

Abstract
The murine molecule dectin-1 (known as the β-glucan receptor in humans) is an immune cell surface receptor implicated in the immunological defense against fungal pathogens. Sequence analysis has indicated that the dectin-1 extracellular domain is a C-type lectin-like domain, and functional studies have established that it binds fungal β-glucans. We report several dectin-1 crystal structures, including a high-resolution structure and a 2.8 Å resolution structure in which a short soaked natural β-glucan is trapped in the crystal lattice. In vitro characterization of dectin-1 in the presence of its natural ligand indicates higher-order complex formation between dectin-1 and β-glucans. These combined structural and biophysical data considerably extend the current knowledge of dectin-1 structure and function, and suggest potential mechanisms of defense against fungal pathogens.
Keywords: immune recognition, fungal pathogen, β-glucan, protein crystallography, C-type lectin-like domain
Dectin-1 is a cell-surface immune receptor for β-glucans, which are major structural cell wall components that are conserved in fungi (Brown and Gordon 2001). Recognition of β-glucans by dectin-1 can trigger phagocytosis of fungal pathogens and protective inflammatory responses. Originally identified as a dendritic cell receptor in the mouse (Ariizumi et al. 2000), dectin-1 is now known to be widely expressed in both mouse and human cells, particularly on monocytes/macrophages and neutrophils (Brown 2006).
Primary sequence analysis indicates that dectin-1 is a 28-kDa type II membrane protein. An extracellular C-type lectin-like domain (CTLD) is connected by a stalk to a transmembrane region, followed by a cytoplasmic tail containing an immunoreceptor tyrosine-based activation (ITAM)-like motif (Ariizumi et al. 2000). First recognized as a calcium-dependent carbohydrate-binding domain, the CTLD fold is also seen in non-calcium-dependent protein recognition interactions (Drickamer 1999). Few of the residues required for calcium coordination in classical CTLDs are conserved in dectin-1. The dectin-1 CTLD has two potential N-linked glycosylation sites, whereas its human homolog, the β-glucan receptor (βGR), has none. In addition to its role in β-glucan binding, it has been suggested that dectin-1 recognizes an endogenous T-cell ligand in a carbohydrate-independent manner (Ariizumi et al. 2000), but there is no evidence as to the nature of this potential ligand.
Dectin-1 recognizes β-1,3 and β-1,6 linked glucans (but not those containing solely β-1,6 linkages) from a variety of sources, including yeast, other fungi, plants, and bacteria (Brown 2006; Palma et al. 2006). Well-characterized ligands include laminarin (a storage polysaccharide found in brown algae) and zymosan (a particulate yeast cell wall extract, composed primarily of β-glucan). β-Glucans can adopt helical and triple-helical formations; their immunological potency is thought to depend on molecular mass, degree of branching, and tertiary structure (Bohn and BeMiller 1995). These features could allow natural β-glucans to form complex three-dimensional arrays, but there is no consensus on what particular structural features contribute to biological activity (Bohn and BeMiller 1995; Herre et al. 2004).
A growing number of fungi previously considered common saprophytes are now being recognized as important human pathogens, and fungal disease recently ranked as the seventh most common cause of infection-related death in the United States (McNeil et al. 2001). This reflects the increasing worldwide prevalence of immunosuppression secondary to HIV infection, the use of lifesaving chemotherapy and organ transplantation, and the use of immunosuppressive agents to treat a variety of diseases. Thus the documented emergence of drug resistance in fungal pathogens is a major public health concern (Cowen et al. 2002), and understanding immune responses against fungi has assumed a new and pressing importance. β-Glucans can potentiate host responses against a wide range of pathogens, and early results of their clinical use in promoting anti-pathogen and antitumor responses are encouraging (Ross et al. 1999; Tzianabos 2000). A detailed consideration of the specific interactions between dectin-1 and β-glucans is clearly of significant biomedical interest.
To investigate β-glucan recognition by dectin-1, we cloned, purified, and crystallized the recognition domain of the murine protein. We describe here the three-dimensional structure of the dectin-1 CTLD at high resolution. In addition, we report a further dectin-1 structure that contains a short soaked β-glucan captured in an acidic environment in the crystal lattice. We also report the results of biophysical characterizations of dectin-1 binding interactions with β-glucan sugars. These structural and functional results allow description of the β-glucan binding site in dectin-1 and suggest a possible mechanism for dectin-1 signaling.
Results
The structure of dectin-1
Table 1 summarizes the X-ray diffraction data collection and structure refinement statistics for four crystals of the extracellular domain of murine dectin-1; two crystals of space group P32 with essentially identical unit cells and two of space group P3221, also with essentially identical unit cells but with one containing β-glucan. The refined crystal structures lack three to four N-terminal residues, and the His-tag in both the N- and C-tagged forms is disordered. Each crystal form contains two monomers (labeled A-chain and B-chain) in the asymmetric unit and the root-mean-squared deviation (RMSD) between A-chains for the P32 and P3221 structures is 0.6 Å (121 equivalent Cα pairs), that between the A- and B-chains of the P32 structures is 0.4 Å (127 equivalent Cα pairs), and between the A- and B-chains of the P3221 structures is 0.2 Å (131 equivalent Cα pairs).
Table 1.
Statistics for crystallographic data collection and refinement

As expected from sequence analysis, the dectin-1 fold is similar to that of other long-form CTLDs (Figs. 1, 2A; Drickamer 1999; Zelensky and Gready 2005), comprising two antiparallel β-sheets (β0-β1-β5-β1′ and β2′-β2-β3-β4-β2″) and two α-helices (α1 and α2). The N and C termini are close together with domain integrity maintained by three disulphide bridges—Cys119-Cys130 (N terminus to β2), Cys147-Cys240 (α1 to β5), and Cys219-Cys232 (β3 to the β4-β5 loop)—stabilizing the long loop region (LLR). The LLR comprises 35 residues in dectin-1 (Arg184–Asn218). Structural superposition of P3221 and P32 monomers reveals that Arg184–Asn208 and Leu216–Asn218 adopt essentially identical conformations and contain two short β-strands (β2′ and β2″). However, the main-chain conformation for Ala209–Leu215 is variable and, in the P3221 crystals, includes a short 310 helix.
Figure 1.

Sequence alignments. Alignment of known dectin-1 sequences, with only the CTLDs shown. Secondary structure elements and solvent accessibilities (blue, accessible; cyan, intermediate; white, buried) of the murine protein are displayed above and below the alignments, respectively. Metal binding residues are boxed and colored yellow. Yellow triangles indicate dectin-1 residues involved in the P3221 asymmetric unit dimer interface, and orange triangles indicate residues mutated by Adachi et al. (2004). The alignment was formatted using ESPript (espript.ibcp.fr/ESPript/ESPript).
Figure 2.
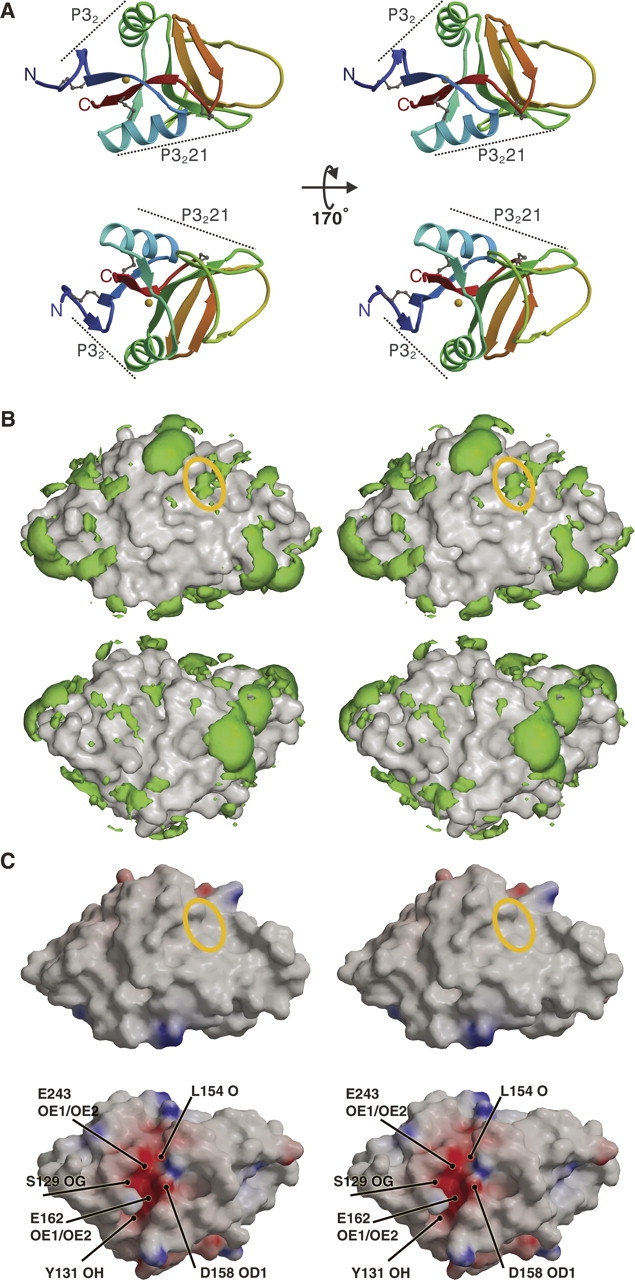
Stereoviews showing surface characteristics of the dectin-1 monomer. (A) Two different ribbon diagram views of the dectin-1 monomer, each colored from blue at the N terminus to red at the C terminus with disulphide linkages shown as gray balls-and-sticks and the metal ion as a golden sphere. Dotted lines indicate surface regions involved in crystallographic dimers. (B) Equivalent orientations as A, showing hydrophobic patches (green) over surface representations of dectin-1. Hydrophobic patches are defined using the program GRID and are shown here as volumes of pseudo-energies contoured at –2.3 kcal mole−1. The solvent accessible regions of Trp221 and His223 are represented by the yellow ellipse. (C) Equivalent orientations as A, showing the electrostatic potential surface of dectin-1 produced using the program GRASP and contoured ±20 kT (blue denotes positive; red, negative potential). The yellow ellipse indicates the solvent accessible regions of Trp221 and His223, and the positions of some of the residues and atoms responsible for the negative potential are indicated.
A structural homology search of the PDB (Protein Data Bank) database performed using MSDFold (Krissinel and Henrick 2004) gave RMSD values for dectin-1 (1.5 Å native P32 A-chain) aligned with various well-known CTLD family members (Table 2). In classical CTLDs, the LLR contains residues responsible for calcium binding, but these residues are absent in dectin-1 and no metal ions stabilize the LLR. Structural superpositions of dectin-1 with the CTLDs from Table 2 demonstrate that, as expected, the LLR is the most variable region of the CTLD structure (Zelensky and Gready 2003), and its conformation does not closely resemble that seen in either classical calcium-dependent carbohydrate-binding CTLDs or those with protein ligands.
Table 2.
RMSD values (obtained from MSDFold) for structural homology of dectin-1 to other CTLDs

For their CTLDs alone, the residue identity between murine dectin-1 and its human homolog βGR (GenBank entry AY026769) is 59.5%, implying that the structure of murine dectin-1 provides a reasonably good model for the structure of the human protein.
Characterization of the dectin-1 surface
Inspection of the dectin-1 surface using GRID highlights several significant areas of hydrophobicity (Fig. 2B), with a number of hydrophobic side chains fully exposed to solvent (Leu120, Tyr141, Ile175, Pro190, Ala198, Phe200, Pro211, and Leu216). Additionally, several hydrophobic side chains are partially exposed to solvent (Leu132, Leu150, Phe163, Trp191, Phe192, Phe199, Phe204, Val206, Val210, Trp221, Tyr228, and Tyr237), and there is a contribution from the aliphatic portion of several long side chains (Lys161, Arg174, Glu188, Gln212, and Glu213). Extended hydrophobic regions are commonly involved in protein–protein interactions, whereas hydrophobic contacts could play a role in β-glucan binding. Adachi et al. (2004) characterized an extensive set of alanine mutations that, when mapped to the dectin-1 structure, are relatively evenly distributed over its surface. Only mutations of Trp221 and His223 showed reduced β-glucan binding, and these side chains were postulated to be critical in the formation of the dectin-1 β-glucan binding site. Indeed both Trp221 and His223 side chains are exposed on the dectin-1 surface, where they could participate directly in ligand binding (Fig. 2), and are conserved in all the dectin-1 homologs sequenced to date. Our structure reveals a shallow surface groove running between the Trp221 and His223 side chains.
Overall analysis of the electrostatic potential surface using GRASP (Fig. 2C) reveals that the putative β-glucan binding groove running between by Trp221 and His223 demonstrates no striking imbalance of charge. This might indicate that β-glucan binding is driven mainly by hydrophobic forces, including stacking with the Trp and His side chains, although electrostatic interactions would be possible with the nitrogen atoms in these side chains. The groove has additional features that could contribute to carbohydrate binding via hydrophobic packing and/or hydrogen bonding, including the side chains of Tyr228 and Gln230.
GRASP analysis does, however, highlight a striking feature of the dectin-1 surface not associated with the Trp221/His223 region: A prominent acidic groove starts between α1 and the β2′-β2″ loop, continues between β1′ and the β0-β1 loop, and finishes between α2 and β1. The negative potential of this region involves contributions from Ser129 OG, Tyr131 OH, Leu154 O, Asp158 OD1/OD2, Glu162 OE1/OE2, Glu194 OE1/OE2, Asp195 OD1/OD2, Glu241 OE1/OE2, and Glu243 OE1/OE2. The groove encompasses a metal binding site located between α2 and β5, close to the C terminus. Metal ion coordination is octahedral, involving Lys156 O, Asp158 OD2, Glu162 OE1, Glu241 OE1, and two water molecules, with interatomic distances ranging from 2.2–2.6 Å (Fig. 3). This site is equivalent to a site observed in the CTLD domains of snake venom coagulation factor IX-binding protein (Mizuno et al. 1999) and the human asialoglycoprotein receptor (Meier et al. 2000), where it is thought to stabilize the CTLD fold. The lower-resolution P32 dectin-1 structure crystallized from a condition containing Mg2+, and the protein buffer from which the P3221 structures derive contained Ca2+. However, the highest-resolution P32 structure did not contain a metal ion since the protein was never exposed to divalent cations. Structural superpositions reveal no conformational variations between the 1.5 Å native and 2.2 Å native P32 dectin-1 structures (i.e., with and without metal bound), with an RMSD of 0.2 Å for 127 equivalent Cα pairs, indicating that metal chelation is not required for correct folding of the polypeptide chain.
Figure 3.

Metal ion binding coordination in dectin-1. Ball-and-stick diagram showing the octahedral metal ion coordination in dectin-1. The distances between the calcium ion and the chelating atoms are as follows: Lys156 O, 2.3 Å; Asp158 OD2, 2.2 Å; Glu162 OE1, 2.2 Å; Glu241 OE1, 2.3 Å; HOH1, 2.2 Å; and HOH2, 2.4 Å.
Refolded dectin-1 CTLDs bind divalent cations and β-glucans
The bioactivity of the refolded dectin-1 used for the structural analysis was confirmed using a flow cytometry–based assay with appropriate controls. This demonstrated that both the N- and C-terminally His-tagged proteins bound β-glucan particles (Fig. 4A). As has been demonstrated previously at the cell surface, this binding did not require metal ions and was inhibited by preincubation with a glucan phosphate inhibitor.
Figure 4.

Biophysical studies of refolded dectin-1. (A) FACS-based assay showing β-glucan binding by refolded dectin-1 constructs. The dark gray region corresponds to dectin-1 binding, the lighter gray line indicates inhibition by glucan phosphate, and the black region corresponds to the negative control. (B) Thermal shift profiles show dectin-1 binds β-glucan and divalent cations. Measurements of dectin-1 alone (blue), with Ca2+/Mg2+ (green), with laminarin (red), and with both Ca2+/Mg2+ and laminarin (orange) are shown. The shift in melting temperature, indicated by the gray lines, reflects the increased energy required to melt the protein in the presence of the various ligands. (C) AUC measurements demonstrate that dectin-1 forms multimeric complexes in the presence of laminarin. Measurements, colored as in B, were taken at 21,000 rpm and 280 nm. In the absence of laminarin (blue and green measurements), the molecular weights suggest monomeric species, whereas in the presence of laminarin (red and orange), there is an obvious increase in molecular weight. Two possible curve fits for the measurements taken with dectin-1 plus laminarin (in red) are shown (for details, see text).
Thermal shift analysis of refolded dectin-1 (Fig. 4B) provided a biophysical demonstration that a divalent cation significantly stabilizes the dectin-1 CTLD, with the observed melting temperature (Tm) increasing from 47.8°C without a metal ion bound to 53.2°C in the presence of 0.5 mM Ca2+/Mg2+. The bioactivity of refolded dectin-1 was further confirmed by the inclusion of laminarin, a natural β-glucan. When laminarin was present, the observed ΔTms were 8.3°C and 9.9°C with and without divalent cations, respectively. Such dramatic increases in ΔTm not only provide clear evidence for interaction between laminarin and dectin-1 but also, because the effect on Tm occurs irrespective of the presence of divalent cations, concur with previous studies demonstrating that β-glucan binding by dectin-1 does not require metal ions (Brown and Gordon 2001).
Monomeric dectin-1 associates with laminarin to form complexes with higher apparent molecular mass
Analysis by dynamic light scattering (DLS) of soluble dectin-1, dectin-1 with divalent cations, and dectin-1 with laminarin gave estimated molecular masses (for the major peaks) of 17.3 kDa, 19.3 kDa, and 81.4 kDa, respectively. Although these results are consistent with the presence of monomeric dectin-1 (16.1 kDa) for the isolated protein and protein/cations, the sample with laminarin (Sigma; ∼7.7 kDa measured by low-angle laser scattering) appears to contain a species of much higher molecular mass. Either several protein molecules are attached to one laminarin monomer or the stoichiometry is lower, but the nonglobular laminarin skews the estimated molecular mass to appear higher than it truly is (the standard curve assumes roughly spherical molecules and is likely to overestimate molecular mass for nonspherical molecules). No interpretable results were obtained with laminarin alone, probably because the unbound molecule is long and flexible in solution.
In order to provide a more definitive answer, analytical ultracentrifugation (AUC) was used to look at a range of concentrations of dectin-1, dectin-1/cations, and an equimolar dectin-1/laminarin mixture (Fig. 4C). The fitted molecular weight for dectin-1 alone and with divalent cations is 15.9 ± 0.9 kDa, consistent with a predominantly monomeric species. In the presence of laminarin, the molecular mass increased to between 77.2 and 92.3 kDa, with evidence of aggregation at higher concentrations, suggestive of complex formation between dectin-1 and laminarin. The laminarin had an approximate molecular weight of 7.7 kDa, equating to a β-glucan polymer of ∼40 β-D-glucose monomers. Fitting the data for dectin-1 and laminarin using an equation assuming tetramerization indicates that the saturated dectin-1:laminarin complex had a weight consistent with a stoichiometry of 4:1. However, a much better fit is obtained when using a cooperative model (Fig. 4C, cf. solid and dotted lines). The coefficient of cooperativity (in this case equivalent to the order of the reaction) calculated from this fit is 2.1 ± 0.5, which does suggest that the cooperativity is experienced as dimers of dectin-1 binding to laminarin rather than a formally tetrameric arrangement. Although the current level of analysis is not capable of providing detailed constants for the various dectin-1–laminarin interactions (involving laminarin monomers with varying numbers of bound dectin-1 monomers), the overall affinity for the total reaction can be expressed as Kd ≈ 0.2 mg/mL (∼0.01 mM). Thus both DLS and AUC clearly indicate the formation of higher-order complexes between dectin-1 and laminarin in vitro.
A dimeric arrangement in the P3221 crystal lattice traps β-glucan
The protein–protein contact in the P3221 asymmetric unit is relatively extensive, comprising a total buried surface area of 1520 Å2 involving Trp140, Tyr141, Lys144, Arg145, Ser148, Gln149, Ser183, Arg184, Asn185, Gln186, Ser187, Pro190, Phe192, Trp193, Glu194, Asp195, Gly196, Ser197, Ala198, Leu216, Asn218, Cys219, Cys232, and Asn233. This crystallographic dimer interface contains seven hydrogen bonds plus hydrophobic interactions. A variety of oligomers have been observed in CTLD crystal structures, not all of which have been verified as existing in vivo (Drickamer 1999; McGreal et al. 2005), and the P3221 arrangement of dectin-1 CTLDs does not resemble any of the previously observed physiological dimers for other members of this family. More importantly, Asn185 has been reported to be glycosylated in murine cells (Kato et al. 2006), a finding that argues that this interface is unlikely to be replicated in vivo. This P3221 asymmetric unit dectin-1 dimer does, however, juxtapose the two dectin-1 monomers such that the acidic groove from each joins up to form a continuous cleft at the interface (Fig. 5A,B). GRASP indicates that this region is predominantly hydrophilic, although analysis by GRID does indicate hydrophobicity associated with Gly151 and the aliphatic regions of the Asp195 and Glu243 side chains.
Figure 5.
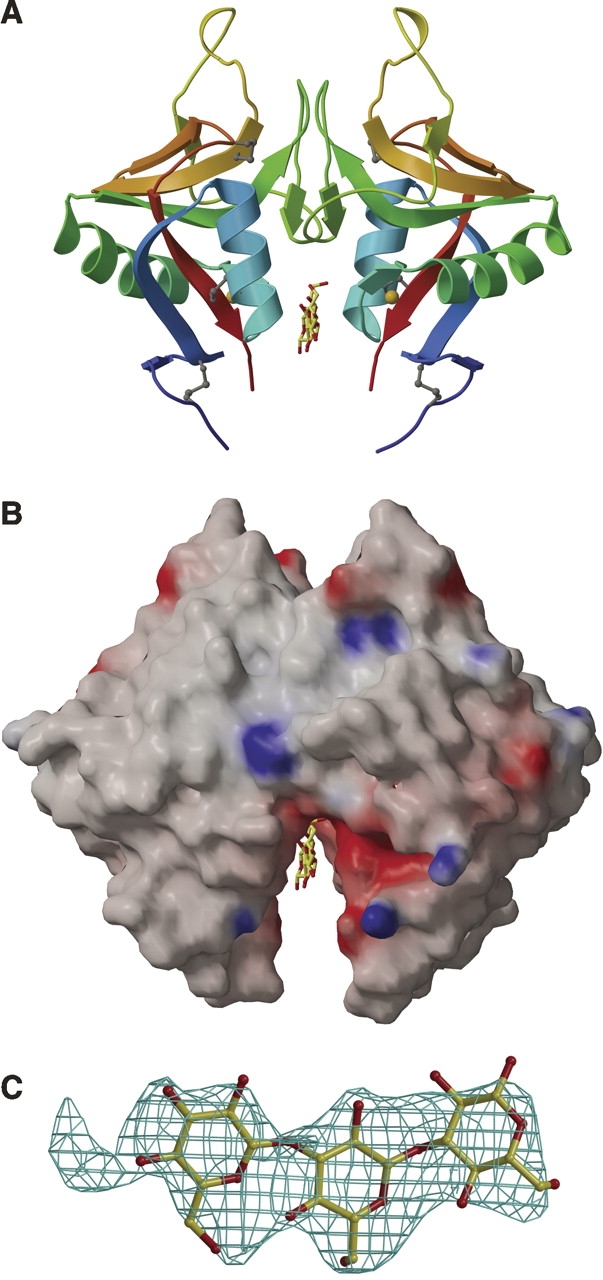
Two dectin-1 monomers form a dimer into which a short β-glucan binds. (A) A cartoon diagram of the dectin-1 P3221 dimer, with each monomer colored from blue at the N terminus to red at the C terminus. Disulphide linkages are shown as gray balls-and-sticks, the metal ion as a golden sphere, and the bound β-glucan as yellow and red balls-and-sticks. (B) Equivalent orientation as A, showing the electrostatic potential surface of the dectin-1 P3221 dimer around the region where β-glucan is observed, produced using GRASP and contoured ±20 kT (blue denotes positive; red, negative potential). Bound β-glucan is shown as yellow and red balls-and-sticks. (C) Electron density for laminaritriose observed in a 2Fo–Fc composite-omit map contoured at 1σ.
Electron density maps calculated for a laminaritriose crystal soak (space group P3221) revealed electron density in this cleft that was not accounted for by the dectin-1 structure. Laminaritriose was modeled into this density (Fig. 5C). The crystallographic dimer forms a symmetrical binding site, such that β-glucan may bind in either orientation across the site with the same chemical groups from each monomer potentially contributing to binding. Since a dimer with this exact geometry is unlikely to form in vivo (because of glycosylation at Asn185) and because the region is distinct from the Trp221/His223 β-glucan binding groove discussed above, we are unable to assign any physiological importance to this dimer. In the mutational analysis reported by Adachi et al. (2004), only Glu194Ala is in this acidic groove; no effect on binding is attributed to this mutation (Adachi et al. 2004). Although the trapping of laminaritriose here may simply be an artifact of crystallization, the presence of this acidic groove may be indicative of a dectin-1 physiological function as yet undefined.
The P32 asymmetric unit contained a dectin-1 dimer that associated via a different interface to that seen in the P3221 asymmetric unit (Fig. 2B). Diffraction data were collected on several P32 crystals soaked with laminaritriose, but no β-glucan was captured in these crystals, possibly because the appropriate dimer arrangement is absent in the P32 crystal lattice and because the solvent content of the P32 crystals was significantly lower than the P3221 crystals. It should also be noted that the reservoir solution for the condition in which laminaritriose bound was pH 4.0. Comparison of predicted side-chain pKa values by PDB2PQR and PropKa (Dolinsky et al. 2004; Li et al. 2005) at pH 4.0 and pH 7.0 suggests that Glu194 and Glu243 would be protonated at low pH. It is unclear whether this protonation is responsible for the β-glucan binding we observe. Notably, His223, highlighted by Adachi et al. (2004) as likely to be involved in β-glucan binding, would be protonated at pH 4.0 and this might explain the lack of binding seen in the Trp221/His223 groove.
Discussion
The work described here suggests potential mechanisms whereby the structure of dectin-1 has evolved to recognize β-glucans. Lectin–carbohydrate recognition can involve a variety of interactions, including nonpolar (hydrophobic) packing and hydrogen bonds (Weis and Drickamer 1996). Our biophysical analyses using a natural β-glucan suggest a role for oligomerization in the mechanism of pathogen recognition and signaling by dectin-1.
The putative β-glucan binding site of dectin-1, as defined by Trp221 and His223, is a shallow groove on the protein surface with some hydrophobic character. It might have been expected that laminaritriose soaked into dectin-1 crystals would bind in this groove, but no binding was evident possibly due to protonation of His223 in the crystallization condition and/or because the ligand is too short. Unfortunately, despite numerous attempts, longer-length β-glucans in complex with dectin-1 proved incompatible with formation of a suitable crystal lattice, probably due to a lack of structural homogeneity. Studies have recently demonstrated that the minimum length of β-glucan that can be detected binding to dectin-1 in solution is a 10- or 11-mer (Palma et al. 2006). It is possible that binding of shorter β-glucans is not detectable because the binding is simply too weak. Nevertheless, simple modeling studies (data not shown) demonstrate that the Trp221/His223 surface groove could easily accommodate a β-glucan chain, and it is possible that this region is part of a binding site that would be occupied by 10- or 11-mer β-glucans.
Our biophysical studies indicate that dectin-1 alone is monomeric in vitro but forms complexes of higher molecular mass in the presence of laminarin. Although no in vivo evidence suggests that dectin-1 oligomerizes, a variety of CTLD oligomers have been observed in vitro and in vivo, and it is formally possible that dectin-1 can form quarternary structures. If a fungal pathogen displaying a naturally multivalent array of long β-glucans encounters an immune cell, oligomerization of dectin-1 could increase avidity, leading to lattice formation (Weis and Drickamer 1996) and clustering of the intracellular domains, providing a simple mechanism by which the presence of a fungal pathogen is signaled to the host cells. Although it is possible that dectin-1 monomers are binding to β-glucan independently of each other, the attractiveness of a dectin-1 oligomerization model is enhanced by the proposal that Syk kinase binds to the cytoplasmic tails of two proximate dectin-1 monomers as part of a novel signaling pathway (Rogers et al. 2005). It might be that these proximate tails actually belong to oligomers formed by the binding of multiple dectin-1 monomers close to each other on the same long β-glucan chain.
Binding of long oligosaccharide β-glucans by dectin-1 is not dependent on metal ions. Although our crystal structures reveal that dectin-1 monomers do contain a metal binding site, this site is distinct from the classical sites seen in those CTLDs that bind monosaccharides or very short oligosaccharides. A metal ion in an equivalent position to that of the dectin-1 site has been postulated to fulfill a structural role in other CTLDs (Zelensky and Gready 2005), and there are several cases of calcium ions stabilizing extracellular protein domains. Our thermal shift studies of dectin-1 reveal a significant change in Tm in the presence of a divalent cation, and we believe that stabilization of the dectin-1 CTLD fold is indeed enhanced by the metal ion. The residues involved in metal coordination are conserved in all dectin-1 homologs sequenced to date, and it is formally possible that, in addition to its structural role, this site may have an unidentified physiological function.
The role of CTLDs in immune recognition is currently the subject of intense investigation. Of these CTLDs, dectin-1 has emerged as the central receptor in the immune recognition of fungal β-glucans, an event that promotes phagocytosis of fungal pathogens and drives a pro-inflammatory immune response. Thus molecular characterization of the structure and function of dectin-1 and its interaction with β-glucans is a direct route to a fundamental understanding of the protective immune response against fungal pathogens. The work presented here provides a basis for further studies aimed at understanding fungal pathogenesis and for the rational design of immunomodulatory therapy based on β-glucans.
Materials and Methods
Dectin-1 production
Two constructs of dectin-1 CTLDs (amino acids 113–244 with N- and C-terminal histidine tags, respectively) were produced as insoluble proteins in Escherichia coli. Both proteins were refolded according to previously described protocols (Brown et al. 2002), with refolded protein concentrated and purified by gel filtration. Gel analysis of the major peak (∼15 kDa, consistent with a monomeric molecule) showed only one band and expected molecular weights were confirmed by mass spectrometry.
Crystallization
Over 300 nL-scale sitting-drop crystallization experiments were set up at 21°C using a Cartesian Technologies Microsys MIC4000 (Genomic Solutions) (Walter et al. 2003). Hexagonal N-tagged dectin-1 crystals (grown from 0.2 M di-ammonium hydrogen citrate, 20% PEG 3350) diffracted to 1.5 Å (Table 1, 1.5 Å native). Similar C-tagged dectin-1 crystals (grown from 0.2 M potassium chloride, 0.05 M sodium cacodylate at pH 6.5, 0.1 M magnesium acetate, 10% PEG 8000) diffracted to 2.2 Å (Table 1, 2.2 Å native). Further trials used dectin-1 with 1 mM CaCl2 and either laminarin (Sigma) or synthetic β-glucan (a β-1,3 linked decasaccharide kindly provided by Professor D.L. Williams, East Tennessee State University, Johnson City, TN). Trials using laminarin gave no crystals. Trials using the synthetic β-glucan yielded hexagonal crystals with a previously unseen morphology, grown from 2 M sodium chloride and 0.1 M sodium acetate (pH 4.6). These crystals diffracted to 2.25 Å, but unfortunately, no carbohydrate was evident in the electron density (Table 1, 2.25 Å native).
Some of the remaining crystals from these trials were used in soaking experiments. Laminaritriose powder (prepared at >95% purity by enzymic digestion of natural β-glucans by Megazyme International Ireland Ltd.) was sprinkled onto sitting-drops containing crystals and allowed to dissolve (van Bueren et al. 2004). After soaking for up to 3 wk, data were collected in-house. After soaking, a crystal (initially grown from 2 M sodium chloride, 0.1 M citric acid at pH 4.0) diffracted to 2.8 Å and was found to contain laminaritriose bound to dectin-1 (Table 1, 2.8 Å complex).
Data collection
Crystals were cryoprotected in perfluoropolyether (Lancaster) and flash cooled to 100 K in liquid nitrogen. All X-ray diffraction data were autoindexed, integrated, and scaled with the HKL program package (Otwinowski and Minor 1997). Three data sets for uncomplexed crystals were collected at the European Synchrotron Radiation Facility (ESRF); two for a P32 space group (two molecules per asymmetric unit, estimated solvent content 42%) and one for a P3221 space group (two molecules per asymmetric unit, estimated solvent content 67%). Data sets for crystals soaked with β-glucan were collected in-house using a MicroMax micro-focus rotating anode X-ray generator (40 kV, 20 mA) with either VariMax-HR or Blue optics (Osmic) and Mar345 imaging plate detectors. The data set collected for the dectin-1–laminaritriose complex was also for a P3221 space group (two molecules per asymmetric unit, estimated solvent content 67%). Details of these data sets are given in Table 1.
Structure determination and refinement
Crystallographic refinement used CNS (Brünger et al. 1998) and REFMAC5 (Murshudov et al. 1997), with O (Jones et al. 1991) and Coot (Emsley and Cowtan 2004) used for manual checking and building with reference to 2Fo-Fc and Fo-Fc electron density maps. ARP/wARP (Perrakis et al. 1999) was used for automatic building and both ARP/wARP and CNS were used for water picking. Ion binding sites were identified from strong positive peaks in Fo-Fc electron density maps.
The structure was solved by molecular replacement using the CaspR Web server (http://igs-server.cnrs-mrs.fr/Caspr2/index.cgi/) with six models (1K9J, 1FM5, 1HQ8, 1JA3, 1MPU, and 1E87) from the Protein Data Bank (www.pdb.org). The top solution, a truncated homology model comprising amino acids 116–171, 179–199, and 217–244, had an RTest of 40.6% and RWork of 34.6%.
This model was refined against the 1.5 Å native P32 data set, and residues 172–178 were built manually. This incomplete structure was used in EPMR (Kissinger et al. 1999) to phase the 2.2 Å native P32 data set. Following rigid body refinement, residues 200–216 were built manually. The complete model was refined against the 1.5 Å native P32 data set.
One monomer from the 1.5 Å native P32 model was used in CaspR against the 2.25 Å native P3221 data. The top solution had an RTest of 33.6% and RWork of 28.4%. The 209–215 loop had a different conformation and was rebuilt. Although this data set was collected on a crystal derived from trials using dectin-1 and the synthetic β-glucan, no carbohydrate was evident in the electron density. In the final refinement stages, it was noted that each monomer had an associated portion of difference electron density in a hydrophobic environment. The most appropriate candidate to explain this, a fragment of Triton X-100 (TTX, used in the inclusion body preparation), fitted the electron density well and was refined.
In-house data sets were solved by molecular replacement using MOLREP (Vagin and Teplyakov 1997). Following rigid-body refinement of a molecular replacement solution in a P3221 data set, an unmodeled portion of electron density was evident. The dimensions corresponded well with those of the soaked-in laminaritriose, which was thus built in and refined. The B-factors of the β-glucan were high compared with the protein B-factors (Table 1). There are several plausible reasons for this. The β-glucan binding site has twofold symmetry, and the bound sugar could be present in two possible orientations. Also, the site is probably not fully occupied. Given the limited resolution of the data, we modeled only one β-glucan orientation, and we fixed the occupancy at 1.0. Stereochemical and refinement statistics are given in Table 1.
β-Glucan binding assays
Flow cytometry based assays were performed with and without glucan phosphate inhibitor (Williams et al. 1991). Briefly, 5 μg mL−1 of purified protein was incubated with 25 μg of β-glucan beads in PBS at 4°C for 1 h. If required, glucan phosphate (500 μg mL−1) was incubated with the protein for 20 min prior to the addition of glucan beads. Samples were washed thoroughly, and the bound dectin-1 quantitated by flow cytometery, following detection with a murine monoclonal penta-His antibody (Qiagen) and phycoerythrin-labeled anti-mouse IgG (BD-Pharmingen). The negative control was the 16-kDa domain 11 of the insulin-like growth factor 2 receptor, which was expressed, purified, and refolded in the same manner as the dectin-1 CTLDs (Brown et al. 2002).
Thermal shift assays were performed using a real-time PCR machine (Lo et al. 2004). Reactions of 25 μL containing 0.5 mg/mL dectin-1, Sypro Orange (Molecular Probes), and either buffer alone, laminarin (1.25 mg/mL; Sigma, molecular mass ∼7.7 kDa, a polymer of ∼40 β-D-glucose monomers), or divalent cations (Ca2+ and Mg2+, 0.5 mM) were heated stepwise from 20°C to 90°C. Changes in fluorescent intensity were measured at excitation/emission wavelengths of 490/575 nm. The midpoint of the unfolding transition approximates to the Tm of a protein. A Boltzmann sigmoid fit was fitted to the melt curves using the MSExcel XLFit plugin (www.idbs.com).
DLS and AUC
DLS samples, prepared in 50 mM Tris (pH 8), 150 mM NaCl at protein concentrations of 1 mg/mL, were added to a quartz sample cell in a DynaPro99 DLS instrument (Protein Solutions Inc.). Scattering measurements taken at 20°C were analyzed using the DYNAMICS software package (Protein Solutions Inc.), and molecular mass was estimated by comparison to a standard curve.
Sedimentation equilibrium experiments were performed in a Beckman Optima XL-I analytical ultracentrifuge as previously described (Harkiolaki et al. 2003). Dectin-1 alone, with divalent cations, and with an equimolar amount of laminarin in 50 mM Tris (pH 8), 150 mM NaCl were used at concentrations ranging from 0.1–0.4 mg/mL; centrifuged at 12,000, 15,000, or 21,000 rpm at 20°C; and imaged using absorbance optics at 254-, 280-, and 300-nm wavelengths. Data at 280 nm and 21,000 rpm are presented as representative (Fig. 4C); lower speeds were used to control for sample aggregation. The sample distributions measured at equilibrium were fitted with the program ULTRASPIN (ultraspin.mrc-cpe.cam.ac.uk) using a single-species equation. Plotting the resulting apparent whole-cell weight-average molecular weights against the sample concentration in each experiment then indicates the associational behavior of the protein. Nonassociating species were fit with a straight line to give their Mw at the Y-axis free of the effects of nonideality. Association was modeled as a tetramerization with dectin-1, each monomer accounting for a quarter of the total weight of laminarin, using the equation
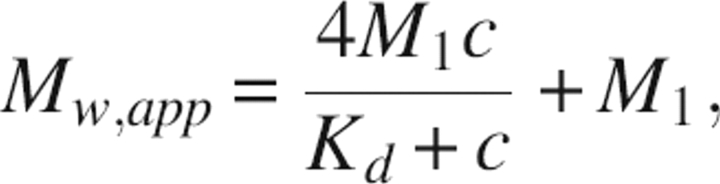 |
where Mw,app is the apparent molecular weight at concentration c, M1 is the monomeric weight of dectin-1 plus a quarter of the weight of laminarin, and Kd is the equilibrium constant of dissociation. Cooperativity in the observed association was modeled using the modified equation
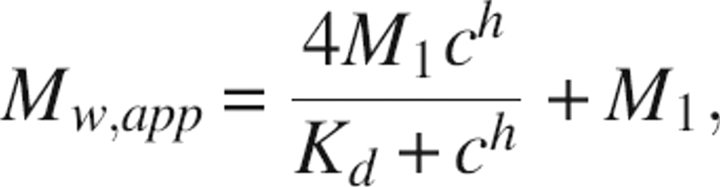 |
where h is the coefficient of cooperativity (as in the Hill equation in steady-state kinetics).
Structural analyses and visualization
Structural superpositions were performed with SHP (Stuart et al. 1979), and protein–protein interfaces were evaluated using the Protein-Protein Interaction Server (Jones and Thornton 1996). Surface hydrophobicity was assessed using the program GRID (Goodford 1985), and electrostatic potential surfaces were displayed using GRASP (Nicholls et al. 1991). Figures were produced using Bobscript (Esnouf 1999) and MolScript (Kraulis 1991).
Accession codes
Atomic coordinates and structure factors have been deposited in the Protein Data Bank, Rutgers University, New Brunswick, New Jersey (www.rcsb.org/), with accession codes 2BPD (1.5 Å native, P32), 2BPH (2.2 Å native, P32), 2BPE (2.25 Å native, P3221), and 2CL8 (2.8 Å complex, P3221 with laminaritriose).
Acknowledgments
We thank G. van Boxel and the staff of the ESRF and EMBL Outstation in Grenoble for help during data collection at beamlines ID-14.1, ID-14.2, and ID-14.3; J. Nettleship for mass spectrometry; J. Dong for assistance with computation; and M. Crispin for helpful discussions. AUC experiments were performed in the AUC facility established by the BBSRC and the Wellcome Trust in the Glycobiology Institute of the University of Oxford, managed by Russell Wallis. Thermal shift assays were undertaken in the Department of Biochemistry and Biophysics at Stockholm University with the help of U. Ericsson. This work was supported by Cancer Research UK and the Wellcome Trust. C.O. is an MRC Senior Clinical Fellow, R.J.C.G. is a Royal Society University Research Fellow, S.G. is a Glaxo-Wellcome Professor of Cellular Pathology, G.D.B. is a Wellcome Trust International Senior Research Fellow in Biomedical Science in South Africa, and E.Y.J. is a Cancer Research UK Principal Research Fellow.
Footnotes
Reprint requests to: E. Yvonne Jones, Division of Structural Biology, Wellcome Trust Centre for Human Genetics, University of Oxford, Roosevelt Drive, Headington, Oxford, OX3 7BN, UK; e-mail: yvonne@strubi.ox.ac.uk; fax: 44-1865-287547.
Abbreviations: AUC, analytical ultracentrifugation; βGR, human β-glucan receptor; BGC, β-glucan; CTLD, C-type lectin-like domain; DLS, dynamic light scattering; TTX, Triton X-100.
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.072791207.
References
- Adachi Y., Ishii, T., Ikeda, Y., Hoshino, A., Tamura, H., Aketagawa, J., Tanaka, S., and Ohno, N. 2004. Characterization of β-glucan recognition site on C-type lectin, dectin 1. Infect. Immun. 72: 4159–4171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ariizumi K., Shen, G.L., Shikano, S., Xu, S., Ritter, R., Kumamoto, T., Edelbaum, D., Morita, A., Bergstresser, P.R., and Takashima, A. 2000. Identification of a novel, dendritic cell-associated molecule, dectin-1, by subtractive cDNA cloning. J. Biol. Chem. 275: 20157–20167. [DOI] [PubMed] [Google Scholar]
- Bohn J.A. and BeMiller, J.N. 1995. (1–3)-β-D-glucans as biological response modifiers: A review of structure-functional activity relationships. Carbohydr. Polym. 28: 3–14. [Google Scholar]
- Brown G.D. 2006. Dectin-1: A signalling non-TLR pattern-recognition receptor. Nat. Rev. Immunol. 6: 33–43. [DOI] [PubMed] [Google Scholar]
- Brown G.D. and Gordon, S. 2001. Immune recognition: A new receptor for β-glucans. Nature 413: 36–37. [DOI] [PubMed] [Google Scholar]
- Brown J., Esnouf, R.M., Jones, M.A., Linnell, J., Harlos, K., Hassan, A.B., and Jones, E.Y. 2002. Structure of a functional IGF2R fragment determined from the anomalous scattering of sulfur. EMBO J. 21: 1054–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brünger A.T., Adams, P.D., Clore, G.M., DeLano, W.L., Gros, P., Grosse-Kunstleve, R.W., Jiang, J.S., Kuszewski, J., Nilges, M., Pannu, N.S., et al. 1998. Crystallography and NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. D54: 905–921. [DOI] [PubMed] [Google Scholar]
- Cowen L.E., Anderson, J.B., and Kohn, L.M. 2002. Evolution of drug resistance in Candida albicans . Annu. Rev. Microbiol. 56: 139–166. [DOI] [PubMed] [Google Scholar]
- Dolinsky T.J., Nielsen, J.E., McCammon, J.A., and Baker, N.A. 2004. PDB2PQR: An automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. 32: W665–W667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drickamer K. 1999. C-type lectin-like domains. Curr. Opin. Struct. Biol. 9: 585–590. [DOI] [PubMed] [Google Scholar]
- Emsley P. and Cowtan, K. 2004. Coot: Model-building tools for molecular graphics. Acta Crystallogr. D60: 2126–2132. [DOI] [PubMed] [Google Scholar]
- Esnouf R.M. 1999. Further additions to MolScript version 1.4, including reading and contouring of electron-density maps. Acta Crystallogr. D55: 938–940. [DOI] [PubMed] [Google Scholar]
- Goodford P.J. 1985. A computational procedure for determining energetically favorable binding sites on biologically important macromolecules. J. Med. Chem. 28: 849–857. [DOI] [PubMed] [Google Scholar]
- Harkiolaki M., Lewitzky, M., Gilbert, R.J.C., Jones, E.Y., Bourette, R.P., Mouchiroud, G., Sondermann, H., Moarefi, I., and Feller, S.M. 2003. Structural basis for SH3 domain-mediated high-affinity binding between Mona/Gads and SLP-76. EMBO J. 22: 2571–2582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herre J., Gordon, S., and Brown, G.D. 2004. Dectin-1 and its role in the recognition of β-glucans by macrophages. Mol. Immunol. 40: 869–876. [DOI] [PubMed] [Google Scholar]
- Jones S. and Thornton, J.M. 1996. Principles of protein-protein interactions derived from structural studies. Proc. Natl. Acad. Sci. 93: 13–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones T.A., Zou, J.Y., Cowan, S.W., and Kjeldgaard, M. 1991. Improved methods for building protein models in electron-density maps and the location of errors in these models. Acta Crystallogr. A47: 110–119. [DOI] [PubMed] [Google Scholar]
- Kato Y., Adachi, Y., and Ohno, N. 2006. Contribution of N-linked oligosaccharides to the expression and functions of β-glucan receptor, dectin-1. Biol. Pharm. Bull. 29: 1580–1586. [DOI] [PubMed] [Google Scholar]
- Kissinger C.R., Gehlhaar, O.K., and Fogel, D.B. 1999. Rapid automated molecular replacement by evolutionary search. Acta Crystallogr. D55: 484–491. [DOI] [PubMed] [Google Scholar]
- Kraulis P.J. 1991. MOLSCRIPT: A program to produce both detailed and schematic plots of protein structures. J. Appl. Crystallogr. 24: 946–950. [Google Scholar]
- Krissinel E. and Henrick, K. 2004. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr. D60: 2256–2268. [DOI] [PubMed] [Google Scholar]
- Li H., Robertson, A.D., and Jensen, J.H. 2005. Very fast empirical prediction and rationalization of protein pKa values. Proteins 61: 704–721. [DOI] [PubMed] [Google Scholar]
- Lo M.C., Aulabaugh, A., Jin, G., Cowling, R., Bard, J., Malamas, M., and Ellestad, G. 2004. Evaluation of fluorescence-based thermal shift assays for hit identification in drug discovery. Anal. Biochem. 332: 153–159. [DOI] [PubMed] [Google Scholar]
- McGreal E.P., Miller, J.L., and Gordon, S. 2005. Ligand recognition by antigen-presenting cell C-type lectin receptors. Curr. Opin. Immunol. 17: 18–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNeil M.M., Nash, S.L., Hajjeh, R.A., Phelan, M.A., Conn, L.A., Plikaytis, B.D., and Warnock, D.W. 2001. Trends in mortality due to invasive mycotic diseases in the United States, 1980–1997. Clin. Infect. Dis. 33: 641–647. [DOI] [PubMed] [Google Scholar]
- Meier M., Bider, M.D., Malashkevich, V.N., Spiess, M., and Burkhard, P. 2000. Crystal structure of the carbohydrate recognition domain of the H1 subunit of the asialoglycoprotein receptor. J. Mol. Biol. 300: 857–865. [DOI] [PubMed] [Google Scholar]
- Mizuno H., Fujimoto, Z., Koizumi, M., Kano, H., Atoda, H., and Morita, T. 1999. Crystal structure of coagulation factor IX-binding protein from habu snake venom at 2.6 Å: Implication of central loop swapping based on deletion in the linker region. J. Mol. Biol. 289: 103–112. [DOI] [PubMed] [Google Scholar]
- Murshudov G.N., Vagin, A.A., and Dodson, E.J. 1997. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53: 240–255. [DOI] [PubMed] [Google Scholar]
- Nicholls A., Sharp, K.A., and Honig, B. 1991. Protein folding and association—insights from the interfacial and thermodynamic properties of hydrocarbons. Proteins 11: 281–296. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z. and Minor, W. 1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276: 307–326. [DOI] [PubMed] [Google Scholar]
- Palma A.S., Feizi, T., Zhang, Y.B., Stoll, M.S., Lawson, A.M., Diaz-Rodriguez, E., Campanero-Rhodes, M.A., Costa, J., Gordon, S., Brown, G.D., et al. 2006. Ligands for the β-glucan receptor, dectin-1, assigned using “designer” microarrays of oligosaccharide probes (neoglycolipids) generated from glucan polysaccharides. J. Biol. Chem. 281: 5771–5779. [DOI] [PubMed] [Google Scholar]
- Perrakis A., Morris, R., and Lamzin, V.S. 1999. Automated protein model building combined with iterative structure refinement. Nat. Struct. Biol. 6: 458–463. [DOI] [PubMed] [Google Scholar]
- Rogers N.C., Slack, E.C., Edwards, A.D., Nolte, M.A., Schulz, O., Schweighoffer, E., Williams, D.L., Gordon, S., Tybulewicz, V.L., Brown, G.D., et al. 2005. Syk-dependent cytokine induction by dectin-1 reveals a novel pattern recognition pathway for C type lectins. Immunity 22: 507–517. [DOI] [PubMed] [Google Scholar]
- Ross G.D., Vetvicka, V., Yan, J., Xia, Y., and Vetvickova, J. 1999. Therapeutic intervention with complement and β-glucan in cancer. Immunopharmacology 42: 61–74. [DOI] [PubMed] [Google Scholar]
- Stuart D.I., Levine, M., Muirhead, H., and Stammers, D.K. 1979. Crystal structure of cat muscle pyruvate kinase at a resolution of 2.6 Å. J. Mol. Biol. 134: 109–142. [DOI] [PubMed] [Google Scholar]
- Tzianabos A.O. 2000. Polysaccharide immunomodulators as therapeutic agents: Structural aspects and biologic function. Clin. Microbiol. Rev. 13: 523–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vagin A. and Teplyakov, A. 1997. MOLREP: An automated program for molecular replacement. J. Appl. Crystallogr. 30: 1022–1025. [Google Scholar]
- van Bueren A.L., Morland, C., Gilbert, H.J., and Boraston, A.B. 2004. Family 6 carbohydrate binding modules recognize the non-reducing end of β-1,3-linked glucans by presenting a unique ligand binding surface. J. Biol. Chem. 280: 530–537. [DOI] [PubMed] [Google Scholar]
- Walter T.S., Diprose, J., Brown, J., Pickford, M., Owens, R.J., Stuart, D.I., and Harlos, K. 2003. A procedure for setting up high-throughput nanolitre crystallization experiments. I. Protocol design and validation. J. Appl. Crystallogr. 36: 308–314. [Google Scholar]
- Weis W.I. and Drickamer, K. 1996. Structural basis of lectin-carbohydrate recognition. Annu. Rev. Biochem. 65: 441–474. [DOI] [PubMed] [Google Scholar]
- Williams D.L., McNamee, R.B., Jones, E.L., Pretus, H.A., Ensley, H.E., Browder, I.W., and Di Luzio, N.R. 1991. A method for the solubilization of a (1-3)-β-D-glucan isolated from Saccharomyces cerevisiae . Carbohydr. Res. 219: 203–213. [DOI] [PubMed] [Google Scholar]
- Zelensky A.N. and Gready, J.E. 2003. Comparative analysis of structural properties of the C-type-lectin-like domain (CTLD). Proteins 52: 466–477. [DOI] [PubMed] [Google Scholar]
- Zelensky A.N. and Gready, J.E. 2005. The C-type lectin-like domain superfamily. FEBS J. 272: 6179–6217. [DOI] [PubMed] [Google Scholar]