

Abstract
D-Glucose/D-Galactose-binding protein (GGBP) mediates chemotaxis toward and active transport of glucose and galactose in a number of bacterial species. GGBP, like other periplasmic binding proteins, can exist in open (ligand-free) and closed (ligand-bound) states. We report a 0.92 Å resolution structure of GGBP from Escherichia coli in the glucose-bound state and the first structure of an open, unbound form of GGBP (at 1.55 Å resolution). These structures vary in the angle between the two structural domains; the observed difference of 31° arises from torsion angle changes in a three-segment hinge. A comparison with the closely related periplasmic receptors, ribose- and allose-binding proteins, shows that the GGBP hinge residue positions that undergo the largest conformational changes are different. Furthermore, the high-quality data collected for the atomic resolution glucose-bound structure allow for the refinement of specific hydrogen atom positions, the assignment of alternate side chain conformations, the first description of CO2 trapped after radiation-induced decarboxylation, and insight into the role of the exo-anomeric effect in sugar binding. Together, these structures provide insight into how the hinge-bending movement of GGBP facilitates ligand binding, transport, and signaling.
Keywords: hinge motion, atomic resolution, chemotaxis, exo-anomeric effect, radiation damage
Periplasmic binding proteins (PBPs) constitute a large family of bacterial receptors that recognize a variety of small molecule ligands. These soluble proteins can serve as intermediary receptors for transport (via ABC transport systems), chemotaxis, and quorum sensing (Tam and Saier 1993; Chen et al. 2002). All PBPs consist of two α/β globular domains connected by a hinge (Fukami-Kobayashi et al. 1999). This “Venus flytrap” architecture is also present in intracellular bacterial proteins, such as the Lac repressor, and in many eukaryotic receptors, including certain G-protein-coupled receptors, ion channels, and GABA receptors (Felder et al. 1999).
PBPs exist in open and closed forms. As demonstrated by small-angle scattering and cross-linking studies, the open form predominates in the absence of ligands (Careaga et al. 1995; Shilton et al. 1996). Ligand binding induces a large-scale hinge-bending motion that clamps the ligand within the binding cleft and stabilizes the closed form. Thus, ligand binding shifts the equilibrium from largely open to largely closed forms. Most, though not all (Neiditch et al. 2005), membrane-bound receptors or transporters are thought to recognize the closed state.
Two members of the pentose/hexose sugar-binding protein subclass of periplasmic binding proteins (Tam and Saier 1993), ribose-binding protein (RBP), and allose-binding protein (ALBP), have been the object of structural studies; closed ligand-bound structures have been determined as well as multiple distinct open structures (Mowbray and Cole 1992; Bjorkman and Mowbray 1998; Chaudhuri et al. 1999; Magnusson et al. 2002). Analysis of these structures indicates that the open-to-closed transition occurs via classical hinge-bending motions that consist of large torsion changes in a few residues, which ultimately confer a global change in domain orientation. Three conserved polypeptide segments constituting the hinge region move similarly in both ALBP and RBP. The locations of key water molecules involved in hydrogen bonding interactions throughout the hinge region are also conserved (Magnusson et al. 2002).
Glucose/galactose-binding protein (GGBP) is another member of the sugar-binding protein subclass of PBPs. Like RBP and ALBP, GGBP is an α/β protein composed of two globular Rossman fold domains joined by three peptide segments three to four amino acids long (Vyas et al. 1983, 1987). GGBP mediates D-glucose and D-galactose transport via the MglABC transporter (Anraku 1968) (it is the MglB component). It also facilitates chemotaxis to these sugars; the closed, ligand-bound form interacts with the Trg chemoreceptor of Escherichia coli (Hazelbauer and Adler 1971). Structures of E. coli GGBP and Salmonella typhimurium GGBP have been solved in both the D-glucose- and D-galactose-bound forms (Vyas et al. 1983, 1988, 1994; Mowbray et al. 1990; Zou et al. 1993). A closed, unliganded structure of GGBP from S. typhimurium has also been described (Flocco and Mowbray 1994), although an open, unliganded structure of GGBP has not. These closed structures have facilitated the design of multivalent GGBP ligands that can effect chemotaxis (Gestwicki et al. 2000; Gestwicki and Kiessling 2002), and the use of GGBP as a glucose biosensor (Salins et al. 2001). GGBP has also been used as a platform for engineering binding proteins with novel ligand specificities (Looger et al. 2003). Insight into the conformational changes that facilitate binding could further such applications. Moreover, structural studies of GGBP could augment current understanding of carbohydrate recognition and facilitate the design of inhibitors of proteins that share this general architecture. Herein, we describe the atomic resolution structure of glucose-bound GGBP and a 1.55 Å resolution structure of an open sugar-free form of GGBP.
Results
Features of the high-resolution glucose-bound structure
The ultra-high-resolution structure of a glucose-bound form of GGBP was solved by molecular replacement and refined against 0.92 Å resolution data (Table 1). These data yielded electron density maps in which N, O, and C atoms could be experimentally distinguished (Fig. 1A). The high quality of the data also allowed anisotropic B-value refinement and explicit CH and NH hydrogen placement (Supplemental Table S1). The Rcrystal and Rfree are 11% and 13.0%, respectively, for the final refined model (Table 1; Fig. 2). With 309 amino acids, GGBP is one of only two non-enzyme proteins with >300 amino acids to have a structure refined to better than 1.0 Å resolution (Wang et al. 1997).
Table 1.
Data collection and refinement statistics
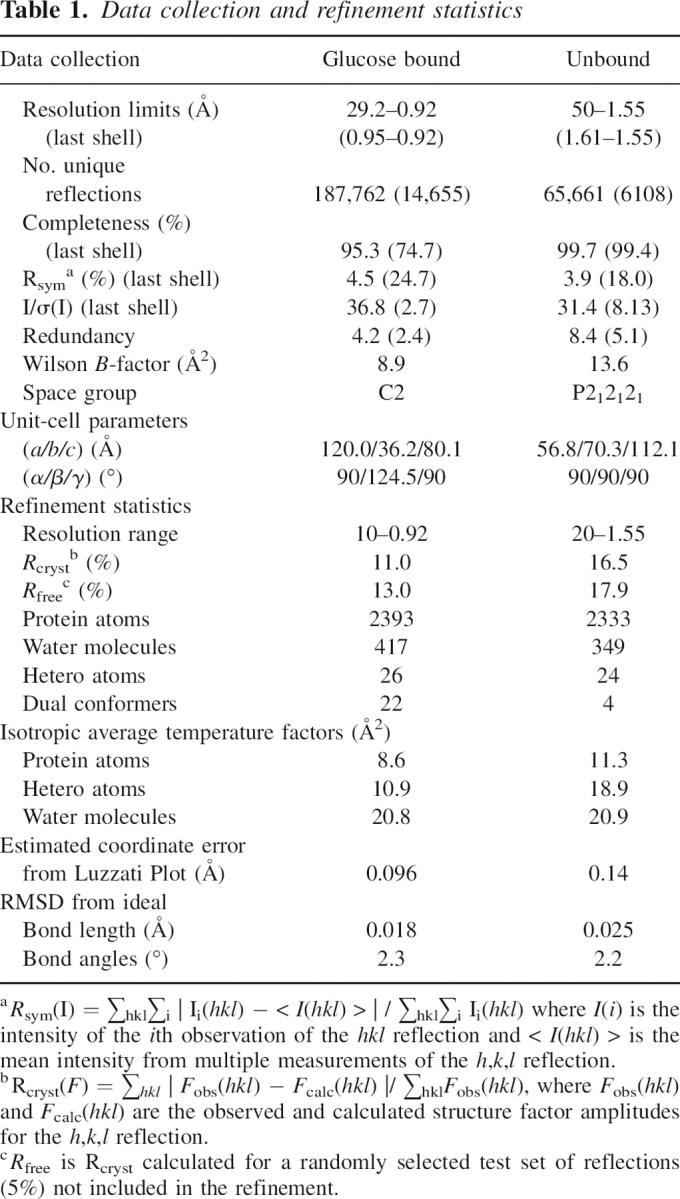
Figure 1.

Representative 2Fo–Fc electron density of segment one in the GGBP hinge region superimposed upon the final refined model. The 0.92 Å resolution glucose-bound structure (contoured at 2.0 σ) (A) and the 1.55 Å resolution structure of the unliganded protein (contoured at 2.0 σ) (B) highlight the overall map quality for both structures as well as the dramatic torsion angle changes at residues 109 and 111.
Figure 2.

Overall structures of GGBP. (A) The 0.92 Å resolution GGBP structure (blue ribbon) bound to glucose (red oxygen and gray carbon spheres) and calcium ion (gold sphere) with the previously solved E. coli glucose-bound structure (PDB accession code 2GBP, red ribbon). The N- and C-terminal domains as well as the three-segment hinge (H) that connects them are labeled. (B) The superposition of the C-terminal domains of the glucose-bound (blue) and open 1.55 Å resolution (green) GGBP structures reveals the 31° hinge opening movement.
Twenty-two dual conformers are present in the glucose-bound model. These presumably represent conformational heterogeneity introduced by cryopreservation (Dunlop et al. 2005) and functional switches in side chain positions. Most dual conformers were easily modeled and were located on the solvent-exposed surfaces of the protein. The electron density at Glu149 defied straightforward interpretation as a simple dual conformer. Refinement led to improved maps that revealed synchrotron radiation-induced decarboxylation of the glutamate side chain on conformer B (Burmeister 2000); strong electron density was observed only to Cγ, with weaker density at the Cɛ position and beyond (Fig. 3A). The carboxyl group of the Glu149B side chain was thus assigned occupancy of 25%. Notably, a linear density feature remained near the carboxyl group of Glu149B. This density is consistent with that of a released CO2 molecule, which was added to the model at 25% occupancy (Fig. 3A). The extended electron density near water 503 was interpreted as a positional shift due to the proximal side chain decarboxylation and was assigned dual occupancy. Radiation damage is common in structures determined using synchrotron radiation, and side chain decarboxylation has been noted previously. However, we are unaware of other radiation-induced decarboxylations in which the released carbon dioxide molecule remains well-ordered.
Figure 3.
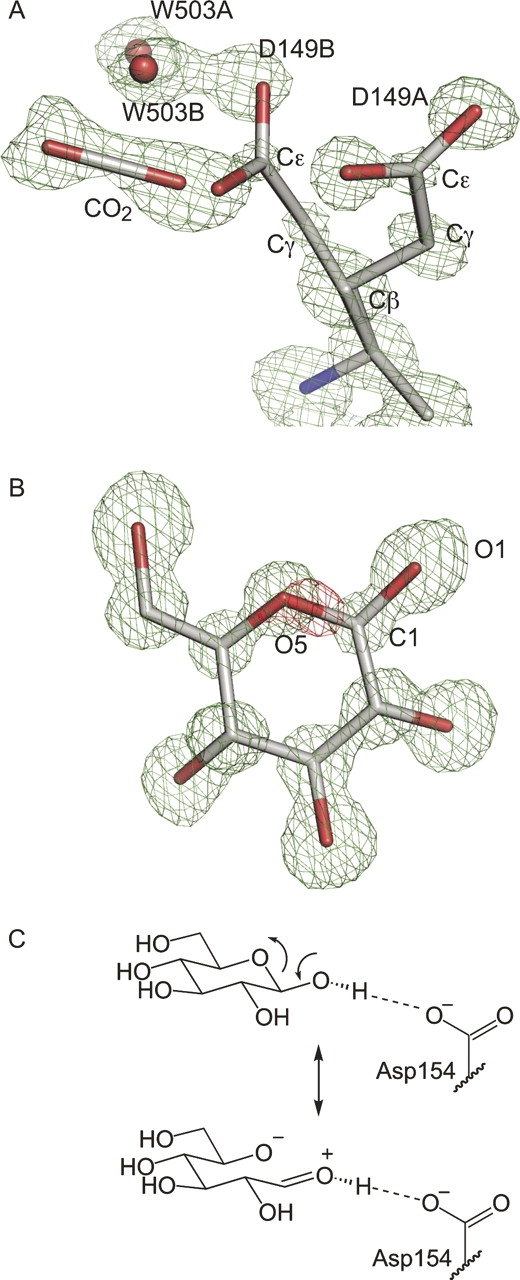
Interpretation of the 0.92 Å resolution glucose-bound structure. (A) Decarboxylation of Glu149 is apparent in this Fo–Fc omit electron density map (contoured at 3σ), calculated without residues 148–150, CO2, W503, or glucose. Electron density is clear for Glu149A and for the B conformer through Cγ. Decarboxylation releases CO2 and causes a rearrangement of water 503 (red spheres). (B) Glucose fits the omit density (green, contoured to 3σ); however, Fo–Fc difference density with the glucose model included (red, contoured at −5σ) indicates a subtle electronic difference between the real structure and the refined model at the O5–C1 bond. (C) This resonance structure is associated with the exo-anomeric effect.
Glucose recognition
The position of the D-glucose ring in the 0.92 Å resolution structure closely matches that of the 1.9 Å resolution E. coli GGBP structure (Vyas et al. 1988). The relatively minor changes in hydrogen bond distances observed in the current ultra-high-resolution refinement underscore the high quality of the previously reported model. The bound glucose molecule is exclusively in the β-conformation, as has been observed with both glucose-bound and galactose-bound E. coli GGBP structures (Vyas et al. 1988, 1994). The preference for only the β-anomer in crystal structures is intriguing; the GGBP-binding site geometry and comparisons with other sugar-binding PBPs suggest that either anomer can bind (Miller et al. 1980, 1983; Vyas et al. 1988). Indeed, Vyas and coworkers have previously predicted that all sugar-binding PBPs should bind to both α- and β-anomers (Vyas et al. 1994). We sought to determine experimentally which anomer binds GGBP in solution using nuclear magnetic resonance (NMR). Using 13C-labeled glucose we were able to confirm that only the β-anomer of glucose is bound in solution, as was observed in our crystal structure (Supplemental Fig. S1). Contrary to previous reports, our results indicate that GGBP has a strong preference for binding to the β-anomer of glucose both in solution and in the crystalline state.
This anomeric preference is interesting in light of another striking feature of the electron density distribution in the bound glucose. Substantial negative electron density was observed in the Fo–Fc Fourier difference maps observed between the O5 and C1 positions of the glucose ring. This difference density persisted when higher resolution data were added in each subsequent round of refinement (Fig. 3B). One explanation for this sizeable negative electron density feature at the position within glucose that undergoes ring opening and closing is that the bound form is lacking an O5–C1 bond. Attempts to model the ring as open, however, failed to improve this negative difference density. It is intriguing to speculate that this negative electron density arises from the unique electronics of the carbohydrate ligand. A ring-opened resonance form contributes to the structure of carbohydrates in the β-conformation; this electronic contribution has been referred to as the exo-anomeric effect (Fig. 3C; Lemieux et al. 1969; Lemieux and Koto 1974; Bitzer et al. 2005). This stabilizing electronic effect could account for the unusual lack of electron density in this bond.
Comparison with other glucose-bound GGBP structures
Subtle differences are revealed upon comparison of the three glucose-bound GGBP structures now available. Our 0.92 Å resolution structure is open ∼9° compared with the known E. coli GGBP structure (Vyas et al. 1988; Fig. 2A) and opened ∼5° compared with the S. typhimurium GGBP model (Mowbray et al. 1990). These minor movements can likely be attributed to the constraints placed upon GGBP by crystal-packing interactions. As the previously reported E. coli GGBP structure was solved using data collected at room temperature, it is also possible that subtle shifts in the protein are caused by cryoprotection (Dunlop et al. 2005). GGBP has previously been shown to adopt a range of conformations in solution, even in the closed ligand-bound state (Careaga et al. 1995). The main differences in these closed states are limited to the hinge region connecting the rigid N and C domains.
Open, unliganded GGBP structure
To understand how the hinge of GGBP facilitates opening and closing, we determined the structure of an open form of unliganded GGBP. Novel crystals of ligand-free GGBP were obtained in the orthorhombic space group P212121. Phasing by molecular replacement, manual fitting to high-quality electron density maps, and refinement against 1.55 Å resolution data (Fig. 1B; Table 1) led to the final model of the open form of GGBP (Fig. 2B). This structure is the first of an open, ligand-free species.
Although this unbound structure does not contain a carbohydrate ligand, a sodium citrate complex was observed within the open binding cleft. Cit 311 coordinates Na+310 in a tridentate manner: The Cα and Cγ of the citrate ion make hydrophobic contacts with the indole ring of Trp183 in the C-terminal domain, and Na+310 is also coordinated by the carbonyl of Asn15 (Fig. 4). Based on B-values and Fo–Fc difference density analysis, the citrate/sodium complex was assigned 50% occupancy. The location of the citrate/sodium complex between Trp183 and Phe16 is noteworthy because these two residues engage in hydrophobic stacking interactions with the sugar ring in the glucose- and galactose-bound closed structures. We suspect the small molecule wedge in the binding cleft may be thermodynamically important for crystal growth, as replacing sodium citrate in the mother liquor with other salts still yields crystals, but their rate of growth is slower (data not shown).
Figure 4.

Stereo view of the sodium citrate complex in the binding cleft of unliganded GGBP. Sodium ion 310 (purple sphere) is coordinated by citrate, two water molecules (red spheres), and Asn15. This complex is centered between two aromatic residues, Phe16 and Trp183, which form the N- and C-terminal domain faces, respectively, for hydrophobic stacking with carbohydrate ligands.
Hinge motions in GGBP
The major conformational differences between the unliganded and glucose-bound structures lie in the three hinge regions separating the N- and C-terminal domains: segments one (residues 109–111), two (residues 253–256), and three (residues 293–296). The hinge regions are defined based on their homology with those previously described for RBP and ALBP (Magnusson et al. 2002). A comparison of Cα-based torsion angles (Fig. 5) confirmed that these regions differ significantly between the glucose-bound and ligand-free structures (Flocco and Mowbray 1995). The most significant torsion angle differences are in hinge segment one and are a manifestation of dramatic ϕ and ψ changes in residues 109 and 111 (Figs. 1, 6). Segments two and three also show significant torsion angle differences. Minor additional conformational changes due to crystal packing differences are observed in some surface-exposed non-hinge residues. In the final 10 residues, the Cα-based torsion angle differences are due to conformational flexibility of the C-terminal tail.
Figure 5.

Cα torsion angle differences between the glucose-bound and open unliganded GGBP structures. Major changes in Cα torsion angles exist in the three hinge segments (gray shading) joining the N- and C-terminal domains (overlined).
Figure 6.

Torsion angle adjustment within hexose-binding PBPs leads to domain closure. (A) Comparison of the sums of Δϕ + Δψ for open and closed forms of GGBP (black), RBP (gray), and ALBP (white) suggests that hinge movement differs among these members of the sugar-binding PBP family. The unliganded GGBP structure, which is 31° open, and the 33°- and 43°-opened forms of ALBP (PDB code: 1GUD, chain B) and RBP (PDB code: 1URP, chain A) were compared with those of their respective closed forms (PDB codes: 1RPJ and 2DRI) to give Δϕ + Δψ values for each residue in the hinge region. Residue numbers (1–3, 1–4, and 1–4) correspond to analogous residues in segments one, two, and three. These are residues G109-T-Y111, T253-V-L-N256, and V293-P-Y-V296 in GGBP; A102-S-D104, T232-I-A-Q235, and V263-D-L-K266 in RBP; and T111-T-D113, 244T-V-A-Q247, and V281-D-S-I284 in ALBP. (B) The hinge axis for GGBP does not recapitulate the hinge axis of either RBP or ALBP, as calculated using DynDom and displayed on the ribbon representation of the closed forms of each protein.
Least squares superpositioning of the N-terminal domains of both structures (amino acids 2–108 and 257–292) and the C-terminal domains of both structures (amino acids 112–252 and 297–306) yields root mean square displacements (rmsd) for all Cα atoms of 0.275 Å and 0.395 Å, respectively, indicating that these domains behave as rigid bodies during opening and closing. Compared with our ligand-bound form, this ligand-free GGBP is opened 31°, with the axis of rotation nearly intersecting the α-carbons of residues 115 and 255 (Fig. 6B). That this “effective hinge axis” is in close proximity to those residues that undergo the greatest conformational changes suggests a mechanical hinge motion converts the open and closed forms (Hayward 1999). The 9° “breathing” axis identified by comparing the previously solved E. coli glucose-bound GGBP with our high-resolution model differs from the effective hinge axis of GGBP and those of RBP and ALBP (Fig. 6B).
Comparison with other PBPs
We sought to compare the opening/closing movement of GGBP with that of other PBPs with the goal of discerning patterns in hinge motion. RBP and ALBP were chosen because of their high sequence identity (28%) and structural homology with GGBP. Additionally, as a consequence of the availability of multiple three-dimensional structures, their hinge-bending motions can be analyzed in-depth (Bjorkman and Mowbray 1998; Magnusson et al. 2002). The magnitude of torsion angle changes is one measure of hinge motions. Changes in ϕ and ψ angles between open and closed structures of GGBP, RBP, and ALBP were summed for structurally analogous residues within the hinge segments (Fig. 6A). Large Δϕ + Δψ values, which do not cancel (and are thus in the same direction), indicate major torsion angle changes that contribute to hinge opening (Trakhanov et al. 2005). In hinge segment one, GGBP exhibits dramatic changes in Δϕ + Δψ at all three key residues. In contrast to GGBP, RBP exhibits a more modest change at the first position of hinge segment one upon opening, and, in ALBP, there are relatively minor changes at all three positions. Similarly, no pattern in hinge opening among GGBP, RBP, and ALBP can be seen in segment three; Δϕ + Δψ values in this region vary substantially for each protein at each position. Interestingly, similarities in hinge movement among the three proteins are discernable for segment two. Δϕ + Δψ values are smaller overall in segment two than in segments one or three. From this analysis, it is clear that individual torsion angle changes in GGBP compared with ALBP or RBP are different. These differences are especially apparent in segment one.
We also compared the hinge axes of all three proteins (Fig. 6B). As suggested by the dramatic differences in segment one torsion angles, this analysis revealed that GGBP has a unique hinge axis. Whereas RBP and ALBP have hinge axes nearly parallel to the plane of their sugar ligands, the GGBP axis is almost perpendicular to the plane of the glucose ring. Thus, our data demonstrate that, even among these closely related periplasmic carbohydrate-binding proteins, hinge motion varies.
In the closed structures of all three proteins, two water molecules relevant for hinge movement are conserved (Magnusson et al. 2002). One of these, W1, appears to act as a molecular “ball bearing” in ALBP and RBP; segments two and three pivot around it during opening or closing (Bjorkman and Mowbray 1998; Magnusson et al. 2002). The open structure of GGBP reveals an analogous water; however, pivoting of the hinge around this water molecule is limited (Supplemental Fig. S2). The second hinge water molecule (W2) identified in ALBP and RBP behaves differently in those two proteins. In RBP, this bridging water molecule is released upon hinge opening to be replaced by direct hydrogen bonding between segments one and three (Bjorkman and Mowbray 1998). In open, unliganded GGBP, the water molecule corresponding to W2 also has been pushed out of the hinge region. The W2 water bridge that spans segments one and three in the closed forms is replaced by direct hydrogen bonding in the open form (Thr110 OH to Val293 NH and O) (Supplemental Fig. S2). We conclude that in all three proteins hydrogen bonding interactions involving water molecules are integral to hinge motions. The malleable nature of these interactions makes water molecules well-suited for their role in the transition from open to closed forms of PBPs.
Discussion
In this report we present two new structures of the glucose/galactose-binding protein. The 0.92 Å resolution structure of E. coli GGBP provides a view of the glucose-bound protein that is extremely accurate. This structure, which was determined from a different crystal form than has been previously characterized, is in agreement with published data, yet the high-resolution electron density maps illuminate new aspects of ligand-bound GGBP.
The high-resolution electron density maps allowed direct visualization of synchrotron radiation-induced decarboxylation at Glu149. It is becoming increasingly clear that even with the protection afforded by cryo-cooling of crystals, high-energy synchrotron sources can lead to secondary radiation damage to proteins via photoelectrons and reactive oxygen species. In addition to decarboxylation of aspartate and glutamate side chains, breakage of covalent bonds (in particular, disulfide bonds) and oxidation of methionine side chains have been observed (Burmeister 2000). In some cases, the effects of radiation damage have been harnessed. For example, experimental phasing techniques that rely on the time-dependent change in electron density at heavy metal sites have been developed (Banumathi et al. 2004). In addition, X-ray-dependent reduction of substrates can be exploited to initiate reactions within crystals (Schlichting et al. 2000). Nevertheless, the typical consequence of radiation damage is a diminution in the overall quality of crystallographic data. In some cases, structures with different ligands in the active site can be altered by radiation damage, thus obscuring biochemical interpretation (Matsui et al. 2002; Parthasarathy et al. 2003). What is thus remarkable in our structure is that a CO2 molecule could be modeled into the electron density near the affected Glu149 (Fig. 3A). To our knowledge, this is the first incidence of a CO2 molecule remaining sufficiently well-ordered after decarboxylation for its position to be refined. Such a result has implications for future structural analysis. For example, in studying crystalline decarboxylase enzymes, it may be possible to generate reaction intermediates or products with the clever use of X-rays.
One unique feature is the negative Fo–Fc difference electron density observed between the C1 and O5 positions in the glucose ring (Fig. 3B). Although we cannot rule out that this feature arises from uncharacterized radiation damage, we speculate that it is due to the exo-anomeric effect. The electron density maps and refined atomic positions indicate the bound glucose ligand is in the cyclized form; still, the ring-opened resonance form contributes to the electron distribution. The exo-cyclic O1 lone pair can donate electron density into the C1–O5 σ* orbital while decreasing electron density in the C1–O5 bond; this interaction, which stabilizes the gauche glycosidic conformation, is referred to as the exo-anomeric effect (Fig. 3C; Lemieux et al. 1969; Lemieux and Koto 1974; Bitzer et al. 2005). The hydrogen bond between the anomeric hydroxyl group and OD1 of Asp154, which is positioned 2.7 Å away (Fig. 3C), should increase the importance of the ring-opened resonance form (Vyas et al. 1994). In GGBP, as in many other carbohydrate-binding proteins, stacking interactions between aromatic side chains and carbohydrate ligands (Trp183 and Phe16 in GGBP) play a significant role in binding (Vyas et al. 1988). We speculate that the cationic character of the exo-anomeric resonance form of glucose could be further stabilized by cation-π-type interactions with an aromatic side chain (Zacharias and Dougherty 2002).
Exo-anomeric stabilization of a ring-opened resonance form can occur only with the β-anomer of carbohydrates. This structure (Fig. 3B) and 13C NMR studies (Supplemental Fig. S1) confirm that only the β-anomer of glucose is bound to GGBP. Notably, the only previously reported instance of negative electron density at the C1–O5 bond was that observed in a strained glucose ring in the 0.94 Å resolution structure of an inactive inverting glycosidase (Guerin et al. 2002). Further structural investigations of carbohydrate-bound proteins may illuminate the underlying basis for this intriguing aspect of our results. A better understanding of the role of stereoelectronics in carbohydrate–protein binding may allow one to exploit these characteristics when designing inhibitors or probes.
The 1.55 Å ligand-free structure allows direct visualization of an open form of GGBP; this form has previously been characterized only by indirect methods including disulfide trapping, small-angle scattering, and F-19 NMR (Luck and Falke 1991a,b; Careaga et al. 1995; Shilton et al. 1996). Those data, along with results indicating that PBPs can adopt multiple conformations in the crystalline state (Sack et al. 1989; Bjorkman and Mowbray 1998; Magnusson et al. 2002, 2004; Trakhanov et al. 2005), suggest that GGBP and other PBPs can sample a variety of open forms in solution. Despite this apparent conformational heterogeneity, for RBP, ALBP, leucine-binding protein, and leucine/isoleucine/valine-binding protein the multiple open and closed forms of a given protein lie along a shared trajectory (Bjorkman and Mowbray 1998; Magnusson et al. 2002, 2004; Trakhanov et al. 2005). In GGBP, the three-segment hinge restricts the axis of rotation during opening and closing. Thus, we predict that other open forms of GGBP will be related by domain motions about a similar hinge axis (Fig. 6B).
The hinge motions of the three members of the hexose/pentose PBP subfamily appear similar at first glance—they all achieve the same end (closing or opening) via a three-segmented hinge with only minute changes observed in the N and C domains. Gross differences can be observed, however, in the opening motions of GGBP when compared with RBP and ALBP due to unique location and magnitude of torsion angle changes throughout the hinge (Fig. 6A). The differences in hinge movement between GGBP and RBP or ALBP can be attributed to steric effects. For instance, Thr111 in segment one of ALBP exhibits limited motion due to steric interference and hydrogen bonding constraints (Magnusson et al. 2002). This residue moves only slightly (−3° ϕ and 1° ψ) upon conversion from the closed to the open form. The analogous hinge residue in GGBP, Gly109, is more flexible and shows significant motion (+21° ϕ and +44° ψ) as the protein moves to the open form (Fig. 6A).
One key issue raised by the unliganded GGBP structure is how the open conformation is stabilized in the absence of ligand. Maltose-binding protein, for example, has two extensive areas of contact that form a “balancing interface” and maintain the open form (Telmer and Shilton 2003). In contrast, no obvious contacts stabilize the open unliganded form of GGBP. Crystal-packing interactions and the presence of the citrate wedge in the ligand-binding cleft apparently provide the small free energy difference needed to hinder the structural transitions between otherwise thermodynamically equivalent states of GGBP. The presence of a sodium ion coordinated by the hinge residues may also restrict hinge motion. Finally, it seems likely that the hydrophilic solvent molecules that fill the binding site in the absence of carbohydrate serve as a barrier to adopting the closed conformations of GGBP (and other PBPs) in the absence of ligand.
The two new high-resolution structures of GGBP illustrate the reversible conformational changes that allow GGBP to toggle between an inactive state and its signaling-active counterpart; the latter communicates glucose/galactose levels in the periplasm to the chemotaxis receptor Trg. Detailed information about the transition between these two forms provides a useful tool for designing ligands that can trap such proteins in one state or the other or alter the ability of these proteins to undergo large conformational changes. Inhibitors that can bind to nonsignaling states along the conserved opening trajectories of proteins with this architecture might serve as effective antagonists. Inhibitors of bacterial PBPs that act in quorum sensing, such as LuxP (Chen et al. 2002), could possibly serve as antimicrobial agents. In addition to bacterial targets, many clinically important eukaryotic receptors such as the AMPA-sensitive glutamate receptor (Armstrong and Gouaux 2000) share a similar architecture with PBPs; thus, they may also be amenable to such a strategy for inhibitor design. We envision that small-molecule PBP inhibitors have the potential to act as tools for biotechnology or as leads for novel human therapeutics.
Materials and Methods
Protein expression and purification
GGBP was purified from E. coli strain HB929 (kindly provided by G. Hazelbauer, U. Missouri-Columbia) using the osmotic shockate method (Anraku 1968; Gestwicki et al. 2001). GGBP was further purified with a Q-Sepharose Fast Flow column (Amersham) in a 0–0.25 M NaCl gradient in 10 mM Tris-HCl buffer pH 8.3. Purified protein was then exhaustively dialyzed against 2 M guanidine-HCl, 25 mM Tris-HCl pH 8.0, and 1 mM EDTA. This protein was refolded by further dialysis in 10 mM Tris-HCl pH 7.3, 1 mM CaCl2 and concentrated.
Crystallization and structure determination of glucose-bound GGBP
All crystals were obtained using the hanging-drop vapor diffusion method (McPherson 1982) with drops consisting of 1.5 μL of mother liquor and 1.5 μL of GGBP (25 mg/mL). Initial crystallization conditions were identified by sparse matrix screening using commercially available kits (Hampton Research) and were optimized to yield large, single, rod-shaped crystals using 25% PEG 4000, 5% glycerol, and 100 mM Hepes pH 7.8, at 4°C. The glucose-bound crystals were in the monoclinic space group C2 (Table 1). A glucose analog was present in the GGBP mother liquor at 1 mM; however, it was residual glucose from the purification process that was identified in electron density maps. Crystals were swiped through mother liquor supplemented with 20% glycerol immediately before flash cooling in liquid nitrogen. Data were collected on the MAR 165 CCD detector on beamline ID14B at the Advanced Photon Source. Separate low- and high-resolution sweeps were measured at a wavelength of 0.83 Å and integrated and merged using HKL2000 (Table 1; Otwinowski and Minor 1997). Phases were determined by molecular replacement using AmoRe (Navaza 1994) with the Salmonella typhimurium glucose-bound structure (Protein Data Bank Accession code: 3GBP) as the starting model. Structure refinement was performed using the programs CNS and SHELX97 (Sheldrick and Schneider 1997; Brunger et al. 1998). Five percent of the data were set aside for calculation of Rfree (Brunger 1992); the same randomly flagged reflections were used in both CNS and SHELX. Simulated annealing and individual B-factor refinement were performed in CNS using 20 to 2.5 Å resolution data. Annealing, B-factor refinement, and manual fitting using XFIT (McRee 1999) were reiterated in shells of increasing resolution with data to 2.0, 1.4, and 1.2 Å. Three hundred water molecules were added in XFIT, and the locations of obvious dual conformers were noted for subsequent addition to the model in SHELX.
The refined 1.2 Å resolution structure from CNS was used as the starting model for conjugate gradient least-squares (CGLS) minimization in SHELX. Supplemental Table S1 illustrates the improvement in R factor in subsequent rounds of CGLS refinement. The final model contains hydrogen atoms added to all atoms requiring them except hydroxyl and amino groups. Hydrogen atoms were not added to these groups, as they are difficult to position accurately and could not be visualized with hydrogen omit maps. The final Rcrystal was 11.0% with a final Rfree of 13.0%.
Crystallization and structure determination of unliganded GGBP
Crystals of unliganded GGBP were obtained in hanging drops using 25 mg/mL GGBP. Optimized mother liquor for these crystals was based on an initial crystallization trial using a commercial sparse matrix sampling kit (Nextal). A mother liquor of 2.0 M ammonium sulfate and 0.05 M sodium citrate dehydrate, pH 6.0 yielded clumps of rod-shaped crystals that were used in a microseeding experiment to yield single rod- and prism-shaped crystals with dimensions of 0.2 mm × 0.2 mm × 0.2 mm. Cryo-cooling in liquid nitrogen was achieved using 2.0 M sodium malonate as a cryoprotectant.
A 3.0 Å resolution data set was collected from a small crystal on a Proteum CCD detector, with X rays generated by a Microstar rotating anode (Bruker AXS). Images were processed with Proteum software. Molecular replacement was successfully performed using PHASER (Storoni et al. 2004; McCoy et al. 2005) with the N- and C-terminal domains from the high-resolution glucose-bound structure as separate search models. A 1.55 Å data set collected from a larger crystal of unliganded GGBP was phased with these molecular replacement phases, and the structure was refined using CNS (Brunger et al. 1998) with simulated annealing and individual B-factor refinement cycled with manual fitting in XFIT. Three sodium ions, identified by octagonal coordination geometry and the short (2.3–2.6 Å) distance to the coordinating ligands, were included in the final model. In neither the unliganded nor glucose-bound structure was electron density detected for residues 1 or 307–309; these residues are not in the models.
Cα-based torsion angles were calculated using the program ca_torsion.f (Flocco and Mowbray 1995). Figures were generated with PyMOL (http://www.pymol.org). Root mean squared XYZ displacement was calculated using TOP (Lu 2000) in the CCP4i suite (Potterton et al. 2003). Rotational angles and axes were calculated using DynDom (Hayward and Berendsen 1998; Hayward 1999).
NMR spectroscopy
13C-NMR spectra were obtained on a Bruker DMX 750 with Cryoprobe. GGBP/glucose sample contained 0.75 mM GGBP and 0.67 mM 13C-glucose. The glucose-only sample contained 0.8 mM 13C-glucose. Both samples contained 50 mM sodium phosphate pH 7 in D2O, with 2,2-Dimethyl-2-silapentane-5-sulfonate sodium salt for reference. Spectra were obtained with proton decoupling. Splitting of C1 carbon resonances results from the adjacent 13C atom of glucose (C2). Peak assignment of α- and β- anomeric resonances was confirmed by comparison with standard spectra from the Human Metabolome Database (http://www.hmdb.ca).
Acknowledgments
We thank Dr. Ken Satysur, Dr. Matthew Benning, and the APS BioCars staff for crystallographic assistance and expertise. We also thank Dr. George Sheldrick for assistance with SHELX. We thank Todd Gruber for performing the NMR experiments. This study made use of the National Magnetic Resonance Facility at Madison, which is supported by NIH grants P41RR02301 (BRTP/NCRR) and P41GM66326 (NIGMS). Research funding was provided by the National Institutes of Health (GM59984, L.L.K.) and the W.M. Keck Foundation (K.T.F.). M.J.B. was supported by an NIH Molecular Biosciences Training Grant (GM07215) and a Steenbock Fellowship from the UW-Madison Department of Biochemistry. Coordinates and structure factors have been deposited with the Brookhaven Protein Data Bank (Berman et al. 2000) with the identification codes 2FVY and 2FW0 for the glucose-bound and open ligand-free forms, respectively.
Footnotes
Supplemental material: see www.proteinscience.org
Reprint requests to: Katrina T. Forest, 420 Henry Mall, Madison, WI 53706-1521, USA; e-mail: forest@bact.wisc.edu; fax: (608) 262-9865; or Laura L. Kiessling, 1101 University Ave., Madison, WI 53706-1521, USA; e-mail: kiessling@chem.wisc.edu; fax: (608) 265-0764.
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.062707807.
References
- Anraku Y. 1968. Transport of sugars and amino acids in bacteria. I. Purification and specificity of the galactose- and leucine-binding proteins. J. Biol. Chem. 243: 3116–3122. [PubMed] [Google Scholar]
- Armstrong N. and Gouaux, E. 2000. Mechanisms for activation and antagonism of an AMPA-sensitive glutamate receptor: Crystal structures of the GluR2 ligand binding core. Neuron 28: 165–181. [DOI] [PubMed] [Google Scholar]
- Banumathi S., Zwart, P.H., Ramagopal, U.A., Dauter, M., and Dauter, Z. 2004. Structural effects of radiation damage and its potential for phasing. Acta Crystallogr. 60D: 1085–1093. [DOI] [PubMed] [Google Scholar]
- Berman H.M., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T.N., Weissig, H., Shindyalov, I.N., and Bourne, P.E. 2000. The Protein Data Bank. Nucleic Acids Res. 28: 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bitzer R.S., Barbosa, A.G., da Silva, C.O., and Nascimento, M.A. 2005. On the generalized valence bond description of the anomeric and exo-anomeric effects: An ab initio conformational study of 2-methoxytetrahydropyran. Carbohydr. Res. 340: 2171–2184. [DOI] [PubMed] [Google Scholar]
- Bjorkman A.J. and Mowbray, S.L. 1998. Multiple open forms of ribose-binding protein trace the path of its conformational change. J. Mol. Biol. 279: 651–664. [DOI] [PubMed] [Google Scholar]
- Brunger A.T. 1992. Free R-Value—A novel statistical quantity for assessing the accuracy of crystal-structures. Nature 355: 472–475. [DOI] [PubMed] [Google Scholar]
- Brunger A., Adams, P., Clore, M., Gros, P., Nilges, M., and Read, R. 1998. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. D54: 905–921. [DOI] [PubMed] [Google Scholar]
- Burmeister W.P. 2000. Structural changes in a cryo-cooled protein crystal owing to radiation damage. Acta Crystallogr. 56A: 328–341. [DOI] [PubMed] [Google Scholar]
- Careaga C.L., Sutherland, J., Sabeti, J., and Falke, J.J. 1995. Large-amplitude twisting motions of an interdomain hinge: A disulfide trapping study of the galactose–glucose binding protein. Biochemistry 34: 3048–3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhuri B.N., Ko, J., Park, C., Jones, T.A., and Mowbray, S.L. 1999. Structure of D-allose binding protein from Escherichia coli bound to D-allose at 1.8 Å resolution. J. Mol. Biol. 286: 1519–1531. [DOI] [PubMed] [Google Scholar]
- Chen X., Schauder, S., Potier, N., Van Dorsselaer, A., Pelczer, I., Bassler, B.L., and Hughson, F.M. 2002. Structural identification of a bacterial quorum-sensing signal containing boron. Nature 415: 545–549. [DOI] [PubMed] [Google Scholar]
- Dunlop K.V., Irvin, R.T., and Hazes, B. 2005. Pros and cons of cryocrystallography: Should we also collect a room-temperature data set? Acta Crystallogr. 61D: 80–87. [DOI] [PubMed] [Google Scholar]
- Felder C.B., Graul, R.C., Lee, A.Y., Merkle, H.P., and Sadee, W. 1999. The Venus flytrap of periplasmic binding proteins: An ancient protein module present in multiple drug receptors. AAPS PharmSci 1: E2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flocco M.M. and Mowbray, S.L. 1994. The 1.9 Ångstrom X-ray structure of a closed unliganded form of the periplasmic glucose/galactose receptor from Salmonella typhimurium . J. Biol. Chem. 269: 8931–8936. [PubMed] [Google Scholar]
- Flocco M.M. and Mowbray, S.L. 1995. C α-based torsion angles: A simple tool to analyze protein conformational changes. Protein Sci. 4: 2118–2122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukami-Kobayashi K., Tateno, Y., and Nishikawa, K. 1999. Domain dislocation: A change of core structure in periplasmic binding proteins in their evolutionary history. J. Mol. Biol. 286: 279–290. [DOI] [PubMed] [Google Scholar]
- Gestwicki J.E. and Kiessling, L.L. 2002. Inter-receptor communication through arrays of bacterial chemoreceptors. Nature (London) 415: 81–84. [DOI] [PubMed] [Google Scholar]
- Gestwicki J.E., Strong, L.E., and Kiessling, L.L. 2000. Tuning chemotactic responses with synthetic multivalent ligands. Chem. Biol. 7: 583–591. [DOI] [PubMed] [Google Scholar]
- Gestwicki J.E., Strong, L.E., Borchardt, S.L., Cairo, C.W., Schnoes, A.M., and Kiessling, L.L. 2001. Designed potent multivalent chemoattractants for Escherichia coli . Bioorg. Med. Chem. 9: 2387–2393. [DOI] [PubMed] [Google Scholar]
- Guerin D.M.A., Lascombe, M.B., Costabel, M., Souchon, H., Lamzin, V., Beguin, P., and Alzari, P.M. 2002. Atomic (0.94 Ångstrom) resolution structure of an inverting glycosidase in complex with substrate. J. Mol. Biol. 316: 1061–1069. [DOI] [PubMed] [Google Scholar]
- Hayward S. 1999. Structural principles governing domain motions in proteins. Proteins 36: 425–435. [PubMed] [Google Scholar]
- Hayward S. and Berendsen, H.J. 1998. Systematic analysis of domain motions in proteins from conformational change: New results on citrate synthase and T4 lysozyme. Proteins 30: 144–154. [PubMed] [Google Scholar]
- Hazelbauer G.L. and Adler, J. 1971. Role of galactose-binding protein in chemotaxis of Escherichia coli toward galactose. Nat. New Biol. 230: 101–104. [DOI] [PubMed] [Google Scholar]
- Lemieux R.U. and Koto, S. 1974. The conformational properties of glycosidic linkages. Tetrahedron 30: 1933–1944. [Google Scholar]
- Lemieux R.U., Pavia, A.A., Martin, J.C., and Watanabe, K.A. 1969. Solvation effects on conformational equilibrium studies related to methyl glycopyranosides. Can. J. Chem. 47: 4427–4439. [Google Scholar]
- Looger L.L., Dwyer, M.A., Smith, J.J., and Hellinga, H.W. 2003. Computational design of receptor and sensor proteins with novel functions. Nature 423: 185–190. [DOI] [PubMed] [Google Scholar]
- Lu G. 2000. TOP: A new method for protein structure comparisons and similarity searches. J. Appl. Crystallogr. 33: 176–183. [Google Scholar]
- Luck L.A. and Falke, J.J. 1991a. 19F NMR studies of the D-galactose chemosensory receptor. 1. Sugar binding yields a global structural change. Biochemistry 30: 4248–4256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luck L.A. and Falke, J.J. 1991b. Open conformation of a substrate-binding cleft: 19F NMR studies of cleft angle in the D-galactose chemosensory receptor. Biochemistry 30: 6484–6490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnusson U., Chaudhuri, B.N., Ko, J., Park, C., Jones, T.A., and Mowbray, S.L. 2002. Hinge-bending motion of D-allose-binding protein from Escherichia coli: Three open conformations. J. Biol. Chem. 277: 14077–14084. [DOI] [PubMed] [Google Scholar]
- Magnusson U., Salopek-Sondi, B., Luck, L.A., and Mowbray, S.L. 2004. X-ray structures of the leucine-binding protein illustrate conformational changes and the basis of ligand specificity. J. Biol. Chem. 279: 8747–8752. [DOI] [PubMed] [Google Scholar]
- Matsui Y., Sakai, K., Murakami, M., Shiro, Y., Adachi, S., Okumura, H., and Kouyama, T. 2002. Specific damage induced by X-ray radiation and structural changes in the primary photoreaction of bacteriorhodopsin. J. Mol. Biol. 324: 469–481. [DOI] [PubMed] [Google Scholar]
- McCoy A.J., Grosse-Kunstleve, R.W., Storoni, L.C., and Read, R.J. 2005. Likelihood-enhanced fast translation functions. Acta Crystallogr. 61D: 458–464. [DOI] [PubMed] [Google Scholar]
- McPherson A. 1982. Preparation and analysis of protein crystals. John Wiley & Sons, Inc, New York.
- McRee D.E. 1999. XtalView/Xfit—A versatile program for manipulating atomic coordinates and electron density. J. Struct. Biol. 125: 156–165. [DOI] [PubMed] [Google Scholar]
- Miller D.M., Olson, J.S., and Quiocho, F.A. 1980. The mechanism of sugar binding to the periplasmic receptor for galactose chemotaxis and transport in Escherichia coli . J. Biol. Chem. 255: 2465–2471. [PubMed] [Google Scholar]
- Miller D.M., Olson, J.S., Pflugrath, J.W., and Quiocho, F.A. 1983. Rates of ligand binding to periplasmic proteins involved in bacterial transport and chemotaxis. J. Biol. Chem. 258: 3665–3672. [PubMed] [Google Scholar]
- Mowbray S.L. and Cole, L.B. 1992. 1.7 Å X-ray structure of the periplasmic ribose receptor from Escherichia coli . J. Mol. Biol. 225: 155–175. [DOI] [PubMed] [Google Scholar]
- Mowbray S.L., Smith, R.D., and Cole, L.B. 1990. Structure of the periplasmic glucose/galactose receptor of Salmonella typhimurium . Receptor 1: 41–53. [PubMed] [Google Scholar]
- Navaza J. 1994. AMoRe: An automated package for molecular replacement. Acta Crystallogr. 50A: 157–163. [Google Scholar]
- Neiditch M.B., Federle, M.J., Miller, S.T., Bassler, B.L., and Hughson, F.M. 2005. Regulation of LuxPQ receptor activity by the quorum-sensing signal autoinducer-2. Mol. Cell 18: 507–518. [DOI] [PubMed] [Google Scholar]
- Carter Jr, C.W. and Sweet, R.M. 1997. Processing of X-ray diffraction data collected in oscillation mode. In Macromolecular Crystallography, Pt A, pp. 307–326. Academic Press, San Diego, CA.
- Parthasarathy S., Eaazhisai, K., Balaram, H., Balaram, P., and Murthy, M.R. 2003. Structure of Plasmodium falciparum triose-phosphate isomerase-2-phosphoglycerate complex at 1.1-Å resolution. J. Biol. Chem. 278: 52461–52470. [DOI] [PubMed] [Google Scholar]
- Potterton E., Briggs, P., Turkenburg, M., and Dodson, E. 2003. A graphical user interface to the CCP4 program suite. Acta Crystallogr. 59D: 1131–1137. [DOI] [PubMed] [Google Scholar]
- Sack J.S., Trakhanov, S.D., Tsigannik, I.H., and Quiocho, F.A. 1989. Structure of the L-leucine-binding protein refined at 2.4 Å resolution and comparison with the Leu/Ile/Val-binding protein structure. J. Mol. Biol. 206: 193–207. [DOI] [PubMed] [Google Scholar]
- Salins L.L.E., Ware, R.A., Ensor, C.M., and Daunert, S. 2001. A novel reagentless sensing system for measuring glucose based on the galactose/glucose-binding protein. Anal. Biochem. 294: 19–26. [DOI] [PubMed] [Google Scholar]
- Schlichting I., Berendzen, J., Chu, K., Stock, A.M., Maves, S.A., Benson, D.E., Sweet, R.M., Ringe, D., Petsko, G.A., and Sligar, S.G. 2000. The catalytic pathway of cytochrome p450cam at atomic resolution. Science 287: 1615–1622. [DOI] [PubMed] [Google Scholar]
- Sheldrick G.M. and Schneider, T.R. 1997. SHELXL: High-resolution refinement. Methods Enzymol. 277: 319–343. [PubMed] [Google Scholar]
- Shilton B.H., Flocco, M.M., Nilsson, M., and Mowbray, S.L. 1996. Conformational changes of three periplasmic receptors for bacterial chemotaxis and transport: The maltose-, glucose/galactose- and ribose-binding proteins. J. Mol. Biol. 264: 350–363. [DOI] [PubMed] [Google Scholar]
- Storoni L.C., McCoy, A.J., and Read, R.J. 2004. Likelihood-enhanced fast rotation functions. Acta Crystallogr. 60D: 432–438. [DOI] [PubMed] [Google Scholar]
- Tam R. and Saier, M.H. 1993. Structural, functional, and evolutionary relationships among extracellular solute-binding receptors of bacteria. Microbiol. Rev. 57: 320–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telmer P.G. and Shilton, B.H. 2003. Insights into the conformational equilibria of maltose-binding protein by analysis of high affinity mutants. J. Biol. Chem. 278: 34555–34567. [DOI] [PubMed] [Google Scholar]
- Trakhanov S., Vyas, N.K., Luecke, H., Kristensen, D.M., Ma, J., and Quiocho, F.A. 2005. Ligand-free and -bound structures of the binding protein (LivJ) of the Escherichia coli ABC leucine/isoleucine/valine transport system: Trajectory and dynamics of the interdomain rotation and ligand specificity. Biochemistry 44: 6597–6608. [DOI] [PubMed] [Google Scholar]
- Vyas N.K., Vyas, M.N., and Quiocho, F.A. 1983. The 3 Ångstrom resolution structure of a D-galactose-binding protein for transport and chemotaxis in Escherichia coli . Biochemistry 80: 1792–1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vyas N.K., Vyas, M.N., and Quiocho, F.A. 1987. A novel calcium-binding site in the galactose-binding protein of bacterial transport and chemotaxis. Nature 327: 635–638. [DOI] [PubMed] [Google Scholar]
- Vyas N.K., Vyas, M.N., and Quiocho, F.A. 1988. Sugar and signal-transducer binding-sites of the Escherichia coli galactose chemoreceptor protein. Science 242: 1290–1295. [DOI] [PubMed] [Google Scholar]
- Vyas N.K., Vyas, M.N., and Quiocho, F.A. 1994. Crystallographic analysis of the epimeric and anomeric specificity of the periplasmic transport/chemosensory protein receptor for D-glucose and D-galactose. Biochemistry 33: 4762–4768. [DOI] [PubMed] [Google Scholar]
- Wang Z., Luecke, H., Yao, N., and Quiocho, F.A. 1997. A low energy short hydrogen bond in very high resolution structures of protein receptor-phosphate complexes. Nat. Struct. Biol. 4: 519–522. [DOI] [PubMed] [Google Scholar]
- Zacharias N. and Dougherty, D.A. 2002. Cation-π interactions in ligand recognition and catalysis. Trends Pharmacol. Sci. 23: 281–287. [DOI] [PubMed] [Google Scholar]
- Zou J.Y., Flocco, M.M., and Mowbray, S.L. 1993. The 1.7 Ångstrom refined X-ray structure of the periplasmic glucose galactose receptor from Salmonella typhimurium . J. Mol. Biol. 233: 739–752. [DOI] [PubMed] [Google Scholar]