EF-2 and EF-4), and vice versa, in CaM (Nakayama et al. 1992) is consistent with the hypothesis that a progenitor EF-hand gave rise to an EF-hand domain that, in turn, evolved into a two-domain progenitor EF-hand protein. Furthermore, EF-1 has evolved to associate with EF-2, and EF-3 has evolved to associate with EF-4. Thus, in a structural and evolutionary sense, the “even–odd” arrangement of EF-2 and EF-3 in CaM2/3 is reversed from that of native CaM. The predominantly unfolded state of Ca2+-free CaM2/3 and the poor Ca2+ affinity of folded CaM2/3 are likely consequences of this reversed arrangement.
Ca2+ affinity and the position of aromatic residues
CaM2/3 binds Ca2+ with micromolar affinity at EF-3, yet millimolar affinity at EF-2. Similar divergent Ca2+-dissociation constants have been observed with homodimeric peptides corresponding to EF-3 and EF-4 of TnC, as well as EF-3 of CaM (Reid 1987a; Kay et al. 1991; Shaw et al. 1991; Franchini and Reid 1999). Heterodimeric peptide models of TnC EF-3 and EF-4 and calbindin D9k EF-1 and EF-2 have improved Ca2+ binding to both sites when compared to homodimers (Finn et al. 1992; Shaw and Sykes 1996), albeit with lower affinity than in their respective native proteins (Leavis et al. 1978; Finn et al. 1992; Shaw and Sykes 1996). Clearly, the particular pairing of EF-hands has an important effect on Ca2+ affinity. In the cases of the EF-hand homodimers and CaM2/3, Ca2+ affinity of at least one binding site is compromised because of this nonnative EF-hand pairing.
The factors that establish the Ca2+ affinity and cooperativity of EF-hands have been investigated in numerous studies (Linse and Forsen 1995; Nelson et al. 2002). In particular, the structural changes associated with Ca2+ binding have been quantified in terms of interhelical angles (Kuboniwa et al. 1995) and through difference distance matrices, which reveal relative alterations in protein packing (Nelson and Chazin 1998). The conformation of the short antiparallel β-sheet has also been proposed as a determinant of the positioning of Ca2+-coordinating groups in the two EF-hands that it bridges (Grabarek 2006). However, as emphasized by Chazin and colleagues, the EF-hand domain should be viewed as a globally cooperative structural unit (Nelson et al. 2002). Each of these parameters reflects the same collective, thermodynamically driven response of an EF-hand domain to changes in its Ca2+ occupancy. Therefore, all aspects of the nonnative pairing of EF-hands in CaM2/3 may play a role in its altered Ca2+ binding relative to native CaM.
With this caveat in mind, a potentially important insight into the Ca2+-binding properties of EF-hand domains is provided by considering a consequence of the “even–odd” arrangement of EF-2 and EF-3 in CaM2/3. A key determinant of EF-hand domain stability is the association of nonpolar residues on neighboring helices from paired EF-hands to form a hydrophobic core (Shaw and Sykes 1996). Of particular note in Ca2+-ligated CaM is the edge-to-face (T-shaped) stacking of four aromatic side chains from two pairs of conserved phenylalanines or tyrosines located on the first and fourth helices in each EF-hand domain (Babu et al. 1988). As summarized in Figure 1, the two aromatic residues in the first helix are found four and one residue(s) before the Ca2+-binding segment of the N-terminal EF-hand. The remaining two aromatic residues in the fourth helix are located at Ca2+-chelating loop position 10 and just after position 12 of the Ca2+-binding segment of the C-terminal EF-hand. The energetically favorable stacking of these conserved aromatic side chains (McGaughey et al. 1998) bridges the first and fourth helices in each domain of Ca2+-ligated CaM (as well as in almost all EF-hand domains) (Nakayama et al. 1992). Together with the loop sequence that covalently links the second and third helices, this serves to “close both ends” of an EF-hand domain (Fig. 7A).
Figure 7.

Schematic representations showing the relative positions of conserved aromatic residues and the inter-EF-hand loop in (A) the C-domain of CaM, (B) an EF-hand homodimer (such as TnC3), and (C) CaM2/3. Helices are cylinders and the aromatic residues involved in aromatic stacking interactions are represented as hexagons. In the CaM domain, the aromatic residues stack on each other opposite the loop between EF-hands, and may help position the EF-hands for high-affinity, cooperative Ca2+ binding. In the EF-hand homodimer, the aromatic stacking interactions cannot form, whereas with CaM2/3, the aromatic stacking interactions occur, but in a position adjacent to the loop between EF-hands. This may lead to low affinity for binding the second Ca2+ by these nonnative EF-hand domains. An EF-hand heterodimer would look like A, but without the loop between EF-hands. (D) The superimposition of CaM2/3 (dark gray) onto the C-domain of Ca2+-ligated CaM (light gray). The structures are arranged such that EF-3 and EF-4 of CaM are superimposed on EF-2 and EF-3 (labeled) of CaM2/3, respectively. As a result, the loop regions overlap, whereas the aromatic residues do not. The Ca2+ ions of CaM2/3 are black spheres.
Based on the structure and Ca2+-binding properties of nonnative species such as CaM2/3, we speculate that the Ca2+ affinity and cooperativity within a given EF-hand domain are dependent in part on the position of the conserved aromatic residues with respect to the loop between its constituent EF-hands. In the cases of the Ca2+-ligated EF-hand homodimers, such stacking interactions cannot occur because the monomers fold upon each other in an antiparallel orientation (Fig. 7B; Kay et al. 1991; Shaw et al. 1992). Aromatic stacking interactions can form in the correct position in EF-hand heterodimers, but the covalent loop between EF-hands is absent. This may correlate with heterodimers having higher Ca2+ affinity than exhibited by the homodimers, yet lower than in their respective native proteins (Finn et al. 1992; Shaw and Sykes 1996). In Ca2+-ligated CaM2/3, aromatic residues from EF-2 and EF-3 still associate to form a hydrophobic core that is roughly superimposable with those of the Ca2+-ligated CaM domains (Fig. 5C). However, owing to the reversed “even–odd” EF-hand pairing of CaM2/3, these aromatic residues are located on the second and third helices (D and E), rather than the first and fourth as in the N- and C-domains of CaM. Thus, the conserved aromatic residues of CaM2/3 form stacking interactions positioned adjacent to the linker sequence between EF-2 and EF-3 (Fig. 7C) and cannot “close” its structure in the same manner as seen with the CaM domains (Fig. 7A). This nonnative packing arrangement may prevent CaM2/3 from adopting a conformation compatible with high-affinity Ca2+ binding at EF-2.
The arrangement of conserved aromatic residues in each domain of CaM also changes in the transition from the Ca2+-free to Ca2+-ligated structures. The hallmark of this transition is a reduction of the angles between adjacent helices of each domain upon Ca2+ binding (Table 2). These angles are large in Ca2+-free CaM because of the nearly antiparallel arrangement of helices, and upon Ca2+ binding, the angles decrease such that the adjacent helices in Ca2+-ligated CaM are nearly perpendicular to each other (Kuboniwa et al. 1995; Zhang et al. 1995). Accompanying this helical rearrangement, the second aromatic residue in the first EF-hand of each domain (i.e., F19 in the N-domain, F92 in the C-domain) shifts inward to join the three remaining conserved aromatic side chains that interact in Ca2+-free CaM (F16/F65/F68 and F89/Y138/F141) (Fig. 5C; Kuboniwa et al. 1995; Schumacher et al. 2004). This hydrophobic core rearrangement may contribute to the mechanism for the transmission of cooperativity from one EF-hand to another because, as shown in Figure 1, the conserved aromatic residues immediately border or are part of Ca2+-chelating segments of each EF-hand in a domain. In CaM2/3, F65 is displaced with respect to its equivalent position in Ca2+-ligated CaM, stacking on F89 in a face-to-face (parallel displaced) fashion rather than in a T-shaped pattern (Figs. 5C and 7D; Babu et al. 1988). This displacement, along with its reversed position relative to the loop between EF-hands, may contribute further to the dramatic reduction of the Ca2+ affinity of EF-2 within the nonnative context of CaM2/3.
Materials and Methods
Construction of the CaM2/3 gene
The gene encoding CaM2/3 (residues 46–113 of human CaM) was constructed using a modified version of the technique of mutually priming long oligonucleotides using the following primers encoding portions of CaM2/3 and a Factor Xa protein cleavage site (underlined), as well as DNA restriction sites NcoI and HindIII (italicized) (Uhlmann 1988):
AGACCATGGGAATTGAGGGACGCGCTGAACTGCAGGACATGATTAACGAAGTAGACGCTGACGGTAACGGCACAATAGAT;
CCTCTTCGGAGTCAGTGTCTTTCATTTTGCGCGCCATCATGGTCAGAAATTCCGGGAAATCTATTGTGCCGTTACCGTCAGC;
AAAATGAAAGACACTGACTCCGAAGAGGAAATCCGTGAGGCCTTCCGTGTTTTCGACAAAGACGGTAACGGTTATATATC;
CCTAAGCTTGTTATTAACCCAGGTTAGTCATAACGTGACGCAGTTCAGCGGCCGATATATAACCGTTACCGTCTTTGTCG.
Initially, the first two and last two mutually priming oligonucleotides were annealed to each other and extended in separate reactions with a MJ Research Peltier thermal cycler (PTC-200) using a gradient annealing thermal cycler program. The mutually priming products of each reaction were then mixed for a second round of PCR to produce the full CaM2/3 gene. The resulting gene was purified and ligated into the pGEM-T (Promega) vector, and subcloned into the pET28a+ vector (Novagen) using the HindIII restriction site.
Expression and purification of unlabeled, 15N-labeled, and 15N/13C-labeled CaM2/3
Unlabeled His6-tagged CaM2/3 was expressed using Escherichia coli BL21 Star (λDE3) grown in Luria broth, and induced with 1 mM isopropyl-β-D-thiogalactopyranoside (IPTG) (Sigma) for 1.75 h, according to established methods (Brent 1998). Uniformly 15N-labeled CaM2/3 was expressed in M9 media supplemented with 1 g/L 99% 15NH4Cl (Cambridge Isotope Laboratories) and 1 g/L Celtone-N (Stable Spectral Isotopes). Uniformly 15N/13C-labeled CaM2/3 was expressed in M9 media containing 4 g/L of 99% 13C6-glucose (Cambridge Isotope Laboratories), 1 g/L 99% 15NH4Cl, and 1.5 g/L Celtone-CN (Muchmore et al. 1989; Reilly and Fairbrother 1994). For both 15N-CaM2/3, and 15N/13C-CaM2/3, protein expression was induced at OD600 ∼ 0.7 with 1 mM IPTG, followed by growth for 1 h at 37°C. The resulting cultures were centrifuged at 4°C, and the pellets were stored at −80°C.
To purify labeled or unlabeled CaM2/3, cells from the thawed pellets were lysed with Bugbuster (Novagen) according to the manufacturer's instructions. The cell lysates were clarified by centrifugation and initially purified with HIS-Select (Sigma) immobilized metal affinity chromatography resin according to the manufacturer's instructions. Fractions corresponding to the His6-tagged CaM2/3 were pooled, applied to BioGel P-6DG (Bio-Rad) desalting resin, and eluted with 50 mM Tris (pH 7.5) and 100 mM NaCl. Fractions containing the imidazole-free fusion peptide were pooled, CaCl2 (Fisher) was added to a concentration of 10 mM, and Factor Xa (Novagen) was added to a concentration of 2 U/mL. Cleavage of His-tagged CaM2/3 proceeded for 24 h at room temperature. Factor Xa-cleaved CaM2/3 was further purified according to previously described methods (Reid 1987b).
Peptide hydrolysis followed by amino acid analysis was performed to confirm the identity and to measure the concentration of the purified CaM2/3. Amino acid analysis was performed with the internal standard carboxymethylcysteine at The Hospital for Sick Children Advanced Protein Technology Center (Toronto, Canada).
The molecular weights and purity of all peptides were confirmed using analytical liquid chromatography mass spectrometry on a Micromass Quattro microelectrospray mass spectrometer. The masses of the natural abundance or uniformly labeled CaM2/3 samples did not deviate significantly from the expected values, and in all cases, the purity was >90% by peak area.
Ca2+-binding studies
CaM2/3 samples were decalcified in preparation for structural and Ca2+ titration studies according to previously described methods (Vorherr et al. 1990). Circular dichroism (CD) monitored titrations were performed on a Jasco J720 spectropolarimeter to record the change in ellipticity at 222 nm with increasing Ca2+ concentration at 25°C. The scanning speed was 50 nm/min, the bandwidth 1 nm, the response 4 sec, and the number of accumulations 4. Samples were loaded in a 1-mm pathlength 110-QS Hellma quartz cell. The initial concentrations of the Ca2+-free peptides were 40 μM in 0.3 mL of 20 mM MOPS (Sigma) and 50 mM KCl in Ca2+-free water at pH 7.4. A total volume of 10 μL of a 2.0 mM CaCl2 standard was added in 1-μL aliquots. An additional 7 μL of a 20 mM Ca2+ standard, as well as 1 μL of a 1 M Ca2+ standard, were added producing an ∼100-fold molar excess of Ca2+ at the end of the titration. The Ca2+ concentrations of the standard Ca2+ solutions were determined by titration, as described previously (Procyshyn and Reid 1994). A far-UV CD scan was recorded from 200 nm to 250 nm at the beginning and end of each titration. The experiments were repeated four times, and the data were fit to a two Ca2+-binding site model using the program CaLigator (Andre and Linse 2002).
NMR spectral assignments of Ca2+-ligated CaM2/3
Uniformly labeled 15N- and 13C/15N-CaM2/3 protein samples were prepared for NMR spectroscopic analysis by dissolving the freeze-dried peptide in 10 mL of an NMR buffer composed of 20 mM Tris-d11 (C.I.L.), 50 mM KCl, and 10% D2O (C.I.L.) at pH 7.4, without or with 10 mM CaCl2. The pH of the protein solution was measured, readjusted to pH 7.4 without correction for isotope effects, and concentrated using a Centricon plus-20 centrifugal filter (Amicon) to a final volume of 500 μL.
NMR experiments were performed at 25°C using Varian Unity 500 MHz and Inova 600 MHz spectrometers. Data were processed with Felix 2000 (Accelrys, Inc.) or NMRpipe (Delaglio et al. 1995) and analyzed using Sparky (Goddard and Kneeler 1999). Initially, sensitivity-enhanced gradient 15N-HSQC spectra (Kay 1995) were recorded with 0.37 mM Ca2+-free 15N-CaM2/3, to which increasing amounts of Ca2+ were titrated. All remaining spectra were recorded in the presence of 10 mM CaCl2 to ensure >90% saturation of both EF-2 and EF-3. The protein backbone connectivity was established using sensitivity-enhanced gradient 15N-HSQC, HNCACB, and CBCA(CO)NH spectra (Sattler et al. 1999), recorded with 0.5 mM 13C/15N-CaM2/3. The assignment of resonances from most of the remaining main-chain and aliphatic side-chain 1H, 13C, and 15N nuclei were obtained from HNCO, C(CO)TOCSY-NH, H(CCO)TOCSY-NH, and HCCH-TOCSY spectra (Sattler et al. 1999). The resonances from aromatic side chains were obtained using 13C-HSQC, CβHδ, and CβHɛ experiments (Yamazaki et al. 1993).
Structure calculations for Ca2+-ligated CaM2/3
Structure calculations were performed using ARIA/CNS v1.2 with torsion angle dynamics (Brunger et al. 1998; Linge et al. 2003). Distance restraints were obtained from a 1H–15N–1H NOESY-HSQC spectrum (τm = 150 msec) recorded with 0.25 mM 15N-CaM2/3, and from a simultaneous aliphatic/amide 1H–13C/15N–1H NOESY-HSQC spectrum (τm = 150 msec), an aromatic 1H–13C–1H NOESY-HSQC spectrum (τm = 150 msec), and a simultaneous constant-time methyl/amide 1H–13C/15N–1H NOESY-HSQC spectrum (τm = 150 msec) recorded with 0.5 mM 13C/15N-CaM2/3 (Pascal et al. 1994; Jahnke et al. 1995; Zwahlen et al. 1998). Most NOE peaks were manually assigned prior to intensity calibration in ARIA. Backbone φ and ψ dihedral angles were determined from 13Cα, 13Cβ, 13C′, 1Hα, and 15N chemical shifts using the program TALOS (Cornilescu et al. 1999). Weighted 13Cα and 13Cβ secondary chemical shifts, calculated with the program CSI (Yap and Tomomori 1999), TALOS-derived dihedral angles, and a manual consideration of initial structures were used to define 28 hydrogen-bond distance restraints involving residues within helices for subsequent rounds of structure calculations.
Residual dipolar couplings (RDC) were measured with an 1H–15N-IPAP-HSQC spectrum recorded on 15N-CaM2/3 (∼0.3 mM final) diffused into a 7% acrylamide gel (29:1 acrylamide:bisacrylamide stock), dialyzed previously against NMR buffer (Chou et al. 2001a). The gel was cast with a 6-mm diameter and stretched approximately twofold lengthwise upon loading into a 5-mm silanized bottomless NMR tube with a gel stretching apparatus (New Era Enterprises, Inc). Before acquisition of the 1H–15N-IPAP-HSQC with the stretched gel, the 2H signal from the lock 2HO1H was split by 11.3 Hz, and after acquisition it was 10.8 Hz. The resulting 1HN–15N RDC restraints ranged from 14.5 to −16.5 Hz. RDCs from residues with an S 2 < 0.6, and disordered residues in the linker region as well as the C and N termini were excluded from subsequent structural calculations. Axial (D a) and rhombic (R) parameters were estimated by the histogram method, followed by a grid search for minimal SANI violations (Clore et al. 1998). The optimized values were D a = −11 Hz and R = 0.3. The RDC restraints were included after iteration 5 of the ARIA protocol.
Eight distance restraints for chelating residues to bound Ca2+ ions were included after iteration 5 of the ARIA protocol. The chelation points were D58 Oδ1, D60 Oδ1, and E67 Oɛ1 and Oɛ2 in EF-2; and D93 Oδ1, D95 Oδ1, and D97 Oδ1 and E104 Oɛ1 in EF-3. The restraints were based on the reported structure of a Ca2+-ligated CaM–peptide complex (2BBM.pdb) (Ikura et al. 1992) as justified in the Results section.
The final CaM2/3 structures were calculated using a two-step ARIA protocol. An initial round of ARIA (iterations 0–8) was started with an unfolded CaM2/3 polypeptide, along with dihedral angle, and NOE restraints. Hydrogen-bond, Ca2+-binding, and RDC restraints were added from iteration 5 onward. A second full ARIA calculation was performed, using the ending structures and the unambiguous and ambiguous NOE restraint sets from the previous round as the starting parameters. After a final ARIA refinement in a water box using Lennard-Jones potentials, an ensemble of 50 structures was generated, from which the 20 lowest-energy structures were selected for subsequent visualization and analysis using MOLMOL (Koradi et al. 1996), WebLab ViewerPro (Accelrys, Inc.), and PROCHECK-NMR (Laskowski et al. 1996). Overall and per-residue pairwise backbone RMSDs were calculated with MOLMOL and SupPose (Smith 1997), respectively, using the coordinates for 13Cα, 13C′, and 15N atoms. The secondary structure of the ensemble of 20 CaM2/3 structures was defined using the program PROMOTIF (Hutchinson and Thornton 1996). Interhelical angles were calculated using the program Interhlx (Yap 1995). Structural figures were rendered with MOLMOL.
Backbone amide 15N relaxation studies
Amide 15N relaxation parameters were acquired for 15N-CaM2/3 at 25°C using a Varian Unity 500 MHz NMR spectrometer (Farrow et al. 1994). Data points for the T 1 (10–903.2 msec) and T 2 (16.7–167.0 msec) experiments were collected in random order. Steady-state heteronuclear 1H-{15N}NOE spectra were acquired with and without 2 sec of 1H saturation and a total recycle delay of 5 sec. All data were processed with NMRpipe, and the above data points were fit to a single exponential using Sparky to obtain the T 1 and T 2 lifetimes (Goddard and Kneeler 1999). Anisotropic diffusion and model-free order parameters (S 2) were calculated with the program Tensor 2.0 using the lowest-energy structural model of CaM2/3 (Dosset et al. 2000). Residues for which the 1H-{15N}NOE ratios were <0.6, and for which the values of {[(〈R 2〉 − R 2i) / 〈R 2〉] − [(〈R 1〉 − R 1i) / 〈R 1〉]} > 1.5 × (the standard deviation of this difference) were excluded from the diffusion tensor calculations because of the possibility of fast internal motions or chemical exchange broadening (Tjandra et al. 1995).
Data deposition
The atomic coordinates of the CaM2/3 ensemble (PDB ID 2HF5) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/). The NMR chemical shift list (accession code 7190) has been deposited in the BioMagResBank (http://www.bmrb.wisc.edu/).
Acknowledgments
This research was supported by grants from the National Cancer Institute of Canada with funds from the Canadian Cancer Society (to L.P.M.). Instrument support was provided by the Protein Engineering Network of Centers of Excellence, the Canadian Foundation for Innovation, the British Columbia Knowledge Development Fund, the UBC Blusson Fund, and the Michael Smith Foundation for Health Research. Support for T.M.L. was provided by a Merck-Frosst Postgraduate Pharmacy Fellowship Award and The UBC University Graduate Fellowship. We thank Grant Mauk for the use of the CD spectropolarimeter and Federico Rosell for technical assistance with the instrument. We also thank Calvin Yip for performing light scattering experiments. Finally, we thank Gary Shaw, Brian Sykes, and Mitsuhiko Ikura for their helpful comments regarding this manuscript.
Footnotes
Reprint requests to: Ronald E. Reid, Faculty of Pharmaceutical Sciences, Division of Biomolecular and Pharmaceutical Chemistry, University of British Columbia, 2146 East Mall, Vancouver, British Columbia, Canada, V6T 1Z3; e-mail: calm@interchange.ubc.ca; fax: (01) (604) 822-3035.
Abbreviations: CaM, vertebrate calmodulin; CaM3, vertebrate calmodulin EF-3; CaM2/3, residues 46–113 of vertebrate calmodulin fragment including EF-2 and EF-3 (unless stated otherwise, CaM2/3 is in the Ca2+-ligated form); CD, circular dichroism; HSQC, heteronuclear single quantum correlation; IMAC, immobilized metal affinity chromatography; NMR, nuclear magnetic resonance; NOE, nuclear Overhauser effect; NOESY, nuclear Overhauser effect spectroscopy; 〈Rn〉, average transverse or longitudinal relaxation rate; RDC, residual dipolar coupling; RMSD, root mean squared deviation; TnC3, troponin C EF-3; TnC4, troponin C EF-4; TOCSY, total correlation spectroscopy; TR1C, vertebrate calmodulin tryptic fragment (residues 1–77); TR2C, vertebrate calmodulin tryptic fragment (residues 78–148).
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.072777107.
References
- Andre I. and Linse, S. 2002. Measurement of Ca2+-binding constants of proteins and presentation of the CaLigator software. Anal. Biochem. 305: 195–205. [DOI] [PubMed] [Google Scholar]
- Babu Y.S., Bugg, C.E., and Cook, W.J. 1988. Structure of calmodulin refined at 2.2 Å resolution. J. Mol. Biol. 204: 191–204. [DOI] [PubMed] [Google Scholar]
- Biekofsky R.R., Martin, S.R., Browne, J.P., Bayley, P.M., and Feeney, J. 1998. Ca2+ coordination to backbone carbonyl oxygen atoms in calmodulin and other EF-hand proteins: 15N chemical shifts as probes for monitoring individual-site Ca2+ coordination. Biochemistry 37: 7617–7629. [DOI] [PubMed] [Google Scholar]
- Brent R. 1998. Protein expression. In Current protocols in molecular biology (eds. F. Ausubel et al.), pp. 16.10.11–16.21.19. John Wiley & Sons, New York.
- Brunger A.T., Adams, P.D., Clore, G.M., DeLano, W.L., Gros, P., Grosse-Kunstleve, R.W., Jiang, J.S., Kuszewski, J., Nilges, M., Pannu, N.S., et al. 1998. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 54: 905–921. [DOI] [PubMed] [Google Scholar]
- Chattopadhyaya R., Meador, W.E., Means, A.R., and Quiocho, F.A. 1992. Calmodulin structure refined at 1.7 Å resolution. J. Mol. Biol. 228: 1177–1192. [DOI] [PubMed] [Google Scholar]
- Chou J.J., Gaemers, S., Howder, B., Louis, J.M., and Bax, A. 2001a. A simple apparatus for generating stretched polyacrylamide gels, yielding uniform alignment of proteins and detergent micelles. J. Biomol. NMR 21: 377–382. [DOI] [PubMed] [Google Scholar]
- Chou J.J., Li, S., Klee, C.B., and Bax, A. 2001b. Solution structure of Ca2+-calmodulin reveals flexible hand-like properties of its domains. Nat. Struct. Biol. 8: 990–997. [DOI] [PubMed] [Google Scholar]
- Clore G.M., Gronenborn, A.M., and Tjandra, N. 1998. Direct structure refinement against residual dipolar couplings in the presence of rhombicity of unknown magnitude. J. Magn. Reson. 131: 159–162. [DOI] [PubMed] [Google Scholar]
- Cornilescu G., Delaglio, F., and Bax, A. 1999. Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J. Biomol. NMR 13: 289–302. [DOI] [PubMed] [Google Scholar]
- Daragan V.A. and Mayo, K.H. 1997. Motional model analyses of protein and peptide dynamics using 13C and 15N NMR relaxation. Prog. Nucl. Magn. Reson. Spect. 31: 63–105. [Google Scholar]
- Delaglio F., Grzesiek, S., Vuister, G.W., Zhu, G., Pfeifer, J., and Bax, A. 1995. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6: 277–293. [DOI] [PubMed] [Google Scholar]
- Dosset P., Hus, J.C., Blackledge, M., and Marion, D. 2000. Efficient analysis of macromolecular rotational diffusion from heteronuclear relaxation data. J. Biomol. NMR 16: 23–28. [DOI] [PubMed] [Google Scholar]
- Fallon J.L. and Quiocho, F.A. 2003. A closed compact structure of native Ca2+-calmodulin. Structure 11: 1303–1307. [DOI] [PubMed] [Google Scholar]
- Farrow N.A., Muhandiram, R., Singer, A.U., Pascal, S.M., Kay, C.M., Gish, G., Shoelson, S.E., Pawson, T., Forman-Kay, J.D., and Kay, L.E. 1994. Backbone dynamics of a free and phosphopeptide-complexed Src homology 2 domain studied by 15N NMR relaxation. Biochemistry 33: 5984–6003. [DOI] [PubMed] [Google Scholar]
- Finn B.E., Kordel, J., Thulin, E., Sellers, P., and Forsen, S. 1992. Dissection of calbindin D9k into two Ca2+-binding subdomains by a combination of mutagenesis and chemical cleavage. FEBS Lett. 298: 211–214. [DOI] [PubMed] [Google Scholar]
- Franchini P.L. and Reid, R.E. 1999. Investigating site-specific effects of the -X glutamate in a parvalbumin CD site model peptide. Arch. Biochem. Biophys. 372: 80–88. [DOI] [PubMed] [Google Scholar]
- Goddard T.D. and Kneeler, D.G. 1999. SPARKY 3. University of California, San Francisco, San Francisco, CA.
- Grabarek Z. 2006. Structural basis for diversity of the EF-hand calcium-binding proteins. J. Mol. Biol. 359: 509–525. [DOI] [PubMed] [Google Scholar]
- Hutchinson E.G. and Thornton, J.M. 1996. PROMOTIF—A program to identify and analyze structural motifs in proteins. Protein Sci. 5: 212–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikura M., Minowa, O., and Hikichi, K. 1985. Hydrogen bonding in the carboxyl-terminal half-fragment 78–148 of calmodulin as studied by two-dimensional nuclear magnetic resonance. Biochemistry 24: 4264–4269. [DOI] [PubMed] [Google Scholar]
- Ikura M., Clore, G.M., Gronenborn, A.M., Zhu, G., Klee, C.B., and Bax, A. 1992. Solution structure of a calmodulin-target peptide complex by multidimensional NMR. Science 256: 632–638. [DOI] [PubMed] [Google Scholar]
- Jahnke W., Baur, M.G., and Kessler, H. 1995. Improved accuracy of NMR structures by a modified NOESY-HSQC experiment. J. Magn. Reson. Ser. B 106: 86–88. [Google Scholar]
- Kay L.E. 1995. Field gradient techniques in NMR spectroscopy. Curr. Opin. Struct. Biol. 5: 674–681. [DOI] [PubMed] [Google Scholar]
- Kay L.E., Forman-Kay, J.D., McCubbin, W.D., and Kay, C.M. 1991. Solution structure of a polypeptide dimer comprising the fourth Ca2+-binding site of troponin C by nuclear magnetic resonance spectroscopy. Biochemistry 30: 4323–4333. [DOI] [PubMed] [Google Scholar]
- Koradi R., Billeter, M., and Wuthrich, K. 1996. MOLMOL: A program for display and analysis of macromolecular structures. J. Mol. Graph. 14: 51–55. [DOI] [PubMed] [Google Scholar]
- Kuboniwa H., Tjandra, N., Grzesiek, S., Ren, H., Klee, C.B., and Bax, A. 1995. Solution structure of calcium-free calmodulin. Nat. Struct. Biol. 2: 768–776. [DOI] [PubMed] [Google Scholar]
- Laskowski R.A., Rullmannn, J.A., MacArthur, M.W., Kaptein, R., and Thornton, J.M. 1996. AQUA and PROCHECK-NMR: Programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR 8: 477–486. [DOI] [PubMed] [Google Scholar]
- Leavis P.C., Rosenfeld, S.S., Gergely, J., Grabarek, Z., and Drabikowski, W. 1978. Proteolytic fragments of troponin C. Localization of high and low affinity Ca2+ binding sites and interactions with troponin I and troponin T. J. Biol. Chem. 253: 5452–5459. [PubMed] [Google Scholar]
- Lee A.L. and Wand, A.J. 2001. Microscopic origins of entropy, heat capacity, and the glass transition in proteins. Nature 411: 501–504. [DOI] [PubMed] [Google Scholar]
- Li M.X., Gagne, S.M., Tsuda, S., Kay, C.M., Smillie, L.B., and Sykes, B.D. 1995. Calcium binding to the regulatory N-domain of skeletal muscle troponin C occurs in a stepwise manner. Biochemistry 34: 8330–8340. [DOI] [PubMed] [Google Scholar]
- Linge J.P., Habeck, M., Rieping, W., and Nilges, M. 2003. ARIA: Automated NOE assignment and NMR structure calculation. Bioinformatics 19: 315–316. [DOI] [PubMed] [Google Scholar]
- Linse S. and Forsen, S. 1995. Determinants that govern high-affinity calcium binding. Adv. Second Messenger Phosphoprotein Res. 30: 89–151. [DOI] [PubMed] [Google Scholar]
- Linse S., Helmersson, A., and Forsen, S. 1991. Calcium binding to calmodulin and its globular domains. J. Biol. Chem. 266: 8050–8054. [PubMed] [Google Scholar]
- Malmendal A., Evenas, J., Forsen, S., and Akke, M. 1999. Structural dynamics in the C-terminal domain of calmodulin at low calcium levels. J. Mol. Biol. 293: 883–899. [DOI] [PubMed] [Google Scholar]
- McGaughey G.B., Gagne, M., and Rappe, A.K. 1998. pi-Stacking interactions. Alive and well in proteins. J. Biol. Chem. 273: 15458–15463. [DOI] [PubMed] [Google Scholar]
- Moncrief N.D., Kretsinger, R.H., and Goodman, M. 1990. Evolution of EF-hand calcium-modulated proteins. I. Relationships based on amino acid sequences. J. Mol. Evol. 30: 522–562. [DOI] [PubMed] [Google Scholar]
- Muchmore D.C., McIntosh, L.P., Russell, C.B., Anderson, D.E., and Dahlquist, F.W. 1989. Expression and nitrogen-15 labeling of proteins for proton and nitrogen-15 nuclear magnetic resonance. Methods Enzymol. 177: 44–73. [DOI] [PubMed] [Google Scholar]
- Nakayama S., Moncrief, N.D., and Kretsinger, R.H. 1992. Evolution of EF-hand calcium-modulated proteins. II. Domains of several subfamilies have diverse evolutionary histories. J. Mol. Evol. 34: 416–448. [DOI] [PubMed] [Google Scholar]
- Nelson M.R. and Chazin, W.J. 1998. An interaction-based analysis of calcium-induced conformational changes in Ca2+ sensor proteins. Protein Sci. 7: 270–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson M.R., Thulin, E., Fagan, P.A., Forsen, S., and Chazin, W.J. 2002. The EF-hand domain: A globally cooperative structural unit. Protein Sci. 11: 198–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascal S.M., Muhandiram, D.R., Yamazaki, T., Forman-Kay, J.D., and Kay, L.E. 1994. Simultaneous acquisition of 15N- and 13C-edited NOE spectra of proteins dissolved in H2O. J. Magn. Reson. B 103: 197–201. [Google Scholar]
- Procyshyn R.M. and Reid, R.E. 1994. A structure/activity study of calcium affinity and selectivity using a synthetic peptide model of the helix–loop–helix calcium-binding motif. J. Biol. Chem. 269: 1641–1647. [PubMed] [Google Scholar]
- Redfield C. 2004. Using nuclear magnetic resonance spectroscopy to study molten globule states of proteins. Methods 34: 121–132. [DOI] [PubMed] [Google Scholar]
- Reid R.E. 1987a. A synthetic 33-residue analogue of bovine brain calmodulin calcium binding site III: Synthesis, purification, and calcium binding. Biochemistry 26: 6070–6073. [DOI] [PubMed] [Google Scholar]
- Reid R.E. 1987b. Total sequential solid phase synthesis of rabbit skeletal troponin C calcium binding site III. Int. J. Pept. Protein Res. 30: 613–621. [DOI] [PubMed] [Google Scholar]
- Reilly D. and Fairbrother, W.J. 1994. A novel isotope labeling protocol for bacterially expressed proteins. J. Biomol. NMR 4: 459–462. [DOI] [PubMed] [Google Scholar]
- Sattler M., Schleucher, J., and Griesinger, C. 1999. Heteronuclear multidimensional NMR experiments for the structure determination of proteins in solution employing pulsed field gradients. Progr. Nucl. Magn. Reson. Spectrosc. 34: 93–158. [Google Scholar]
- Schumacher M.A., Crum, M., and Miller, M.C. 2004. Crystal structures of apocalmodulin and an apocalmodulin/SK potassium channel gating domain complex. Structure 12: 849–860. [DOI] [PubMed] [Google Scholar]
- Shaw G.S. and Sykes, B.D. 1996. NMR solution structure of a synthetic troponin C heterodimeric domain. Biochemistry 35: 7429–7438. [DOI] [PubMed] [Google Scholar]
- Shaw G.S., Golden, L.F., Hodges, R.S., and Sykes, B.D. 1991. Interactions between paired calcium-binding sites in proteins NMR determination of the stoichiometry of calcium binding to a synthetic troponin-C peptide. J. Am. Chem. Soc. 113: 5557–5563. [Google Scholar]
- Shaw G.S., Hodges, R.S., and Sykes, B.D. 1992. Determination of the solution structure of a synthetic two-site calcium-binding homodimeric protein domain by NMR spectroscopy. Biochemistry 31: 9572–9580. [DOI] [PubMed] [Google Scholar]
- Shuman C.F., Jiji, R., Kerfeldt, K.S., and Linse, S. 2006. Reconstitution of calmodulin from domains and subdomains: Influence of target peptide. J. Mol. Biol. 358: 870–881. [DOI] [PubMed] [Google Scholar]
- Smith J.A. 1997. SupPose. Vanderbilt University, Nashville, TN.
- Tjandra N., Feller, S.E., Richard, W.P., and Bax, A. 1995. Rotational diffusion anisotropy of human ubiquitin from 15N NMR relaxation. J. Am. Chem. Soc. 117: 12562–12566. [Google Scholar]
- Uhlmann E. 1988. An alternative approach in gene synthesis: Use of long selfpriming oligodeoxynucleotides for the construction of double-stranded DNA. Gene 71: 29–40. [DOI] [PubMed] [Google Scholar]
- Vorherr T., James, P., Krebs, J., Enyedi, A., McCormick, D.J., Penniston, J.T., and Carafoli, E. 1990. Interaction of calmodulin with the calmodulin binding domain of the plasma membrane Ca2+ pump. Biochemistry 29: 355–365. [DOI] [PubMed] [Google Scholar]
- Wilson M.A. and Brunger, A.T. 2000. The 1.0 Å crystal structure of Ca2+-bound calmodulin: An analysis of disorder and implications for functionally relevant plasticity. J. Mol. Biol. 301: 1237–1256. [DOI] [PubMed] [Google Scholar]
- Yamazaki T., Forman-Kay, J.D., and Kay, L.E. 1993. Two-dimensional NMR experiments for correlating 13Cβ and 1Hδ/ɛ chemical shifts of aromatic residues in 13C labeled proteins via scalar couplings. J. Am. Chem. Soc. 115: 11054–11055. [Google Scholar]
- Yap K.L. 1995. Interhlx. University of Toronto, Toronto.
- Yap K.L. and Tomomori, C. 1999. CSI. University of Toronto, Toronto.
- Zhang M., Tanaka, T., and Ikura, M. 1995. Calcium-induced conformational transition revealed by the solution structure of apo calmodulin. Nat. Struct. Biol. 2: 758–767. [DOI] [PubMed] [Google Scholar]
- Zwahlen C., Gardner, K.H., Sarma, S.P., Horita, D.A., Byrd, R.A., and Kay, L.E. 1998. An NMR experiment for measuring methyl-methyl NOEs in 13C-labeled proteins with high resolution. J. Am. Chem. Soc. 120: 7617–7625. [Google Scholar]