

Abstract
High-throughput screening of protein–protein and protein–peptide interactions is of high interest both for biotechnological and pharmacological applications. Here, we propose the use of the noncoded amino acids o-nitrotyrosine and p-iodophenylalanine as spectroscopic probes in combination with circular dichroism and fluorescence quenching techniques (i.e., collisional quenching and resonance energy transfer) as a means to determine the peptide orientation in complexes with SH3 domains. Proline-rich peptides bind SH3 modules in two alternative orientations, according to their sequence motifs, classified as class I and class II. The method was tested on an SH3 domain from a yeast myosin that is known to recognize specifically class I peptides. We exploited the fluorescence quenching effects induced by o-nitrotyrosine and p-iodophenylalanine on the fluorescence signal of a highly conserved Trp residue, which is the signature of SH3 domains and sits directly in the binding pocket. In particular, we studied how the introduction of the two probes at different positions of the peptide sequence (i.e., N-terminally or C-terminally) influences the spectroscopic properties of the complex. This approach provides clear-cut evidence of the orientation of the binding peptide in the SH3 pocket. The chemical strategy outlined here can be easily extended to other protein modules, known to bind linear sequence motifs in a highly directional manner.
Keywords: SH3 domains, proline-rich peptides, o-nitrotyrosine, p-iodophenylalanine, protein recognition, noncoded amino acids, fluorescence energy transfer, quenching, circular dichroism
The central role played by molecular interactions in the functioning of all biological systems requires a detailed structural understanding of how recognition takes place (Jones and Thornton 1996; Pawson and Nash 2003). Complete structural determination of protein complexes can, however, be time-demanding, since with the current technical tools it is often necessary to undertake de novo structure determinations even when the structures of all the interacting partners are known individually. It is therefore increasingly important to develop new high-throughput approaches that may allow us to grasp quickly at least the basic features of a complex without its detailed and lengthy structural description.
Here, we discuss the use of suitable spectroscopic probes, such as o-nitrotyrosine (NT) or p-iodophenylalanine (IF), to study molecular interactions in a fast and reliable way. Both IF and NT are expected to decrease protein fluorescence through two distinct mechanisms. In particular, iodine-containing molecules are known to act as collisional quenchers of Trp fluorescence by promoting nonradiative decay of the excited singlet state through intersystem crossing to an excited triplet state (Berlman 1973; Lakowicz 1999). The nonfluorescent NT, on the other hand, which absorbs radiation in a pH-dependent manner in the wavelength range where Tyr and Trp emit fluorescence, has proven to be an efficient acceptor in energy transfer processes, such as those occurring in protein folding (Rischel and Poulsen 1995; Tcherkasskaya and Ptitsyn 1999) and molecular recognition (De Filippis et al. 2006).
We demonstrate that a complementary use of the two probes can be exploited for high-throughput characterization of complexes of Src homology-3 (SH3) domains with their binding peptides. SH3 domains are relatively small protein modules of 50–70 amino acid residues and one of the widespread motifs mostly observed in eukaryotic organisms (Kay et al. 2000; Mayer 2001; Tong et al. 2002). Through specific recognition of proline-rich sequences, they play a crucial role in the formation of multiprotein complexes and networks responsible for signal transduction, cytoskeletal organization, and other cellular processes (Morton and Campbell 1994; Mayer 2001). In humans, mutations in several SH3 domains are known to cause severe malfunctions leading, among others, to inflammatory diseases and to cancer (Smithgall 1995; Dalgarno et al. 1997; Vidal et al. 2001).
Despite even large differences in the sequences of both components, the mode of binding of SH3/peptide complexes is highly reproducible (Larson and Davidson 2000; Di Nardo et al. 2003; Li 2005). In a minimalist approach, two parameters seem to be necessary and sufficient to describe an SH3/peptide complex: the affinity of the interaction and the directionality of the peptide in the binding groove. The target peptides, typically seven to 10 residues long (Zarrinpar and Lim 2000; Musacchio 2002; Li 2005; Rath et al. 2005), adopt a left-handed polyproline II helix conformation (PPII) and sit in a well-defined groove of the SH3 domain that is formed between the so-called n-Src- and RT-loops (Musacchio 2002). Because of the intrinsic symmetry of PPII helices, the peptides can, in principle, be accommodated into the same SH3 cavity in the two possible orientations (Fig. 1; Mayer 2001; Musacchio 2002). Specific consensus motifs, denoted as class I and class II peptides, seemed at first to determine the peptide orientation. However, more recently, several nonstandard sequences that are difficult to be classified as class I or class II binders have been identified (Cestra et al. 1999; Mongiovì et al. 1999; Kang et al. 2000; Tong et al. 2002; Jia et al. 2005), thus suggesting that the orientation cannot be reliably predicted solely from the peptide sequence (Cesareni et al. 2002; Li 2005; Musi et al. 2006). Fast and reliable determination of the affinity and orientation should therefore be sufficient to provide, at least, the general features of the interaction without undergoing complete structure determination of all possible SH3/peptide complexes.
Figure 1.

Schematic representation of the mode of binding of class I and class II peptides into the conserved groove of SH3 domains (adapted from Mayer 2001 and reproduced with permission of the Company of Biologists ©2001). The consensus sequences of the proline-rich peptides are indicated, using X for any amino acid, whereas P and R indicate semiconserved prolines and arginines.
Both IF and NT derivatives are ideally suited for characterizing the binding mode of target peptides to SH3 domains which contain as part of their signature motif a highly conserved Trp (Larson and Davidson 2000; Fernandez-Ballester et al. 2004). The fluorescence quenching effects determined by the amino acid derivatives on the fluorescence signal of the conserved Trp can therefore be used to define the peptide orientation. Additionally, the presence of NT, which has absorption properties totally different from those of other protein amino acids, is expected to generate a unique circular dichroism (CD) signal in the near-UV/Vis spectrum, upon complex formation.
We have chosen here the SH3 domain (Myo3-SH3) from the type I myosin isoform Myo3 from Saccharomyces cerevisiae as a model system. Myo3-SH3 is of potential medical interest, since mutations in the corresponding human orthologs result in Wiskott-Aldrich syndrome, a severe immunodeficiency related to alteration of key cellular processes, such as endocytosis and cytoskeleton assembly (Anderson et al. 1998; Rodal et al. 2003). The solution structure of Myo3-SH3 and a detailed analysis of its binding mode have recently been described (Musi et al. 2006). We show that, short of doing a full structural characterization of the complex, the orientation of Myo3-SH3/peptide complexes can be easily determined by fluorescence and CD through incorporation of IF or NT at the N- or C-terminal ends of the peptide. Our approach can be used as a valuable and efficient tool for obtaining a low-resolution but fast and reliable description of peptide/protein interactions.
Results and Discussion
Strategy of the work
Structural analysis based on representative SH3/peptide complexes from the Protein Data Bank (PDB) (see Materials and Methods) shows that one of the terminal ends of the bound peptide is much closer to the conserved Trp than the other: In class I peptides, the N terminus is within 4.7 ± 0.9 Å (Cβ–Cβ distance) of the conserved Trp, whereas the C terminus is 18.8 ± 1.5 Å away (Fig. 2A). This pattern is inverted in class II peptides, although with slightly larger distance variability. For a class I peptide, we can therefore confidently expect that incorporation of IF or NT at the N-terminal end of the binding peptide should bring these probes close enough to the conserved Trp to specifically quench its fluorescence or, as in the case of NT, generate a new signal in the near-UV/Vis CD spectrum of the SH3/peptide complex. Conversely, when NT or IF is incorporated far from the Trp pocket (i.e., at position 10 or farther), the direct collisional quenching of IF should be unable to operate and the efficiency of Trp-to-NT energy transfer, which is strongly dependent on distance (Wu and Brand 1994; Dos Remedios and Moens 1995; Selvin 1995; Eftink 1997), is expected to be reduced, together with the CD signal of NT. On the other hand, for class II peptides, the position-dependent spectroscopic effects are reversed.
Figure 2.
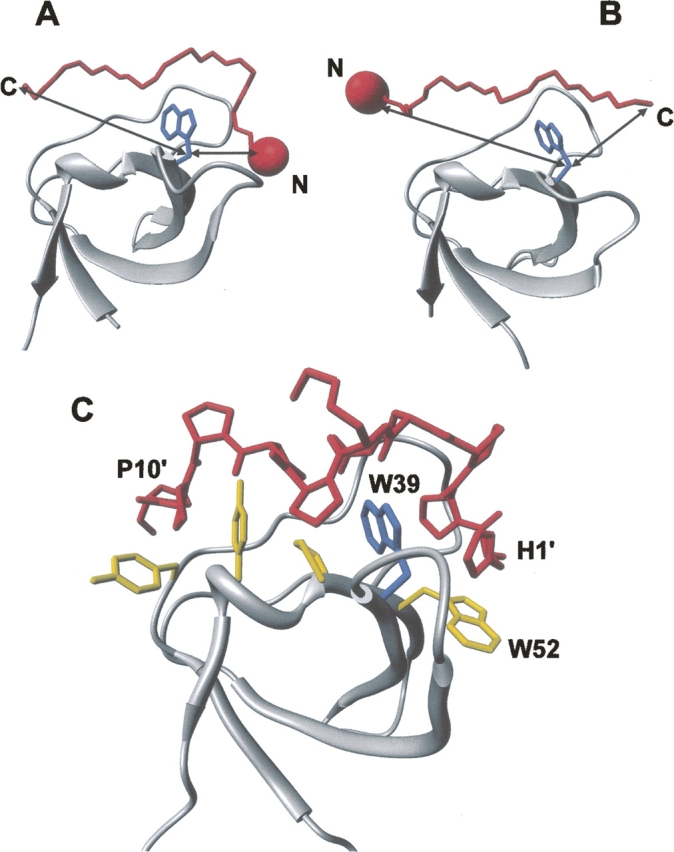
Structural analysis of SH3 complexes to illustrate the strategy proposed. Structures of two representative SH3 domains (ribbon drawing, gray) bound to (A) class I (1abo) (Musacchio et al. 1994) and (B) class II (1cka) (Wu et al. 1995) peptides (Cα trace, red). (Blue) The side chain of the conserved Trp residue in the SH3 domain; (arrows) the distances (Cβ–Cβ atoms) between the peptide termini and Trp. The incorporation of a suitable spectroscopic probe (red sphere), like NT or IF, is expected to perturb the intrinsic fluorescence of the conserved Trp in the SH3 domain, in a way that is strongly dependent on the orientation of the binding peptide on the SH3 surface. For instance, if the probe is incorporated at the N terminus of the ligand peptide, the fluorescence of SH3 is expected to be quenched only by a class I peptide, whereas it should be essentially unaffected if the ligand is a class II peptide. (C) Structure of the P2/Myo3-SH3 complex (Musi et al. 2006), showing relevant interactions of the ligand peptide (stick, red) with the SH3 domain (ribbon drawing, gray). The side chains of key amino acids of Myo3-SH3 are also explicitly shown (stick, yellow), while the conserved Trp39 is shown in blue.
Hence, we tested our strategy on a known model system formed by Myo3-SH3 and the P2 peptide. We have recently reported the solution structure of Myo3-SH3 and demonstrated that the domain recognizes class I SH3 peptides (Musi et al. 2006). We therefore selected one of these peptides, P2, which has high affinity for Myo3-SH3 and for which we have a reliable model in complex with Myo3-SH3 (Musi et al. 2006). A distance (Cβ–Cβ) of 6 Å from the conserved Trp39 to position 1 of P2 was estimated, whereas the peptide C terminus is at ∼22 Å from Trp39 (Fig. 2B). It is important to note that Myo-SH3 contains an additional nonconserved Trp, at position 52. The presence of this extra Trp residue should not, however, alter our analysis, since it is within the Trp pocket and spatially close to Trp39 (Trp39–Trp52, Cβ–Cβ distance 4.9 Å).
Different P2 analogs were synthesized, incorporating either NT or IF in different positions along the P2 sequence (Table 1). Unmodified P2, P2NT1, P2NT10, and P2NT13 were prepared to check the effect of the introduction of NT at the N or C termini of the peptide. Likewise, the analogs P2IF1 and P2IF13, containing IF at positions 1 and 13, were produced to test the effect of IF. In the case of P2NT13 and P2IF13, the P2 sequence was extended C-terminally with the highly flexible segment –G-G-X-G (X = IF or NT) to have the spectroscopic probes outside the P2/SH3 interface, to exclude possible interferences with complex formation. Finally, the unmodified and the NT1 analog of the P4 peptide, P4NT1, were synthesized as negative controls, since P4 does not bind Myo3-SH3 (Musi et al. 2006).
Table 1.
Binding data of P2 and P4 analogs to myosin-3 SH3 domain

Incorporation of NT or IF does not alter the binding mode of P2 to Myo3-SH3
The chemical strategy outlined above requires that incorporation of NT or IF does not alter the binding mode of P2 analogs to Myo3-SH3. To test this assumption, we measured the affinities of NT and IF analogs of P2 by exploiting their ability to quench the fluorescence of Myo3-SH3 (see Figs. 3 and 4). As recently demonstrated for the binding of the Tyr3 NT analog of hirudin to thrombin (De Filippis et al. 2006), quenching of Myo3-SH3 fluorescence by NT analogs (Fig. 3A,B) is predominantly caused by strong resonance energy transfer occurring between the Trp residues of the SH3 domain (i.e., the donor) and NT of P2 peptides (i.e., the acceptor) (Fig. 3C). On the other hand, IF analogs of P2 remarkably decrease Myo3-SH3 fluorescence (Fig. 4A,B) by a specific and direct effect of iodine in the SH3 pocket, by promoting nonradiative decay of the excited state of Trp residues through a collisional mechanism (Berlman 1973; Lakowicz 1999).
Figure 3.

Binding of NT analogs of P2 to Myo3-SH3 monitored by Trp-to-NT fluorescence energy transfer. (A) Fluorescence spectra of Myo3-SH3 (175 nM) in the presence of increasing concentrations of P2NT1 (0–120 μM). (B) Plot of the fluorescence intensity of Myo3-SH3 as a function of P2NT1 concentration (•). As a control, the data relative to free NT (○) are also reported. Protein samples were excited at 295 nm, and fluorescence data was corrected for IFE according to Equation 1 (see Materials and Methods). (C) Superposition of the fluorescence spectrum of Myo3-SH3 (continuous line), obtained after excitation of the sample at 295 nm, with the absorption spectrum of P2NT1 at pH 2 (dotted line) and pH 8 (dashed line). (D) Determination of Kd values of the Myo3-SH3 complexes with NT analogs: P2NT1 (•-•), P2NT10 (▴-▴), and P2NT13 (○-○). Corrected fluorescence intensities were expressed as F 0 − F, where F 0 is the intensity of Myo3-SH3 in the absence of ligand, and the data points fitted by Equation 2 (continuous lines) to yield K d and ΔF max (see Materials and Methods). All measurements were carried out at 25° ± 0.2°C by exciting the protein samples at 295 nm in 5 mM Tris-HCl buffer (pH 8.0) containing 0.1% (w/v) PEG 8000 and 0.2 M NaCl.
Figure 4.

Binding of IF analogs of P2 to Myo3-SH3 monitored by collisional quenching of Myo3-SH3 fluorescence. (A) Fluorescence spectra of Myo3-SH3 (175 nM) in the presence of increasing concentrations of P2IF1 (0–120 μM). (B) Change in the fluorescence intensity of Myo3-SH3 as a function of P2IF1 concentration (•). As a control, the effect of free IF is also reported (○). (C) Plot of the fluorescence intensity of Myo3-SH3 as a function of P2IF13 concentration (▴). (Inset) Fluorescence spectra of Myo3-SH3 (175 nM) in the presence of increasing concentrations of P2IF13. (D) Determination of Kd values of SH3 complexes with IF analogs: P2IF1 (•-•) and P2IF13 (▴-▴). For comparison, the binding data of the unmodified P2 peptide to Myo3-SH3 are also included (▵-▵). All spectra were recorded at 25° ± 0.2°C by exciting the protein samples at 295 nm in 5 mM Tris-HCl buffer (pH 8.0) containing 0.1% (w/v) PEG 8000 and 0.2 M NaCl. Continuous lines represent the best fit of the data points to Equation 2.
Analysis of fluorescence binding data is sometimes complicated by the contribution of the intrinsic emission of the ligand to the fluorescence of the receptor and by unspecific effects, including static and dynamic quenching, as well as the inner filter effect (IFE) (Selvin 1995; Eftink 1997). We carefully analyzed their contribution in the Supplemental material to make sure that these effects do not alter our analysis. In the case of NT peptides, the emission of free NT is negligible at 349 nm (i.e., the wavelength at which the fluorescence signal was measured) (Supplemental Fig. S1), and therefore we conclude that the addition of NT peptides does not interfere with fluorescence measurements. The data shown in Figure 3 and Supplemental Figure S2 suggest that free NT, taken as a suitable model compound, only slightly affects Myo3-SH3 fluorescence by static and dynamic quenching, as expected for nitro-compounds, which, in the absence of energy transfer, are known to quench the fluorescence of aromatic hydrocarbons by a mixed static and dynamic mechanism (Sawicki et al. 1964; Dreeskamp et al. 1975). In addition, the contribution of IFE to the binding data of high-affinity NT analogs (i.e., P2NT1 and P2NT13) is marginal (Supplemental Fig. S2A). In all cases, the fluorescence data were nonetheless corrected for IFE using Equation 1, and the adequacy of the correction method was carefully established (Supplemental Fig. S2B). Unspecific effects can be fully excluded for IF analogs, since IF absorbs weakly in the near-UV region and is essentially nonfluorescent. As a consequence, free IF only marginally reduces Myo3-SH3 fluorescence (Fig. 4B).
The fluorescence data, corrected for IFE in the case of NT analogs, were analyzed assuming a one-site binding mechanism (see Materials and Methods). The excellent fit of the experimental data to Equation 2 is a stringent, albeit indirect, proof of a 1:1 binding stoichiometry (Copeland 2000). The affinity data indicate that incorporation of NT or IF at position 1 or 13 does not change the affinity of the P2 derivatives for Myo3-SH3 (Table 1). This is in agreement with our structural analysis, which suggests that, when at position 1, either NT or IF can be easily accommodated into the Trp pocket with only minimal adjustments of the highly flexible 35–37 segment of Myo3-SH3 (Musi et al. 2006). More generally, our results are consistent with the fact that the Trp pocket of SH3 is quite “tolerant” in binding the target peptides outside the polyproline consensus sequence, and this is also well documented by the high structural variability of the amino acid that in the ligand peptides interacts with the conserved Trp (Musacchio 2002; Li 2005). Likewise, incorporation of NT or IF at position 13 does not interfere with binding, since the modifications involve residues outside the P2/Myo3-SH3 interface. Only when Pro10 was replaced with NT was a twofold decrease of the affinity of P2NT10 observed. This effect is likely caused by the replacement of a structurally constrained Pro (MacArthur and Thornton 1991) with the more flexible NT side chain (Searle and Williams 1992).
Our data concurrently support the assumption that the mutations introduced in P2 do not alter the binding mode of the resulting analogs to the SH3 domain and allow us to interpret confidently the position-dependent effects of the mutations on the intrinsic spectroscopic properties of P2 analogs or on the spectral changes evoked in Myo3-SH3 upon ligand binding.
Probing the binding mode of P2 to Myo3-SH3 by incorporation of NT
Fluorescence resonance energy transfer
The efficiency of energy transfer depends on the donor quantum yield, on the extent of spectral overlap of the emission spectrum of the donor with the absorption spectrum of the acceptor, on the donor–acceptor distance, and on their relative orientation (Lakowicz 1999). However, for a given donor–acceptor pair, the distance factor is predominant (Wu and Brand 1994; Dos Remedios and Moens 1995), and therefore we have exploited here the distance dependence of FRET efficiency to probe the orientation of P2 in complex with Myo3-SH3.
The presence of NT at position 1 of P2 decreases Myo3-SH3 fluorescence up to 90% (Fig. 3A,B), in agreement with the extensive overlap of the emission spectrum of the SH3 domain (i.e., the donor) with the absorption spectrum of P2NT1 (i.e., the acceptor) (Fig. 3C) and with the short donor–acceptor distance in the SH3 complex (<6 Å). Concomitantly, the λmax value is blue-shifted by 7 nm. This latter result parallels those obtained with the unmodified P2 peptide (data not shown), where a similar shift of the λmax was observed, and is consistent with the partial shielding of Trp residues from water upon ligand binding. An alternative possibility is that one of the two Trp residues emits at longer wavelengths and that preferential quenching of the fluorescence of this Trp would result in the blueshift of λmax. However, this possibility is unlikely because the two Trp residues in Myo3-SH3 are exposed to the solvent to a similar extent (Musi et al. 2006), and therefore they are expected to display identical emission properties. In addition, some other SH3 domains, like the N-terminal SH3 of Grb-2 (Vidal et al. 1999), contain only the conserved Trp and still have λmax values very similar to that of Myo3-SH3. Despite the similar affinities of the two complexes, quenching of Myo3-SH3 fluorescence at saturating concentrations of ligand is lower for P2NT13 (Fig. 3D), where NT is far from the Trp pocket (>22 Å). The reduced extent of quenching is a consequence of the weaker efficiency of FRET at longer Trp-to-NT distances and is even more significant if one considers that the unmodified P2 peptide is also able to quench SH3 fluorescence (Fig. 4D). In the P2NT13/Myo3-SH3 complex, the efficiency of energy transfer is reduced, but not abolished, because the Trp-to-NT distances are still within the Förster's distance of the donor–acceptor pair (26 Å) (Steiner et al. 1991).
Taken together, these results are fully consistent with a class I orientation of P2 in binding Myo3-SH3.
Circular dichroism
Further evidence strongly supporting a class I orientation of P2 was obtained by comparison of the near-UV/Vis CD spectra of free and SH3-bound NT-peptides (Fig. 5). The CD spectrum of free SH3 displays an intense positive band centered at 268 nm and a pronounced fine structure in the 285–300-nm range, characteristic of Trp absorption (Strickland 1974). As expected, beyond 305 nm, the signal approaches zero. The spectrum of free P2NT1 presents a marginally weak negative signal in the 300–390-nm range and a low-intensity positive band at 280 nm, assigned to the 1A1g → 1B1u transition of NT (Meloun et al. 1968; De Filippis et al. 2006), which acquires some rotational strength because of the presence of several rigid prolines that stabilize the PPII conformation.
Figure 5.

Binding of NT analogs of P2 to Myo3-SH3 monitored by circular dichroism. CD spectra of free Myo3-SH3 (65 μM, ---) and P2NT1 (168 μM, —) were recorded in 5 mM Tris-HCl buffer (pH 8.0) containing 0.1% (w/v) PEG 8000, 0.2 M NaCl. Myo3-SH3 and P2NT1 were alternatively mixed in the same molar ratio (65 μM:168 μM) to yield ∼85% of bound SH3 (Mix_P2NT1, —). (Inset) Difference spectra (Diff_P2NT1 and Diff_P2NT13) obtained by subtracting the theoretical sum spectra of free SH3 and NT peptides from the corresponding spectra of the experimental complexes (Mix_P2NT1 and Mix_P2NT13). Measurements were carried out at 25° ± 0.2°C in a 0.5-cm quartz cuvette and subtracted for the corresponding baseline. Ellipticity data, θ, were expressed in millidegrees (mdeg), without further normalization.
When SH3 and P2NT1 are mixed to form the noncovalent complex, a new band appears at ∼350 nm. Considering that natural amino acids do not absorb beyond 305–310 nm, this band can solely originate from the 1A1g → 1B2u transition of NT that becomes optically active by induction of chirality of NT upon binding in the Trp pocket of the SH3 domain. Furthermore, the λmax value of the newly generated band suggests that NT binds in the Trp pocket of Myo3-SH3 in its neutral –OH form, which maximally absorbs at 350 nm (Fig. 3C; Meloun et al. 1968). Similar results have been recently reported for the binding of NT analogs of hirudin to thrombin (De Filippis et al. 2006). Strikingly, when NT is incorporated at position 13, in the highly flexible segment –G-G-NT-G and outside the binding interface of P2 with Myo3-SH3, this band is absent (Fig. 5, inset).
These findings therefore provide conclusive support to the class I orientation of P2 to Myo3-SH3 (Musi et al. 2006) and suggest that the combined use of FRET and CD techniques can be successfully exploited for characterizing peptide/protein interactions.
Probing the binding mode of P2 to Myo3-SH3 by incorporation of IF
Although the work carried out with the NT analogs indicates that it is possible to use FRET to determine the orientation of an SH3-binding peptide, some limitations might be envisaged when it is not easy to make reliable assumptions on the relative distance of the N and C termini from the conserved Trp. To overcome these problems, we exploited the effect of IF on the fluorescence signal of Myo3-SH3, since the radius of action of this derivative is much more limited (3–6 Å) than that observed by FRET (Berlman 1973; Selvin 1995).
The incorporation of IF at position 1 of P2 decreases the fluorescence intensity of Myo3-SH3 by ∼50%, with a concomitant blueshift in the λmax value of 6–7 nm (Fig. 4A,B). As for P2NT1 binding, the blueshift in the emission of Myo3-SH3 reflects the less polar environment of Trp39 and Trp52 upon ligand binding, while the strong quenching of fluorescence is caused by the specific and direct contact of the iodine atom of IF in the Trp pocket. Conversely, when IF is incorporated in the fraying C terminus of P2IF13, the quenching effect is remarkably lower and identical to that of the unmodified P2 (Fig. 4C,D).
These results highlight the strong position-dependent effect of IF on the SH3 fluorescence. When located outside the ligand–SH3 interaction surface, IF is too far from the Trp pocket to quench the intrinsic fluorescence of Myo3-SH3, thus providing key information on the orientation of the ligand peptide in the SH3 complex.
Conclusions
The development of new spectroscopic tools for studying protein–protein interactions finds applications that span from structural biology to drug discovery (Hovius et al. 2000). For this purpose, several nonnatural amino acids with physico-chemical properties (e.g., size, polarity, and hydrogen-bonding properties) similar to those of the corresponding natural amino acids, but with distinct spectral features have been used to monitor protein folding and binding processes (Cornish et al. 1994; Twine and Szabo 2003; De Filippis et al. 2004, 2006).
In this study, we have shown that incorporation of NT or IF can be effectively used to obtain fast and reliable information on the affinity and the orientation of SH3-binding peptides, which are the parameters necessary and sufficient to describe, albeit at low resolution, the structural features of peptide/SH3 complexes. While easily introduced in peptides by standard solid-phase synthetic methods, these spectroscopic probes exploit different physical processes (e.g., FRET, chiroptical properties, and direct fluorescence quenching) that can be used in a complementary fashion, as also integrated by comparative modeling. The different spectroscopic effects elicited by NT or IF at different positions along the ligand peptide sequence can be confidently exploited to generate low-resolution models on a large scale with no need of solving the detailed atomic structure of each complex.
Here, we have reported the successful application of our chemical strategy to an SH3/peptide complex, but the same approach may also have vast applications in screening studies of other interacting domains of potential pharmacological interest, in which knowledge of the binding directionality is important (see Li 2005 and citations therein). Finally, we believe that the possibility of approaching the study of complex networks of interactions in a high-throughput fashion will open new avenues to the development of new drugs.
Materials and Methods
Production of Myo3-SH3 and the synthetic analogs of P2 and P4 peptides
Nα-Fmoc derivatives of NT, IF, and other protected amino acids, solvents, and reagents for peptide synthesis were purchased from Bachem AG or Applied Biosystems. Buffers and organic solvents were of analytical grade and purchased from Fluka.
P2 and P4 peptides and their analogs containing NT or IF were manually synthesized by the solid-phase Fmoc method on a chlorotrityl resin (36 mg) (Barlos et al. 1991) derivatized (0.69 μmol/mg) with the C-terminal amino acid (i.e., Pro). The chlorotrityl resin was used to minimize the release of the C-terminal dipeptide occurring through di-ketopiperazine formation (Rothe and Mazànek 1972). For those analogs having a non-Pro residue at the C terminus, a Wang resin (46 mg) derivatized (0.65 μmol/mg) with the Fmoc-amino acid was used. Elongation of the peptide chain was accomplished by a double-coupling cycle at all steps, using HATU as an activator (Carpino 1993) and a fivefold molar excess of protected amino acids. Removal of the Fmoc group was carried out by a 15-min treatment with 20% pyperidine in NMP. Peptide cleavage from the resin and removal of side-chain protecting groups was carried out by treating the peptidyl resin with a mixture of TFA/EDT/H2O (95:2.5:2.5 [v/v]) for 90 min at 0°C. After precipitation with ice-cold diethylether, the crude peptides were purified to homogeneity (>98%) by RP-HPLC on a semipreparative Vydac C18 column (1 × 25 cm, 5 μm particle size), equilibrated in aqueous TFA (0.1%), and eluted with an acetonitrile-TFA (0.1%) gradient from 2% to 35%, at a flow rate of 1.5 mL/min. The absorbance of the effluent was recorded at 226 nm. The chemical identity of the purified peptides was established by high-resolution mass spectrometry on a Mariner ESI-TOF instrument from Perseptive Biosystems. Analyses were conducted according to standard manufacturers’ procedures and gave mass values in agreement with amino acid composition within 50 ppm accuracy.
Myo3-SH3, spanning residues 1122–1190 of S. cerevisiae Myo3, was expressed and purified as previously detailed (Musi et al. 2006).
Determination of peptide/protein concentration
Protein/peptide concentrations were determined by UV absorption (Gill and von Hippel 1989) on a Lambda-2 spectrophotometer from Perkin-Elmer. The molar absorptivity value of Myo3-SH3 was taken as 15,220 M−1 · cm−1 at 280 nm. The concentrations of the synthetic peptides containing NT (P2NT1, P2NT10, P2NT13, P4NT1) or IF (P2IF1, P2IF13) were calculated from the molar absorbance value of NT, 2200 M−1 · cm−1 at 381 nm (Tcherkasskaya and Ptitsyn 1999), and IF, 260 M−1· cm−1 at 280 nm (De Filippis et al. 2002). The concentrations of the unmodified P2 and P4 peptides were estimated by quantitative analysis of amino acid composition, carried out on the corresponding peptide solutions (∼2.5 mg/mL), and prepared by weighting the lyophilized peptides.
Fluorescence measurements
Fluorescence spectra were recorded on a Perkin-Elmer spectrofluorimeter model LS-50B, equipped with a thermostated cell holder. The interaction of P2 and P4 peptides and their analogs (0–120 μM) with Myo3-SH3 (175 nM) was monitored by adding, under gentle magnetic stirring in a 1-cm pathlength cuvette (2 mL), aliquots (2–10 μL) of NT and IF peptide stock solutions (2.5–5 mM) to a solution of Myo3-SH3 (2 mL). At each peptide concentration, Myo3-SH3 samples were equilibrated for 5 min at 25° ± 0.2°C and excited at 295 nm, using an excitation/emission slit of 5 and 10 nm, respectively, and a scan speed of 240 nm/min. The decrease in fluorescence intensity of Myo3-SH3 at the wavelength, where the maximum fluorescence change was observed (i.e., 349 nm for NT peptides and 360 nm for the unmodified P2 and P4 peptides and IF derivatives), was recorded as a function of ligand concentration, [L]. Fluorescence data were corrected for sample dilution (<5% of the final volume) and expressed as ΔF = F 0 − F, where F 0 and F are the fluorescence of Myo3-SH3 in the absence and presence of the ligand, respectively.
For NT peptides, fluorescence data were also corrected for IFE (see below and Supplemental material), since fluorescence intensity is only proportional to the absorbance of the sample up to an optical density of 0.05 units, both at λex and λem (Puchalski et al. 1991; Lakowicz 1999). In our case, IFE becomes significant for ligand concentrations higher than 20 μM. Fluorescence data were corrected using the equation:
where ΔAex and ΔAem are the observed additional absorbances at the excitation (λex = 295 nm) and emission (λem = 349 nm) wavelengths.
For a simple one-site binding mechanism R + L ⇔ RL, the fluorescence intensity, F, of the receptor, R, at a given concentration of ligand, L, is linearly related to the concentration of the complex [RL], according to the equation F = [RL]F bound + [R]free F free. Since [R]free = [R] − [RL], then ΔF = ([RL]/[R])ΔF max (Eftink 1997). The data were interpolated with Equation 2, using the program Origin 6.0 (MicroCal Inc.) to obtain the fitting parameters ΔF max and K d:
where K d is the dissociation constant of the complex, RL, and ΔF max is the maximum fluorescence change at infinite concentration of the ligand, [L]∞. Equation 2 assumes that at equilibrium [L]free ≅ [L]tot, and thus it is valid only when K d ≫ [R], as usually observed for SH3 complexes (Kay et al. 2000).
CD measurements
CD spectra were recorded on a Jasco model J-810 spectropolarimeter. Near-UV/Vis spectra were recorded in a 0.5-cm cell, at a scan speed of 50 nm/min, with a response time of 2 sec, and resulted from the average of six accumulations. After baseline subtraction, ellipticity values, θ, were expressed in millidegrees (mdeg), without further data elaboration.
Computational analysis
Structural analysis of SH3/peptide complexes was performed on 14 PDB entries (1abo, 1a0n, 1azg, 1bbz, 1fyn, 1io6, and 1rlq as examples of class I peptides; 1aze, 1cka, 1ckb, 1efn, 1n5z, 1prm, and 1qwe as examples of class II peptides). For NMR structures, the best-representative model in the NMR ensemble was selected using the program OLDERADO available online at the Web site http://pqs.ebi.ac.uk/pqs-nmr.html. In most cases, the Cβ–Cβ distances were measured from the conserved Trp of the SH3 domain to the N- or C-terminal amino acid of the ligand. In a few SH3 complexes, where the coordinates of the ligand peptides extend far beyond the segment that actually contacts the SH3 surface, the Cβ–Cβ distances were measured from the conserved Trp to the first residue (from the N terminus or from the C terminus, respectively) that is at a distance <6 Å from any atom of the SH3 structure. The structures of P2 analogs in the SH3-bound state were modeled on the 2btt structure (Musi et al. 2006) by keeping unchanged the positions of all atoms in the lowest-energy HADDOCK (Dominguez et al. 2003) structure and substituting His1′ or Pro10′ with the NT and IF side chains.
Electronic supplemental material
The Supplemental material includes Supplemental Figure S1, the effect of NT concentration on the fluorescence intensity of the free amino acid; and Supplemental Figure S2, the correction of Myo3-SH3 fluorescence for the inner filter effect, as a function of P2NT1 and free NT concentration.
Acknowledgments
This work was supported by a EU Grant (QLG2-CT-2001-01663) and a Grant (PRIN-2005) to V.D.F. from the Italian Ministry of University and Scientific Research. We thank D. Dalzoppo and M. Sudol for critically reading the manuscript.
Footnotes
Supplemental material: see www.proteinscience.org
Reprint requests to: Vincenzo De Filippis, Department of Pharmaceutical Sciences, University of Padua, Via F. Marzolo 5, I-35131 Padua, Italy; e-mail: vincenzo.defilippis@unipd.it; fax: 39-049-827-5366.
Abbreviations: Standard single- or three-letter abbreviations were used for natural amino acids; CD, circular dichroism; DIEA, diisopropylethylamine; EDT, ethanedithiol; ESI, electrospray ionization; Fmoc, 9-fluorenyl-methyloxycarbonyl; FRET, fluorescence resonance energy transfer; IF, p-iodophenylalanine; IFE, inner filter effect; HATU, 2-(7-aza-1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate; Myo3, isoform 3 of myosin from the yeast Saccharomyces cerevisiae; Myo3-SH3, SH3 domain of Myo3; NMP, N-methylpyrrolidone; NMR, nuclear magnetic resonance; NT, o-nitrotyrosine; PDB, Protein Data Bank; PPII, type-II polyproline; PEG, polyethylene glycol; ppm, parts per million; SH3, Src homology-3; TFA, trifluoacetic acid, TOF, time of flight; UV/Vis: ultraviolet/visible spectroscopy.
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.062726807.
References
- Anderson B.L., Boldogh, I., Evangelista, M., Boone, C., Greene, L.A., and Pon, L.A. 1998. The Src homology domain 3 (SH3) of a yeast type I myosin, Myo5p, binds to verprolin and is required for targeting to sites of actin polarization. J. Cell Biol. 141: 1357–1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barlos K., Chatzi, O., Gatos, D., and Stavropoulos, G. 1991. 2-Chlorotrityl chloride resin. Studies on anchoring of Fmoc-amino acids and peptide cleavage. Int. J. Pept. Protein Res. 37: 513–520. [PubMed] [Google Scholar]
- Berlman I.B. 1973. Empirical study of heavy-atom collisional quenching of the fluorescence state of aromatic compounds in solution. J. Phys. Chem. 77: 562–567. [Google Scholar]
- Carpino L.A. 1993. 1-Hydroxy-7-azabenzotriazole. An efficient peptide coupling additive. J. Am. Chem. Soc. 115: 4397–4398. [Google Scholar]
- Cesareni G., Panni, S., Nardelli, G., and Castagnoli, L. 2002. Can we infer peptide recognition specificity mediated by SH3 domains? FEBS Lett. 513: 38–44. [DOI] [PubMed] [Google Scholar]
- Cestra G., Castagnoli, L., Dente, L., Minenkova, O., Petrelli, A., Mingine, N., Hoffmuller, U., Schneider-Mergener, J., and Cesareni, G. 1999. The SH3 domains of endophilin and amphiphysin bind to the proline-rich region of synaptojanin at distinct sites that display an unconventional binding specificity. J. Biol. Chem. 274: 32001–32007. [DOI] [PubMed] [Google Scholar]
- Copeland R.A. 2000. Enzymes: A practical introduction to structure, mechanism, and data analysis, 2nd ed. Wiley, New York.
- Cornish V.W., Benson, D.R., Altenbach, C.A., Hideg, K., Hubbell, W.L., and Schultz, P.G. 1994. Site-specific incorporation of biophysical probes into proteins. Proc. Natl. Acad. Sci. 91: 2910–2924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalgarno D.C., Botfield, M.C., and Rickles, R.J. 1997. SH3 domains and drug design: Ligands, structure, and biological function. Biopolymers 43: 383–400. [DOI] [PubMed] [Google Scholar]
- De Filippis V., Colombo, G., Russo, I., Spadari, B., and Fontana, A. 2002. Probing hirudin–thrombin interaction by incorporation of noncoded amino acids and molecular dynamics simulation. Biochemistry 43: 1537–1550. [DOI] [PubMed] [Google Scholar]
- De Filippis V., De Boni, S., De Dea, E., Dalzoppo, D., Grandi, C., and Fontana, A. 2004. Incorporation of the fluorescent amino acid 7-azatryptophan into the core domain 1–47 of hirudin as a probe of hirudin folding and thrombin recognition. Protein Sci. 13: 1489–1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Filippis V., Frasson, R., and Fontana, A. 2006. 3-Nitrotyrosine as a spectroscopic probe for investigating protein–protein interactions. Protein Sci. 15: 976–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Nardo A.A., Larson, S.M., and Davidson, A.R. 2003. The relationship between conservation, thermodynamic stability, and function in the SH3 domain hydrophobic core. J. Mol. Biol. 333: 641–655. [DOI] [PubMed] [Google Scholar]
- Dominguez C., Boelens, R., and Bonvin, A.M. 2003. HADDOCK: A protein–protein docking approach based on biochemical or biophysical information. J. Am. Chem. Soc. 125: 1731–1737. [DOI] [PubMed] [Google Scholar]
- Dos Remedios C.G. and Moens, P.D.J. 1995. Fluorescence resonance energy transfer spectroscopy is a reliable “ruler” for measuring structural changes in proteins Dispelling the problem of the unknown orientation factor. J. Struct. Biol. 115: 175–185. [DOI] [PubMed] [Google Scholar]
- Dreeskamp H., Koch, E., and Zander, M. 1975. On the fluorescence quenching of polycyclic aromatic hydrocarbons by nitromethane. Z. Naturforsch. A 30: 1311–1314. [Google Scholar]
- Eftink M.R. 1997. Fluorescence methods for studying equilibrium macromolecule–ligand interactions. Methods Enzymol. 278: 221–257. [DOI] [PubMed] [Google Scholar]
- Fernandez-Ballester G., Blanes-Mira, C., and Serrano, L. 2004. The tryptophan switch: Changing ligand-binding specificity from type-I to type-II in SH3 domains. J. Mol. Biol. 335: 619–629. [DOI] [PubMed] [Google Scholar]
- Gill S.G. and von Hippel, P.H. 1989. Calculation of protein extinction coefficients from amino acid sequence data. Anal. Biochem. 182: 319–326. [DOI] [PubMed] [Google Scholar]
- Hovius R., Vallotton, P., Wohland, T., and Vogel, H. 2000. Fluorescence techniques: Shedding light on ligand–receptor interactions. Trends Pharmacol. Sci. 21: 266–273. [DOI] [PubMed] [Google Scholar]
- Jia C.Y.H., Nie, J., Wu, C., Li, C., and Li, S.S. 2005. Novel Src homology 3 domain-binding motifs identified from proteomic screen of a Pro-rich region. Mol. Cell. Proteomics 4: 1155–1166. [DOI] [PubMed] [Google Scholar]
- Jones S. and Thornton, J.M. 1996. Principles of protein–protein interactions. Proc. Natl. Acad. Sci. 93: 13–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang H., Freund, C., Duke-Cohan, J.S., Musacchio, A., Wagner, G., and Rudd, C.E. 2000. SH3 domain recognition of a proline-independent tyrosine-based RkxxYxxY motif in immune cell adaptor SKAP55. EMBO J. 19: 2889–2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kay B., Williamson, M.P., and Sudol, M. 2000. The importance of being proline: The interaction of proline-rich motifs in signaling proteins with their cognate domains. FASEB J. 14: 231–241. [PubMed] [Google Scholar]
- Lakowicz J.R. 1999. Principles of fluorescence spectroscopy, 2nd ed. Kluwer Academic/Plenum, New York.
- Larson S.M. and Davidson, A.R. 2000. The identification of conserved interactions within the SH3 domain by alignment of sequences and structures. Protein Sci. 9: 2170–2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S.S. 2005. Specificity and versatility of SH3 and other proline-recognition domains: Structural basis and implications for cellular signal transduction. Biochem. J. 390: 641–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacArthur M.W. and Thornton, J.M. 1991. Influence of proline residues on protein conformation. J. Mol. Biol. 218: 397–412. [DOI] [PubMed] [Google Scholar]
- Mayer B.J. 2001. SH3 domains: Complexity in moderation. J. Cell Sci. 114: 1253–1263. [DOI] [PubMed] [Google Scholar]
- Meloun B., Frič, I., and Šorm, F. 1968. Nitration of tyrosine residues in the pancreatic trypsin inhibitor with tetranitromethane. Eur. J. Biochem. 4: 112–117. [DOI] [PubMed] [Google Scholar]
- Mongiovì A.M., Romano, P.R., Panni, S., Mendoza, M., Wong, W.T., Musacchio, A., Cesareni, G., and Di Fiore, P.P. 1999. A novel peptide–SH3 interaction. EMBO J. 18: 5300–5309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morton C.J. and Campbell, I.D. 1994. SH3 domains: Molecular ‘velcro.’ Curr. Biol. 4: 615–617. [DOI] [PubMed] [Google Scholar]
- Musacchio A. 2002. How SH3 domains recognize proline. Adv. Protein Chem. 61: 211–268. [DOI] [PubMed] [Google Scholar]
- Musacchio A., Saraste, M., and Wilmanns, M. 1994. High resolution crystal structure of tyrosine kinase SH3 domains complexed to proline-rich peptides. Nat. Struct. Biol. 1: 546–551. [DOI] [PubMed] [Google Scholar]
- Musi V., Birdsall, B., Fernandez-Ballester, G., Guerrini, R., Salvatori, S., Serrano, L., and Pastore, A. 2006. New approaches to high-throughput structure characterization of SH3 complexes: The example of myosin-3 and myosin-5 from S. cerevisiae. Protein Sci. 15: 795–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawson T. and Nash, P. 2003. Assembly of cell regulatory systems through protein interaction domains. Science 300: 445–452. [DOI] [PubMed] [Google Scholar]
- Puchalski M.M., Morra, M.J., and von Wandruszka, R. 1991. Assessment of inner filter effect corrections in fluorimetry. Fresenius J. Anal. Chem. 340: 341–344. [Google Scholar]
- Rath A., Davidson, A.R., and Deber, C.M. 2005. The structure of “unstructured” regions in peptides and proteins: Role of the polyproline II helix in protein folding and recognition. Biopolymers 80: 179–185. [DOI] [PubMed] [Google Scholar]
- Rischel C. and Poulsen, F.M. 1995. Modification of a specific tyrosine enables tracing of the end-to-end distance during apomyoglobin folding. FEBS Lett. 374: 105–109. [DOI] [PubMed] [Google Scholar]
- Rodal A.A., Manning, A.L., Goode, B.L., and Drubin, D.G. 2003. Negative regulation of yeast WASP by two SH3 domain-containing proteins. Curr. Biol. 13: 1000–1008. [DOI] [PubMed] [Google Scholar]
- Rothe M. and Mazànek, J. 1972. Side-reactions arising on formation of cyclodipeptides in solid-phase peptide synthesis. Angew. Chem. Int. Ed. Engl. 11: 293. [DOI] [PubMed] [Google Scholar]
- Sawicki E., Stanley, T.W., and Elbert, W.C. 1964. Quenchofluorometric analysis for fluoranthenic hydrocarbons in the presence of other types of aromatic hydrocarbons. Talanta 11: 1433–1441. [Google Scholar]
- Searle A.S. and Williams, D.H. 1992. The cost of conformational order: Entropy changes in molecular association. J. Am. Chem. Soc. 114: 10690–10697. [Google Scholar]
- Selvin P.R. 1995. Fluorescence resonance energy transfer. Methods Enzymol. 246: 300–334. [DOI] [PubMed] [Google Scholar]
- Smithgall T.E. 1995. SH2 and SH3 domains: Potential targets for anti-cancer drug design. J. Pharmacol. Toxicol. Methods 34: 125–132. [DOI] [PubMed] [Google Scholar]
- Steiner R.F., Albaugh, S., and Kilhoffer, M.C. 1991. Distribution separations between groups in an engineered calmodulin. J. Fluoresc. 1: 15–22. [DOI] [PubMed] [Google Scholar]
- Strickland E.H. 1974. Aromatic contributions to circular dichroism spectra of proteins. CRC Crit. Rev. Biochem. 3: 113–175. [DOI] [PubMed] [Google Scholar]
- Tcherkasskaya O. and Ptitsyn, O.B. 1999. Direct energy transfer to study the 3D structure of nonnative proteins: AGH complex in the molten globule state of apomyoglobin. Protein Eng. 12: 485–490. [DOI] [PubMed] [Google Scholar]
- Tong A.H., Drees, B., Nardelli, G., Bader, G.D., Brannetti, B., Castagnoli, L., Evangelista, M., Ferracuti, S., Nelson, B., Paoluzi, S., et al. 2002. A combined experimental and computational strategy to define protein interaction networks for peptide recognition modules. Science 295: 321–324. [DOI] [PubMed] [Google Scholar]
- Twine S.M. and Szabo, A.G. 2003. Fluorescent amino acid analogs. Methods Enzymol. 360: 104–127. [DOI] [PubMed] [Google Scholar]
- Vidal M., Goudreau, N., Cornille, F., Cussac, D., Gincel, E., and Garbay, C. 1999. Molecular and cellular analysis of Grb2 SH3 domain mutants: Interaction with Sos and dynamin. J. Mol. Biol. 290: 717–730. [DOI] [PubMed] [Google Scholar]
- Vidal M., Gigoux, V., and Garbay, C. 2001. SH2 and SH3 domains as targets for anti-proliferative agents. Crit. Rev. Oncol. Hematol. 40: 175–186. [DOI] [PubMed] [Google Scholar]
- Wu P. and Brand, L. 1994. Resonance energy transfer: Methods and applications. Anal. Biochem. 18: 1–13. [DOI] [PubMed] [Google Scholar]
- Wu X., Knudsen, B., Feller, S.M., Zheng, J., Sali, A., Cowburn, D., Hanafusa, H., and Kuriyan, J. 1995. Structural basis for the specific interaction of lysine-containing proline-rich peptides with the N-terminal SH3 domain of c-Crk. Structure 3: 215–226. [DOI] [PubMed] [Google Scholar]
- Zarrinpar A. and Lim, W.A. 2000. Converging on proline: The mechanism of WW domain peptide recognition. Nat. Struct. Biol. 7: 611–613. [DOI] [PubMed] [Google Scholar]