

Abstract
The tumor suppressor protein pp32 is highly overexpressed in many cancers of the breast and prostate, and has also been implicated in the neurodegenerative disease spinocerebellar ataxias type 1 (SCA1). Pp32 is a multifunctional protein that is involved in the regulation of transcription, apoptosis, phosphorylation, and cell cycle progression, the latter through its association with the hyperphosphorylated form of the retinoblastoma tumor suppressor. We have determined the structure of an N-terminal pp32 fragment comprising a capped leucine-rich repeat (LRR) domain, which provides insight into the structural and biochemical properties of the pp32 (Anp32) family of proteins.
Keywords: crystal structure, pp32, Anp32, PHAPI, LRR
Pp32 (Anp32a) belongs to a family of evolutionarily conserved proteins known as the acidic nuclear phosphoprotein family (Anp32a–h). Members of the pp32 family help to modulate cellular signaling and gene expression to regulate the morphology and dynamics of the cytoskeleton, cell adhesion, neuronal development, or cerebellar morphogenesis (Matilla and Radrizzani 2005). Pp32 has been isolated several times, and has therefore been assigned a variety of names: pp32 (phosphoprotein 32) (Malek et al. 1990), Anp32a (acidic nuclear phosphoprotein 32a) (Matilla et al. 1997), PHAPI (putative HLA-associated protein I) (Vaesen et al. 1994), mapmodulin (Ulitzur et al. 1997b), Lanp (leucine-rich nuclear protein) (Matsuoka et al. 1994), and I1 PP2A (inhibitor 1 protein phosphatase 2A) (Li et al. 1995, 1996).
Pp32 is expressed in normal tissues as well as in many breast, pancreas, and prostate cancers (J. Brody, pers. comm.), where it can act as a tumor suppressor (Bai et al. 2001). Pp32 has also been associated with the neurodegenerative disease spinocerebellar ataxias through binding the polyglutamine repeats of Ataxin-1 (Matilla et al. 1997). The pp32 protein contains a nuclear localization signal (NLS, import signal) at the C terminus but can also bind the nuclear export protein Crm1 through the N terminus LRR domain (Brennan et al. 2000), which allows pp32 to exist as a component of both nuclear and cytoplasmic complexes. Nuclear pp32 is found associated with the protein SET (TAF-Iβ) in the INHAT (INHibitor of Acetyl Transferase Activity) complex (Schneider et al. 2004; Seo et al. 2002), which regulates histone modification/transcription. The endoplasmic reticulum (ER)-associated SET complex contains pp32 as one of its components. The SET complex is part of the Granzyme A-mediated apoptosis pathway, and the precise role of pp32 in this complex is unclear (Lieberman and Fan 2003). Interestingly, pp32 can promote apoptosis by accelerating caspase-9 activation after apoptosome formation (Jiang et al. 2003). Both pp32 and SET were initially isolated as potent inhibitors of protein phosphatase 2A (Li and Damuni 1998), a cell cycle regulatory phosphatase. Pp32 also helps regulate cell cycle progression by associating with the hyperphosphorylated form of the retinoblastoma tumor suppressor (Adegbola and Pasternack 2005). Pp32 is also involved in the regulation of mRNA trafficking through an association with Crm1 as part of the HuR complex (Brennan et al. 2000). Since this complex transports the mRNAs of cytokines and proto-oncogenes, it may stabilize factors involved in tumor progression and cell differentiation, such as IL-6 and vascular endothelial growth factor (Nabors et al. 1998). A structural role in cytoskeletal dynamics has also been inferred because the phosphorylated form of pp32 stimulates the localization of the Golgi apparatus in a microtubule and dynein-dependent manner via binding to the microtubule-associated proteins, MAP1, MAP2, and MAP4 (Ulitzur et al. 1997a,b; Itin et al. 1999; Opal et al. 2003).
The pp32 family (Anp32a–h) belongs to the superfamily of proteins containing leucine-rich repeats (LRRs). The LRR is a short motif of 20–29 residues in length that is present in tandem arrays in a variety of cytoplasmic, membrane, and extracellular proteins (Kobe and Kajava 2001). The 28-kDa pp32 protein contains three capped LRR motifs at the N terminus and a highly acidic C terminus containing ∼70% aspartic and glutamic acid residues (Chen et al. 1996). Similar acidic stretches are found in the nucleosome assembly proteins, nucleomorphin, nucleoplasmin, p62, and SET, and in the HMG box proteins, where the acidic domain is likely to be involved in chromatin binding (Matilla and Radrizzani 2005). Acidic stretches are also found in proteins such as tubulin, which are responsible for microtubule association (Matilla and Radrizzani 2005).
The LRR domain is of particular importance, as it is the minimal fragment of pp32 necessary to bind to a hyperphosphorylated form of the retinoblastoma protein (Rb) (Adegbola and Pasternack 2005). In cancers where p16 is inactivated, such as pancreatic cancer, the cyclin D:CD4/6 complex is unregulated and hyperphosphorylated Rb is predominant (Rozenblum et al. 1997). In these cases, hyperphosphorylated Rb both sequesters the pro-apoptotic activity of pp32 and is unable to correctly regulate E2F mediated gene transcription, allowing proliferation of tumor cells (Weinberg 1995; Adegbola and Pasternack 2005). The pp32:Rb protein complex therefore represents a potential target for the design of novel peptides or small molecules that could be used in the treatment of pancreatic cancer, for which there are currently limited treatments available.
Results
We determined the crystal structure of the pp32 N-terminal LRR domain from two crystal forms at 2.4 Å and 2.7 Å resolution, respectively (see Materials and Methods). Data collection and refinement statistics are given in Table 1. The C2 crystal form contains six molecules and the P63 crystal form contains four molecules in the asymmetric unit. The structures were refined with tight noncrystallographic symmetry restraints because of the low data:parameter ratio (see Materials and Methods). The Cα positions of monomers from the C2 and P63 superimpose with an RMSD of ∼0.18 Å, indicating that the structure of the LRR domain is virtually identical in both crystal forms. Unlike the structures of decorin (Scott et al. 2004), biglycan (Scott et al. 2006), and slit (Howitt et al. 2004) which dimerize through the LRR concave surface, the noncrystallographic symmetry in our pp32 crystal forms has no biological significance. The pp32 fragment used in this study is both monomeric by size-exclusion chromatography (see Materials and Methods), and lacks the C-terminal residues which are essential for chemical cross-linking of pp32 dimers and trimers (Ulitzur et al. 1997b). For clarity, further structural analysis is of a monomeric LRR domain from the high-resolution C2 crystal form.
Table 1.
Data collection and refinement statistics

Overall structure
The pp32 LRR domain features a concave surface lined by β-strands and an outer surface formed by either a 310-helix, short α-helix, or extended structure that runs antiparallel to the strands. The LRR domain of pp32, spanning residues 1–149, adopts the canonical LRR curved solenoid shape (∼50 × 25 × 20 Å) and includes three tandem LRRs with flanking regions containing incomplete LRRs that cap the repeats to form a stable structural unit (Fig. 1A,B).
Figure 1.
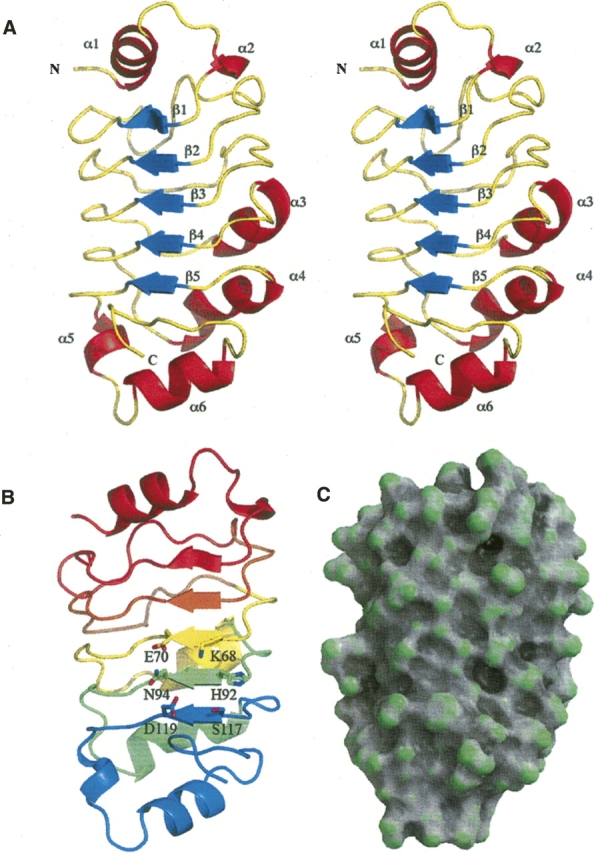
(A) Stereo ribbon representation of the structure of pp32ΔCT illustrating the canonical curved structure of the LRR domain. The ribbon is colored according to secondary structure, with β-strands colored blue, α-helices colored red, and coil colored yellow. (B) A ribbon representation face on to the β-strands which line the concave surface, the ribbons are colored: red (N-terminal cap residues 1–40), orange LRR1 (residues 42–62), yellow LRR2 (residues 63–86), green LRR3 (residues 87–111), and blue (C-terminal cap residues 112–149). Illustrated as sticks are residues K68, E70, H92, N94, S117, and D119, which surround a shallow cleft on the concave surface of the LRR. (C) A surface representation created with GRASP (Baker et al. 2001) and colored according to surface curvature potential to highlight the shallow cleft on the concave surface of the LRR domain.
The N-terminal cap region is comprised of a helix-loop-helix (residues 1–15), followed by an incomplete LRR (residues 16–40) that covers the top of the domain and shields the hydrophobic core residues from solvent. The subsequent LRRs (LRR1: 41–62, LRR2: 63–86, LRR3: 87–111) highlighted in Figure 1B are characteristic of the Sds22-like LRR consensus sequence LxxLxxLxLxxNxIxxIxxLx-x (Kobe and Kajava 2001). The sequences of the individual repeats are shown aligned to this consensus sequence (Fig. 2). The N-terminal capping region contains the first half of the consensus sequence and is an incomplete repeat. LRR1 is also slightly shorter and contains an unusual TxN sequence instead of the LxxN consensus. The central repeats LRR2 and LRR3 show the most conservation, and are followed by a C-terminal capping region that contains almost complete consensus sequences, missing only the last hydrophobic residue of the repeat, which is substituted with tyrosine instead of isoleucine.
Figure 2.

Sequence alignment of the individual LRRs from pp32. The central repeats belong to the SDS22-like class of LRRs.
Structural homologs
A comparative database search using the DALI server (Holm and Sander 1994) revealed a number of structures containing LRRs that are similar to the pp32 structure (Table 2). Of these proteins, the most closely related is the U2A′ spliceosomal factor, a protein containing five LRR (Price et al. 1998). The mRNA export protein, Tap (Liker et al. 2000), was also identified as being structurally similar. Despite limited sequence identity, the U2A′ and Tap LRR domains superimpose with pp32 with an RMSD of 1.8 Å and 2.6 Å, respectively. In particular, a C-terminal motif that caps the LRR domains appears structurally conserved, and is found in several other LRR containing proteins. This C-terminal cap had been previously identified in the Sds22, U2A′, and Tap proteins and comprises an incomplete LRR followed by the structural motif YRxxϕxxxϕPxϕxxLD, where ϕ represents a hydrophobic residue (Ceulemans et al. 1999). Within this motif the aspartic acid (D146 in pp32, D146 in U2A′, and D352 in Tap) form a conserved hydrogen bond to a main-chain nitrogen and a tyrosine side chain (Y131 in pp32, Y131 in U2A′ and Y337 in Tap) that maintains the structural integrity of the cap (Fig. 3).
Table 2.
Structure alignment, DALI statistics (Holm and Sander 1994)

Figure 3.

(A) Ribbon representation to illustrate the structural homology between pp32 (green), U2A′ spliceosomal protein (blue), and Tap mRNA export factor (magenta). (B) as in A, rotated 90°. Conserved and structurally important residues capping the LRR that are part of the YRxxϕxxxϕPxϕxxLD motif are illustrated as sticks.
Putative protein interaction surface
The concave face of LRR proteins is a versatile binding surface that mediates interactions with other proteins. Binding to the concave face of LRR domains has been seen in several complexes including, ribonuclease inhibitor LRR bound to ribonuclease (Kobe and Deisenhofer 1995), internalin LRR complexed with E-cadherin (Schubert et al. 2002), and the LRR domain glycoprotein Ibα bound to the von Willebrand factor (Uff et al. 2002). The pp32 LRR domain interacts with Crm1 (Gallouzi et al. 2001), ataxin-1 (Matilla et al. 1997), hyperphosphorylated Rb (Adegbola and Pasternack 2005), and is also likely to interact with some components of the SET complex (Lieberman and Fan 2003). There are no biochemical or mutational data for the pp32 LRR domain to identify residues on the concave surface that are required for these interactions. We therefore analyzed the surface features of the pp32 LRR domain structure and the sequence conservation between pp32 and its family members (Anp32b–h) in order to identify the potential binding interface. The electrostatic surface potential of the pp32 LRR domain concave face (Suppl. Fig. 1A) shows an overall negative potential with surrounding positive potential at the periphery of the domain, but does not have a distinct charged surface patch that could mediate possible interactions.
The pp32 gene knockout shows no phenotype (Opal et al. 2004), indicating some functional overlap between the pp32 (Anp32) family members. We therefore analyzed the conservation of residues within the pp32 (Anp32) protein family to identify residues that could form a common binding interface. There are many sequences of pp32 (Anp32) proteins in the database from multiple species, a sequence alignment of the human sequences is shown in Figure 4A. The sequence conservation of the human pp32 (Anp32) family has been mapped onto the surface of the pp32 LRR domain structure (Fig. 4 B,C) and clearly indicates that LRR3 and the C-Cap are the most conserved regions within the domain. We note a shallow cleft in the concave surface (Fig. 1C), which is formed by the surface residues K68 and E70 in β3 (LRR2), H92 and N94 in β4 (LRR3), and S117 and D119 in β5 (C-Cap) (Fig. 1B). These residues lining the cleft are highly conserved among the members of the pp32 (Anp32) family (residues highlighted with an asterix in Fig. 4A). In pp32r1 (Anp32c), which is overexpressed in prostate cancer (Kadkol et al. 2001), there is a deletion of four residues in LRR2 and two residue changes (E70R and N94Y) in the residues surrounding the cleft (Fig. 4A). Pp32r1 (Anp32c) does not associate with hyperphosphorylated retinoblastoma (Adegbola and Pasternack 2005), consistant with the possible involvement of these cleft residues in this interaction. Additional mutational analysis of residues on the concave surface of the pp32 LRR will be necessary to establish whether the cleft region mediates interactions with retinoblastoma and other protein ligands, or whether other residues on the pp32 surface are required.
Figure 4.
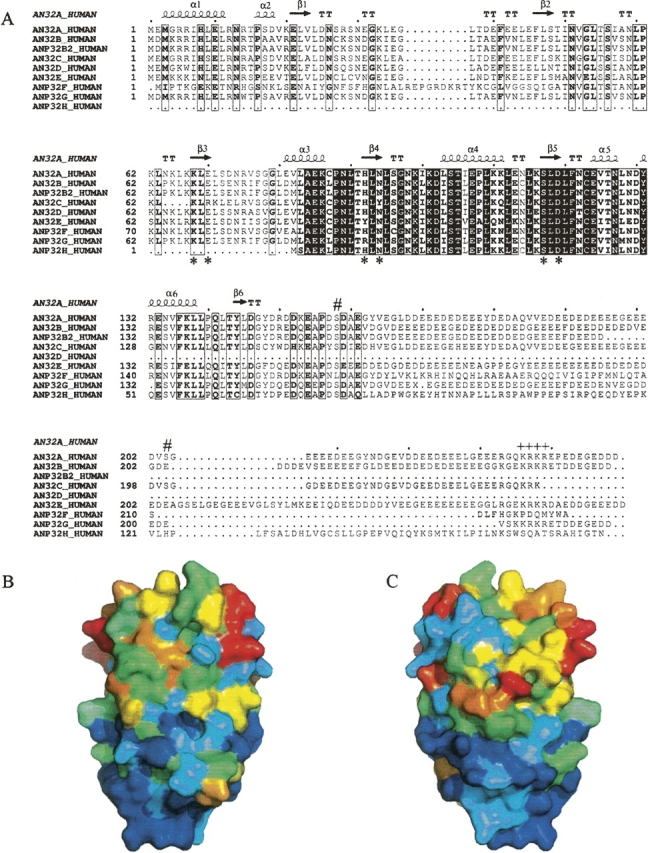
(A) Sequence alignment of human pp32 (Anp32) family homologs a–h. Identical residues from all sequences are boxed black and similar residues are open boxed. The consensus sequence is shown below, and the secondary structure taken from the pp32 (Anp32a) crystal structure is shown above. (*) Denotes the position of the residues K68, E70, H92, N94, S117, and D119 that surround a surface cleft in the pp32 crystal structure. The position of the CKII phosphorylation sites S158 and S204 are indicated with # and the nuclear localization signal KRKR indicated with ++++. (B) Sequence variability of the pp32 (Anp32) family with a view of the concave surface of the pp32 LRR domain. The surface is colored according to sequence variability (derived from the alignment in A) with blue being the most conserved through the spectrum to red being the least conserved. (C) As in B, rotated by 180° viewing the convex surface.
Discussion
The crystal structure of the pp32 LRR domain provides an accurate basis to help us rationalize the structural and functional differences between the family members (Anp32a–h). The Anp32b gene produces two products; Anp32b1, also known as acidic protein rich in leucines (APRIL) (Mencinger et al. 1998), or proliferation-related acidic nuclear protein (PAL31) (Mutai et al. 2000) and a truncated splice variant Anp32b2 which lacks the acidic tail and NLS. Anp32d (pp32r2), Anp32f, and Anp32h also lack the NLS (Fig. 4A highlighted by ++++), and would be expected to be retained in the cytoplasm and lack the nuclear associated functions of pp32. However, it is possible that these pp32 family members could form hetero-oligomers with those containing a consensus NLS. Although not present in this crystallized fragment of pp32, the region directly after the LRR domain contains a stretch of ∼30 amino acids (residues 150–180) that is essential for in vitro tumor suppression (Brody et al. 1999) and INHAT activities (Seo et al. 2002). The pp32 (Anp32a) family sequences diverge more significantly after this region particularly in the tumorigenic variants pp32r1 (Anp32c), pp32r2 (Anp32d), (Kadkol et al. 2001) and Anp32f, g, h, whose role is presently unclear (Fig. 4A). This central region could be structurally important and, like the Skp2 LRR (Schulman et al. 2000), could extend back toward the first LRR packing against the concave surface of the LRR domain. Interestingly, the C-terminal acidic tail (residues 150–249) is required for multimerization (Ulitzur et al. 1997b), and could pack on the concave surface of a neighboring pp32 LRR domain in the formation of pp32 homo/hetero oligomers.
The structure provides insights into the molecular architecture of potential protein interactions mediated by the pp32 LRR domain, such as the interaction with the hyperphosphorylated form of Rb, which is predominant in p16 inactivated pancreatic tumors. The structure also provides the basis to identify possible small molecule binding partners by virtual screening that could be used to disrupt this interaction and be developed as therapeutics. We can also selectively probe the interactions made by pp32 (Anp32a) and other family members through rational site-directed mutagenesis to provide further insight into the structure and function of the pp32 (Anp32) family.
Materials and Methods
Protein expression and purification
The pp32 (Anp32a) gene fragment pp32ΔCT comprising residues 1–149 was amplified by PCR from a full-length clone (RZPD). The fragment was cloned into the pProEX HTb expression vector in frame with an N-terminal His6 tobacco etch virus (TEV) cleavable tag. Cultures of Escherichia coli Rosetta 2 (DE3) (Novagen) containing the pp32 expression construct were grown in LB at 37°C, induced at mid-log phase at 28°C with 0.5 mM IPTG, and harvested after 3–4 h. Cells were lysed using a microfluidizer in Buffer A (25 mM Tris, 500 mM NaCl, 2 mM 2-mercaptoethanol, 10 mM Imidazole pH 8.0) supplemented with DnaseI (Roche) 2 mM MgCl2, 1 mM PMSF, and EDTA free protease inhibitor cocktail (Roche). Lysed cells were then centrifuged at 20,000g before loading onto a 5-mL Hi-Trap chelating column equilibrated in buffer A. After washing with 10 column volumes of buffer A protein was eluted in buffer B (25 mM Tris, 500 mM NaCl, 2 mM 2-mercaptoethanol, 250 mM Imidazole pH 8.0). Protein was dialyzed into TEV cleavage buffer (25 mM Tris, 150 mM NaCl, 2 mM 2-mercaptoethanol, pH 8.0) and the His6 tag removed with His6-TEV protease for 3–4 h at room temperature. Subsequent repurification on a Hi-Trap chelating column removed contaminants and TEV protease. Size-exclusion chromatography, using a Superdex 200 16/60 (Pharmacia) was then used as the final purification step, where the fragment elutes as a monomer compared to standards. Fractions containing pure pp32ΔCT were then pooled and dialyzed into crystallization buffer (25 mM Tris, 150 mM NaCl, 2 mM 2-mercaptoethanol, pH 8.0) before concentrating to 20–40 mg/mL.
Crystallization and data collection
Two plate-like crystal forms of pp32ΔCT were grown at 22°C by vapor diffusion using the hanging-drop method. Crystal form I grew from 1.6–2.0 M Ammonium sulphate, 5% PEG 3350, and 0.1 M sodium citrate buffer, pH 5.5 at a protein concentration of ∼20–30 mg/mL. These crystals of pp32ΔCT belong to the monoclinic space group C2 with unit cell parameters a = 106.5, b = 183.2, c = 67.47 Å, β = 104.4 and six molecules per asymmetric unit.
Crystal form II grew in 15%–25% PEG3350, 0.2 M ammonium citrate, and 0.1 M sodium citrate buffer, pH 5.0–5.5, at a protein concentration of ∼25–40 mg/mL. These crystals of pp32ΔCT belong to the hexagonal space group P63 with unit cell parameters a = b = 105.9, c = 132.4 Å, and four molecules per asymmetric unit.
For both crystal forms, single crystals of pp32ΔCT were transferred briefly to cryoprotectant solutions consisting of mother liquor supplemented with increasing concentrations of glycerol (up to 25%) before being frozen directly with liquid nitrogen. Data were collected at 100K at BioCars beamline 14-BM-C (APS). The data were processed, scaled, and reduced using HKL2000 (Table 1).
Structure determination and refinement
A homology model of the N-terminal LRR domain of pp32 was created using Modeller 8.01 and the coordinates of the spiceosomal protein U2A′ protein (Price et al. 1998) (accession 1A9NA), which was used to solve the structure by molecular replacement with MOLREP (Vagin and Teplyakov 2000). Initially, data from crystal form II were processed in space group P6322 with two molecules in the asymmetric unit; however, this structure would not refine satisfactorily beyond an R free of ∼39%. The correct space group was subsequently identified as P63 with four molecules in the asymmetric unit, in which the NCS molecules mimic pseudo P6322 symmetry. Crystal form I was then solved by molecular replacement with MOLREP (Vagin and Teplyakov 2000) using the coordinates of a monomer from the partially refined P63 structure.
Both structures were refined using REFMAC5 (Murshudov et al. 1997) and model building carried out with the program Coot (Emsley and Cowtan 2004). The progress of the refinement was monitored by the agreement between calculated and observed structure factors for the excluded reflections (R free). Due to limited observations tight NCS restraints were used throughout the refinement process. Final cycles of refinement were carried out using TLS refinement as implemented in REFMAC5 (Winn et al. 2003); with the individual NCS-related molecules as TLS groups, final refinement statistics are shown in Table 1. Coordinates and structure factors have been deposited in the RCSB Protein Data Bank, for both crystal forms (accession 2je0 and 2je1, respectively).
Structural analysis and graphics
Sequences from human Anp32a–h were aligned using ClustalW (Thompson et al. 1994) and plotted using ESPript (Fig. 4A; Gouet et al. 1999). The Shannon entropy was calculated for each residue position for the ClustalW alignment, with H2PDB (http://bio.dfci.harvard.edu/Tools/entropy2pdb.html) and the calculated values transferred to the B-factor column of the PDB file and plotted on the surface (Fig. 3). Surface curvature was calculated with GRASP (Baker et al. 2001) and figures prepared with MolScript (Esnouf 1997) and Raster3D (Fig. 1C; Merritt and Murphy 1994). All other molecular images were generated using PyMol (http://pymol.sourceforge.net/).
Electronic supplemental material
Electrostatic-potential maps were calculated with APBS (Baker et al. 2001) (Suppl. Fig. 1).
Acknowledgments
We thank Jonathan Brody (Thomas Jefferson University Hospital) for critical reading of this manuscript, Michael Love (Johns Hopkins University) for computational support, Robbert Henning, Michael Bolbat, and the staff at BioCARS beamline 14-BM-C (APS, Chicago) for assistance during data collection. The U.S. Department of Energy, Basic Energy Sciences, Office of Science, under Contract No. W-31-109-Eng-38, supports use of the Advanced Photon Source. Use of the BioCARS Sector 14 is supported by the National Institutes of Health, National Center for Research Resources, under grant number RR07707.
Footnotes
Supplemental material: see www.proteinscience.org
Reprint requests to: Cynthia Wolberger, Department of Biophysics and Biophysical Chemistry, Howard Hughes Medical Institute, Johns Hopkins University School of Medicine, 725 N. Wolfe Street, Baltimore, MD 21205-2185, USA; e-mail: cwolberg@jhmi.edu; fax: (410) 614-8648.
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.072803507.
References
- Adegbola O. and Pasternack, G.R. 2005. Phosphorylated retinoblastoma protein complexes with pp32 and inhibits pp32-mediated apoptosis. J. Biol. Chem. 280: 15497–15502. [DOI] [PubMed] [Google Scholar]
- Bai J., Brody, J.R., Kadkol, S.S., and Pasternack, G.R. 2001. Tumor suppression and potentiation by manipulation of pp32 expression. Oncogene 20: 2153–2160. [DOI] [PubMed] [Google Scholar]
- Baker N.A., Sept, D., Joseph, S., Holst, M.J., and McCammon, J.A. 2001. Electrostatics of nanosystems: Application to microtubules and the ribosome. Proc. Natl. Acad. Sci. 98: 10037–10041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan C.M., Gallouzi, I.E., and Steitz, J.A. 2000. Protein ligands to HuR modulate its interaction with target mRNAs in vivo. J. Cell Biol. 151: 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brody J.R., Kadkol, S.S., Mahmoud, M.A., Rebel, J.M., and Pasternack, G.R. 1999. Identification of sequences required for inhibition of oncogene-mediated transformation by pp32. J. Biol. Chem. 274: 20053–20055. [DOI] [PubMed] [Google Scholar]
- Ceulemans H., De Maeyer, M., Stalmans, W., and Bollen, M. 1999. A capping domain for LRR protein interaction modules. FEBS Lett. 456: 349–351. [DOI] [PubMed] [Google Scholar]
- Chen T.H., Brody, J.R., Romantsev, F.E., Yu, J.G., Kayler, A.E., Voneiff, E., Kuhajda, F.P., and Pasternack, G.R. 1996. Structure of pp32, an acidic nuclear protein which inhibits oncogene-induced formation of transformed foci. Mol. Biol. Cell 7: 2045–2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P. and Cowtan, K. 2004. Coot: Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60: 2126–2132. [DOI] [PubMed] [Google Scholar]
- Esnouf R.M. 1997. An extensively modified version of MolScript that includes greatly enhanced coloring capabilities. J. Mol. Graph. Model. 15: 132–134. [DOI] [PubMed] [Google Scholar]
- Gallouzi I.E., Brennan, C.M., and Steitz, J.A. 2001. Protein ligands mediate the CRM1-dependent export of HuR in response to heat shock. RNA 7: 1348–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouet P., Courcelle, E., Stuart, D.I., and Metoz, F. 1999. ESPript: Analysis of multiple sequence alignments in PostScript. Bioinformatics 15: 305–308. [DOI] [PubMed] [Google Scholar]
- Holm L. and Sander, C. 1994. Searching protein structure databases has come of age. Proteins 19: 165–173. [DOI] [PubMed] [Google Scholar]
- Howitt J.A., Clout, N.J., and Hohenester, E. 2004. Binding site for Robo receptors revealed by dissection of the leucine-rich repeat region of Slit. EMBO J. 23: 4406–4412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itin C., Ulitzur, N., Muhlbauer, B., and Pfeffer, S.R. 1999. Mapmodulin, cytoplasmic dynein, and microtubules enhance the transport of mannose 6-phosphate receptors from endosomes to the trans-golgi network. Mol. Biol. Cell 10: 2191–2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X., Kim, H.E., Shu, H., Zhao, Y., Zhang, H., Kofron, J., Donnelly, J., Burns, D., Ng, S.C., Rosenberg, S., et al. 2003. Distinctive roles of PHAP proteins and prothymosin-alpha in a death regulatory pathway. Science 299: 223–226. [DOI] [PubMed] [Google Scholar]
- Kadkol S.S., El Naga, G.A., Brody, J.R., Bai, J., Gusev, Y., Dooley, W.C., and Pasternack, G.R. 2001. Expression of pp32 gene family members in breast cancer. Breast Cancer Res. Treat. 68: 65–73. [DOI] [PubMed] [Google Scholar]
- Kobe B. and Deisenhofer, J. 1995. A structural basis of the interactions between leucine-rich repeats and protein ligands. Nature 374: 183–186. [DOI] [PubMed] [Google Scholar]
- Kobe B. and Kajava, A.V. 2001. The leucine-rich repeat as a protein recognition motif. Curr. Opin. Struct. Biol. 11: 725–732. [DOI] [PubMed] [Google Scholar]
- Li M. and Damuni, Z. 1998. I1PP2A and I2PP2A. Two potent protein phosphatase 2A-specific inhibitor proteins. Methods Mol. Biol. 93: 59–66. [DOI] [PubMed] [Google Scholar]
- Li M., Guo, H., and Damuni, Z. 1995. Purification and characterization of two potent heat-stable protein inhibitors of protein phosphatase 2A from bovine kidney. Biochemistry 34: 1988–1996. [DOI] [PubMed] [Google Scholar]
- Li M., Makkinje, A., and Damuni, Z. 1996. Molecular identification of I1PP2A, a novel potent heat-stable inhibitor protein of protein phosphatase 2A. Biochemistry 35: 6998–7002. [DOI] [PubMed] [Google Scholar]
- Lieberman J. and Fan, Z. 2003. Nuclear war: The granzyme A-bomb. Curr. Opin . Immunol. 15: 553–559. [DOI] [PubMed] [Google Scholar]
- Liker E., Fernandez, E., Izaurralde, E., and Conti, E. 2000. The structure of the mRNA export factor TAP reveals a cis arrangement of a non-canonical RNP domain and an LRR domain. EMBO J. 19: 5587–5598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malek S.N., Katumuluwa, A.I., and Pasternack, G.R. 1990. Identification and preliminary characterization of two related proliferation-associated nuclear phosphoproteins. J. Biol. Chem. 265: 13400–13409. [PubMed] [Google Scholar]
- Matilla A. and Radrizzani, M. 2005. The Anp32 family of proteins containing leucine-rich repeats. Cerebellum 4: 7–18. [DOI] [PubMed] [Google Scholar]
- Matilla A., Koshy, B.T., Cummings, C.J., Isobe, T., Orr, H.T., and Zoghbi, H.Y. 1997. The cerebellar leucine-rich acidic nuclear protein interacts with ataxin-1. Nature 389: 974–978. [DOI] [PubMed] [Google Scholar]
- Matsuoka K., Taoka, M., Satozawa, N., Nakayama, H., Ichimura, T., Takahashi, N., Yamakuni, T., Song, S.Y., and Isobe, T. 1994. A nuclear factor containing the leucine-rich repeats expressed in murine cerebellar neurons. Proc. Natl. Acad. Sci. 91: 9670–9674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mencinger M., Panagopoulos, I., Contreras, J.A., Mitelman, F., and Aman, P. 1998. Expression analysis and chromosomal mapping of a novel human gene, APRIL, encoding an acidic protein rich in leucines. Biochim. Biophys. Acta 1395: 176–180. [DOI] [PubMed] [Google Scholar]
- Merritt E.A. and Murphy, M.E. 1994. Raster3D Version 2.0. A program for photorealistic molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 50: 869–873. [DOI] [PubMed] [Google Scholar]
- Murshudov G.N., Vagin, A.A., and Dodson, E.J. 1997. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53: 240–255. [DOI] [PubMed] [Google Scholar]
- Mutai H., Toyoshima, Y., Sun, W., Hattori, N., Tanaka, S., and Shiota, K. 2000. PAL31, a novel nuclear protein, expressed in the developing brain. Biochem. Biophys. Res. Commun. 274: 427–433. [DOI] [PubMed] [Google Scholar]
- Nabors L.B., Furneaux, H.M., and King, P.H. 1998. HuR, a novel target of anti-Hu antibodies, is expressed in non-neural tissues. J. Neuroimmunol. 92: 152–159. [DOI] [PubMed] [Google Scholar]
- Opal P., Garcia, J.J., Propst, F., Matilla, A., Orr, H.T., and Zoghbi, H.Y. 2003. Mapmodulin/leucine-rich acidic nuclear protein binds the light chain of microtubule-associated protein 1B and modulates neuritogenesis. J. Biol. Chem. 278: 34691–34699. [DOI] [PubMed] [Google Scholar]
- Opal P., Garcia, J.J., McCall, A.E., Xu, B., Weeber, E.J., Sweatt, J.D., Orr, H.T., and Zoghbi, H.Y. 2004. Generation and characterization of LANP/pp32 null mice. Mol. Cell. Biol. 24: 3140–3149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price S.R., Evans, P.R., and Nagai, K. 1998. Crystal structure of the spliceosomal U2B″-U2A′ protein complex bound to a fragment of U2 small nuclear RNA. Nature 394: 645–650. [DOI] [PubMed] [Google Scholar]
- Rozenblum E., Schutte, M., Goggins, M., Hahn, S.A., Panzer, S., Zahurak, M., Goodman, S.N., Sohn, T.A., Hruban, R.H., Yeo, C.J., et al. 1997. Tumor-suppressive pathways in pancreatic carcinoma. Cancer Res. 57: 1731–1734. [PubMed] [Google Scholar]
- Schneider R., Bannister, A.J., Weise, C., and Kouzarides, T. 2004. Direct binding of INHAT to H3 tails disrupted by modifications. J. Biol. Chem. 279: 23859–23862. [DOI] [PubMed] [Google Scholar]
- Schubert W.D., Urbanke, C., Ziehm, T., Beier, V., Machner, M.P., Domann, E., Wehland, J., Chakraborty, T., and Heinz, D.W. 2002. Structure of internalin, a major invasion protein of Listeria monocytogenes, in complex with its human receptor E-cadherin. Cell 111: 825–836. [DOI] [PubMed] [Google Scholar]
- Schulman B.A., Carrano, A.C., Jeffrey, P.D., Bowen, Z., Kinnucan, E.R., Finnin, M.S., Elledge, S.J., Harper, J.W., Pagano, M., and Pavletich, N.P. 2000. Insights into SCF ubiquitin ligases from the structure of the Skp1–Skp2 complex. Nature 408: 381–386. [DOI] [PubMed] [Google Scholar]
- Scott P.G., McEwan, P.A., Dodd, C.M., Bergmann, E.M., Bishop, P.N., and Bella, J. 2004. Crystal structure of the dimeric protein core of decorin, the archetypal small leucine-rich repeat proteoglycan. Proc. Natl. Acad. Sci. 101: 15633–15638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott P.G., Dodd, C.M., Bergmann, E.M., Sheehan, J.K., and Bishop, P.N. 2006. J. Biol. Chem. 281: 13324–13332. [DOI] [PubMed] [Google Scholar]
- Seo S.B., Macfarlan, T., McNamara, P., Hong, R., Mukai, Y., Heo, S., and Chakravarti, D. 2002. Regulation of histone acetylation and transcription by nuclear protein pp32, a subunit of the INHAT complex. J. Biol. Chem. 277: 14005–14010. [DOI] [PubMed] [Google Scholar]
- Thompson J.D., Higgins, D.G., and Gibson, T.J. 1994. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22: 4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uff S., Clemetson, J.M., Harrison, T., Clemetson, K.J., and Emsley, J. 2002. Crystal structure of the platelet glycoprotein Ib(alpha) N-terminal domain reveals an unmasking mechanism for receptor activation. J. Biol. Chem. 277: 35657–35663. [DOI] [PubMed] [Google Scholar]
- Ulitzur N., Humbert, M., and Pfeffer, S.R. 1997a. Mapmodulin: A possible modulator of the interaction of microtubule-associated proteins with microtubules. Proc. Natl. Acad. Sci. 94: 5084–5089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulitzur N., Rancano, C., and Pfeffer, S.R. 1997b. Biochemical characterization of mapmodulin, a protein that binds microtubule-associated proteins. J. Biol. Chem. 272: 30577–30582. [DOI] [PubMed] [Google Scholar]
- Vaesen M., Barnikol-Watanabe, S., Gotz, H., Awni, L.A., Cole, T., Zimmermann, B., Kratzin, H.D., and Hilschmann, N. 1994. Purification and characterization of two putative HLA class II associated proteins: PHAPI and PHAPII. Biol. Chem. Hoppe Seyler 375: 113–126. [DOI] [PubMed] [Google Scholar]
- Vagin A. and Teplyakov, A. 2000. An approach to multi-copy search in molecular replacement. Acta Crystallogr. D Biol. Crystallogr. 56: 1622–1624. [DOI] [PubMed] [Google Scholar]
- Weinberg R.A. 1995. The retinoblastoma protein and cell cycle control. Cell 81: 323–330. [DOI] [PubMed] [Google Scholar]
- Winn M.D., Murshudov, G.N., and Papiz, M.Z. 2003. Macromolecular TLS refinement in REFMAC at moderate resolutions. Methods Enzymol. 374: 300–321. [DOI] [PubMed] [Google Scholar]