

Abstract
Detailed knowledge of how networks of surface salt bridges contribute to protein thermal stability is essential not only to understand protein structure and function but also to design thermostable proteins for industrial applications. Experimental studies investigating thermodynamic stability through measurements of free energy associated with mutational alterations in proteins provide only macroscopic evidence regarding the structure of salt-bridge networks and assessment of their contribution to protein stability. Using explicit-solvent molecular dynamics simulations to provide insight on the atomic scale, we investigate here the structural stability, defined in terms of root-mean-square fluctuations, of a short polypeptide designed to fold into a stable trimeric coiled coil with a well-packed hydrophobic core and an optimal number of intra- and interhelical surface salt bridges. We find that the increase of configurational entropy of the backbone and side-chain atoms and decreased pair correlations of these with increased temperature are consistent with nearly constant atom-positional root-mean-square fluctuations, increased salt-bridge occupancies, and stronger electrostatic interactions in the coiled coil. Thus, our study of the coiled coil suggests a mechanism in which well-designed salt-bridge networks could accommodate stochastically the disorder of increased thermal motion to produce thermostability.
Keywords: molecular dynamics, GROMOS, configurational entropy, folding, coiled coil
Understanding on a molecular basis how proteins of hyperthermophilic organisms maintain stability at high temperatures remains an important challenge, since detailed knowledge of protein stability increases our basic understanding not only of protein structure and function, but also of the origin and progression of several fatal protein misfolding diseases, such as amyloidoses (Dobson 2003). Furthermore, it may suggest rules for the rational design and engineering of proteins that withstand the high temperatures used in many industrial processes (Sanchez-Ruiz and Makhatadze 2001).
Attempts to explain the stability of thermophilic proteins on the basis of sequence or structure alone have proven difficult. Comparison of high-resolution structures of homologous proteins found in mesophilic and thermophilic organisms has nevertheless revealed a variety of potentially stabilizing factors, e.g., rigidity as indicated by B-factor analysis, improved packing in the hydrophobic core, greater density of hydrophobic loops in the protein core, shortening of flexible loops, and increased hydrogen bonding (Jaenicke and Bohm 1998; Cambillau and Claverie 2000; Szilagyi and Zavodszky 2000).
Recent reviews (Karshikoff and Ladenstein 2001; Kumar and Nussinov 2001) conclude, however, that the most striking difference between mesophilic and thermophilic proteins is an increased number of surface salt-bridge networks. Numerous studies suggest that favorable electrostatic interactions strongly depend on optimal salt-bridge location and geometry and on interaction of the salt-bridge residues with other residues in the protein. These studies indicate that, while isolated salt bridges provide only marginal stabilization at room temperature, reduced desolvation at high temperatures favors the formation of salt bridges incorporated in networks (de Bakker et al. 1999; Karshikoff and Ladenstein 2001; Kumar and Nussinov 2001). High-resolution structures of thermophilic proteins evidence multiple configurations of charged-residue side chains involved in salt bridges. The implied interaction patterns suggest that the networks are dynamic rather than static (Kumar and Nussinov 2001).
Avoiding the limitations of models based on static protein structures, explicit solvent molecular dynamics (MD) simulations provide a straightforward and powerful means to explore temperature-dependent conformational changes of a protein at atomic spatial and temporal scales. Complete free-energy calculations including solvent degrees of freedom are, however, beyond their scope. Recent MD simulations of the structural stability and folding of peptides and small proteins under reversible conditions reproduce measured average properties, instilling confidence that the conformational distributions are reasonably realistic (Daura et al. 1998a; Duan and Kollman 1998; Schäfer et al. 1998; Takano et al. 1999; Hummer et al. 2001; Simmerling et al. 2002; Snow et al. 2002). Few explicit solvent MD simulations have assessed the temperature dependence of salt-bridge interactions (de Bakker et al. 1999; Thomas and Elcock 2004). These simulations, which studied charged-residue contact frequencies, distance distributions, and selected contributions to free energy, indicated that salt-bridge interactions are indeed suited to promoting structural stability to proteins at high temperatures.
Continuing with this structural approach, we report here explicit solvent MD simulations of the de novo, 17-residue peptide ccβ-p as a coiled coil of three α-helices and as an α-helical monomer at three temperatures. The X-ray structure of ccβ-p, displayed in Supplemental Figure S1, supplied the initial coordinates of the peptides in the three-stranded α-helical coiled-coil trimer configuration (Kammerer et al. 2004). Experimentally, the Arg 8 to Lys mutant switches the oligomerization state from the coiled coil to a dimer (Kammerer and Steinmetz 2006). The monomer, which cannot be observed experimentally due to its propensity for trimer formation, serves therefore as a standard of comparison for the coiled coil and may provide insight into trimer formation. Simulations of monomer and trimer at 278°K and 330°K portray 50 ns; simulation of the coiled coil at 370°K, 100 ns. Composed of the amino acid sequence, S-IRELEAR-IRELELR-IG, ccβ-p was designed to fold into a stable coiled coil of parallel and in-register α-helices with a well-packed hydrophobic core and an optimal number of intra- and interhelical surface salt bridges to facilitate network formation while minimizing electrostatic repulsion (Kammerer et al. 2004). These characteristics are shared by thermostable proteins and thus make ccβ-p a suitable model system to investigate with explicit-solvent MD simulations the role of extended salt-bridge networks on protein thermostability. Analysis of the simulations relates the salt bridges to hydrogen bonds, secondary structure and root-mean-square fluctuations (RMSF) of the backbone atoms from initial and mean structures. Complementing these conventional analyses are computations of configurational entropies of the backbone and side chains of the monomer and trimer helices and of configurational entropies due to the correlation between pairs of side chains.
Results
Comparison of the atom-positional root-mean-square deviations from the initial structure at 278°K and 330°K shown in Figure 1 suggests the structural stability of the trimer in the simulations. Figure 2 evidences a clearer indication; the RMSFs about simulated mean configurations of the trimer backbone and side-chain atoms are essentially the same at the two temperatures.
Figure 1.

Backbone atom-positional root-mean-square deviations from initial crystallographic structure (Supplemental Fig. S1) and secondary structure of ccβ-p coiled-coil trimer at 278°K (left) and 330°K (right). For secondary structure, green denotes α-helix, red H-bonded turn, and cyan β-bridge.
Figure 2.

Atom-positional RMSFs of backbone (left) and side-chain (right) atoms from mean configurations of the ccβ-p coiled-coil monomer and trimer at 278°K (solid line) and 330°K (dotted line). For the trimer the averages of the three helices are shown.
Further evidence of structural stability is provided by analysis of the salt bridges. In contrast to the crystal structure (Supplemental Fig. S1), in which five out of eight charged side chains are directly or indirectly involved in crystal contacts, the networks of salt bridges anticipated in the design of ccβ-p are a prominent feature of the trimer simulations in water. Three interhelical and seven intrahelical salt bridges appear uniformly during the 50-ns time course of the simulations at both 278°K and 330°K (see Fig. 3; Supplemental Fig. S2). The salt-bridge networks, illustrated in Figure 3, comprise the interactions of Arg3 with Glu4 and Glu6, of Arg8 with Glu4, Glu11, Glu6′, and Glu13′, of Arg10 with Glu6 and Glu13, and the interactions of Arg15 with Glu11 and Glu13′ (the prime discriminates equivalent residues between clockwise-related monomers when viewed from the N termini down the superhelical axis). The mean occupancies of the salt bridges displayed in Figure 4 and Table 1 show that the interactions, with two exceptions, are approximately symmetric among the three helices, which are initially identical in structure. Five of seven intrahelical and two of three interhelical salt bridges evidence greater overall occupancies in the trimer at the higher temperature, indicating stronger interactions. The salt bridges between Arg10 and Glu6 and between Arg8 and Glu6′ constitute two notable exceptions to this pattern. The range of the medians is less at the higher temperature and, thus, the symmetry among helices greater. The three i to i − 4 salt bridges exhibit higher occupancies than the remaining intrahelical salt bridges. Reflecting their dominance are the secondary structures of the trimer, consisting almost exclusively of α-helices with minor occurrences of β-bridge and H-bonded turns at the N termini (Fig. 1), and the pattern of intrahelical hydrogen bonds between main-chain atoms: 11 i to i − 4 bonds constitute between 70% and 90% of the configurations in each helix of the trimer at both temperatures (Supplemental Fig. S2). The interhelical salt bridges tend to exhibit higher occupancies than the intrahelical salt bridges.
Figure 3.

Simulated structures of ccβ-p trimer (2.7 ns) and monomer (38.8 ns) illustrating intrahelical and interhelical salt-bridge networks. In the trimer networks involve interactions of the glutamate residues with Arg3, Arg8, Arg10, and Arg15. In the monomer simultaneous salt bridges of Arg8 with Glu4 and Glu13 occur at 278°K.
Figure 4.

Median occupancies of salt bridges from Table 1. The triangles (apex down) denote the monomer at 278°K, the triangles (apex up) the monomer at 330°K, the three squares the helices of the trimer at 278°K, and the three diamonds the helices of the trimer at 330°K.
Table 1.
Occurrences (%) of dominant salt bridges of ccβ-p

Structural stability of the trimer is supported by analysis of potential energy (Table 2). The electrostatic energy between water and trimer increases slightly with temperature, indicating a weakening of the interaction due to decreasing solvation by water at higher temperature. The electrostatic energy among the atoms of the trimer decreases with temperature, which is consistent with strengthening of the interaction and increased configurational stability.
Table 2.
Means (standard deviations obtained by block averaging 5-ns intervals) of the energetic contributions in kJ/mol for the simulated trajectories of ccβ-p at 278°K and 330°K

Displaying the limit of thermostability, the trimer unfolds at 370°K differently in each helix. All interhelical salt bridges begin to disperse after 20 ns (Supplemental Fig. S3). Of the intrahelical i to i − 4 salt bridges, only two salt bridges between Arg8 and Glu4 and one salt bridge between Arg10 and Glu6 persist for the entire simulation of 100 ns. The secondary structure of all three helices shows a distinct transition from the dominant α-helical configurations after 16 ns. In two of the helices, the N-terminal heptad retains α-helical configurations longer than the C-terminal heptad, reflecting the persistence of the salt bridges between Arg8 and Glu4. Bends and extended structures dominate the unfolded configurations, with fewer occurrences of β-sheets and H-bonded turns. In two helices, π-helical configurations appear briefly.
Not observed experimentally due to its propensity to form the trimer, the ccβ-p monomer nevertheless offers a standard with which to compare simulations of the trimer. Figure 2 shows that the atom-positional RMSFs of the monomer, in contrast to those of the trimer, increase between 278°K and 330°K. This increase reflects the unfolding of the monomer at 330°K evident in the secondary structure (Fig. 5), which shows helical structure vanishing within 20 ns in a manner similar to the unfolding of the trimer at 370°K.
Figure 5.

Secondary structure of the ccβ-p monomer and atom-positional root-mean-square deviations of the backbone atoms from their positions in the initial crystal structure at 278°K (left) and 330°K (right). Green denotes α-helix, red H-bonded turn, yellow 310-helix, orange bend, cyan β-bridge, violet extended strand, and blue π-helix.
Analysis of the salt bridges reveals additional differences between monomer and trimer. Seven corresponding salt bridges occur in monomer and trimer, but the monomer displays systematically smaller occupancies than the trimer. Moreover, the occupancies of six of the bridges decrease with increased temperature in the monomer (Fig. 4; Table 1), whereas increases predominate in the trimer. Of the three i to i − 4 salt bridges associated with i to i − 4 hydrogen bonds (Supplemental Fig. S5) and helical structure in trimer and monomer, two, between Arg10 and Glu6 and between Arg15 and Glu11, decrease markedly with temperature, vanishing completely with the helical structure at 330°K (Supplemental Fig. S5). The third salt bridge between Arg8 and Glu4 evidences little influence on the helical structure.
Analysis of potential energy (Table 2) confirms the instability of the monomer. The electrostatic energy between water and monomer decreases slightly with temperature, consistent with a stronger interaction and more solvation as the exposed surface of the peptide increases during unfolding. The electrostatic energy among atoms of the monomer increases with temperature, indicating the weakening of the interaction as the monomer unfolds.
In addition to the seven salt bridges common to trimer and monomer, an i to i + 5 salt bridge between Arg8 and Glu13 emerges in the monomer at both temperatures and persists until the end of the simulations (Supplemental Fig. S5). This salt bridge coexists with the salt bridge between Arg8 and Glu4. Figure 3 shows that the side chains of Glu4, Arg8, and Glu13 can simultaneously accommodate the two salt bridges involving Arg8. These salt bridges appear at the analysis resolution of 0.5 ps to mediate the coexistence of several pairs of i to i − 4 and i − 5 hydrogen bonds, and associated mixed configurations of α- and π-helical secondary structure. Occurring very rarely in the trimer, the salt bridge between Arg8 and Glu13 in the monomer may presage the transformation to an interhelical interaction involving its interhelical analog Glu13′ in the trimer, suggesting a role of these residues in ccβ-p coiled-coil formation. Recently demonstrated to play an essential role in trimer formation of short, parallel coiled coils, this interhelical interaction is seen in the crystal structure of ccβ-p as well as in the structures of many other three-stranded coiled-coil domains (Kammerer et al. 2005).
Additional insight into the role of the salt bridges in the stability of the trimer and monomer of ccβ-p is provided by the analysis of configurational entropies shown in Table 3. In contrast to the atom-positional RMSFs, the configurational entropies of the backbone and side-chain atoms increase with temperature for the helices of the trimer and for the monomer. The increases for the monomer are substantially greater than for the trimer helices. The average entropy per atom of the backbones is noticeably smaller than the average entropy per atom of the side chains. Since the atom-positional RMSFs constitute the diagonal of the covariance matrix, the increase with temperature in configurational entropy while the RMSFs remain constant implies a decrease with temperature of correlations in the trimer helices.
Table 3.
Total internal and rotational configurational entropies (ir) of the side chains (sc) and complete backbone (bbe) of the monomer (ccβ-p) and the trimer (ccβ-p_a,b,c)

The configurational entropies per atom of the side chains are comparable for monomer and trimer. The entropies per atom of charged-residue side chains, i.e., the arginine and glutamate side chains involved in networks of salt bridges (Table 4), are distinctly less than those of the neutral-residue side chains (Table 5). We remark that the entropies per atom of all side chains (Table 3) are smaller than those of individual charged or neutral side chains because the former include the correlations among the side chains. Analysis also shows that pairs of glutamate and arginine residues involved in the salt bridges of the trimer evidence a much smaller decrease with temperature in magnitude of configurational entropies associated with correlation than analogous pairs of the monomer (Table 6); the magnitude of the correlation between Arg15 and Glu11 even increases with temperature in the trimer. Pairs of neutral residues involved in hydrophobic interactions evidence the same phenomena (Table 5). Moreover, the magnitudes of the correlations are systematically greater than those of the charged-residue pairs, and the magnitudes of three pairs near the C terminus are larger (Table 7).
Table 4.
Sum of internal and rotational configurational entropies (ir) of the side chains involving the charged residues, Arg and Glu, of the monomer (ccβ-p) and the trimer (ccβ-p_a,b,c)

Table 5.
Sum of internal and rotational configurational entropies (ir) of the side chains involving the neutral residues of the monomer (ccβ-p) and the trimer (ccβ-p_a,b,c)

Table 6.
Contributions of correlations (Equation 3) between intrahelical charged residue pairs to configurational entropies

Table 7.
Contributions of correlations between intrahelical neutral residue pairs to configurational entropies

The marked decreases of correlations and salt-bridge occupancies, as well the increases of entropy and RMSF with temperature in the monomer are expected, because it unfolds. The constancy or decrease of RMSF, the increase in salt-bridge occupancies, and the slight decrease in solute potential energy of the trimer indicate structural thermostability. That the correlations decrease and configurational entropies increase concurrently with these changes may appear at first paradoxical. The participation of the salt bridges in networks suggests a resolution. The creation of interhelical salt bridges implied by the formation of the trimer presents each charged residue with more accessible bridges than the monomer. The greater exposure to electrostatic fields due to desolvation facilitates increased occupation frequencies while increased thermal motion of the charged residues reduces pair correlations. The subtle interplay of these effects suggests the following proposition: The trimer, in contrast to the monomer, is conformationally stable at higher temperatures because the salt-bridge networks accommodate stochastically the disorder produced by thermal motion.
Discussion
Using MD simulations, we investigated the de novo, 17-residue peptide ccβ-p as a coiled coil of three α-helices and as an α-helical monomer at three temperatures: 278°K, 330°K, and 370°K. The intention of the design was the production of a stable coiled coil. However, the networks of salt bridges contributing to the structural stability of the coiled coil have also been proposed to promote configurational stability in thermophilic proteins, which we observe in the simulations of the trimer. This observation suggested the trimer as a model system for theoretical investigation of thermostability at the atomic scale; a complete free-energy calculation including solvent degrees of freedom, which would be required to establish thermodynamic stability, is beyond the scope of the simulations.
Three measures, atom-positional RMSFs, salt-bridge occupancies, and electrostatic interaction energies, support our hypothesis. The RMSFs of the trimer backbone and side-chain atoms, which provide the structural definition of thermostability, are essentially identical at 278°K and 330°K, whereas the fluctuations of the monomer backbone and side-chain atoms increase with temperature. The mean occupancies of the intrahelical salt bridges are systematically higher in the trimer than in the monomer. Five of seven intrahelical and two of three interhelical salt bridges evidence greater occupancy in the trimer at higher temperature, whereas six of eight monomer salt bridges show decreased occupancy at higher temperature. The interhelical salt bridges tend to exhibit higher occupancies than the intrahelical salt bridges, suggesting their contribution to the conformational stability of the trimer. Finally, the potential energies due to electrostatic interactions reflect stronger interaction among the atoms of the trimer and weaker interaction between the trimer and water at 330°K than at 278°K, whereas the monomer exhibits the opposite effect. These measures are consistent with previous explicit solvent MD simulations which indicated that desolvation at higher temperatures permits electrostatic interactions to emerge through salt bridges as a dominant factor in protein thermostability.
To supplement these structural measures, we investigated the configurational entropies of the three trimer helices and of the monomer. Insufficient convergence of the total trimer entropy and the restriction of entropy calculations to nondiffusive degrees of freedom, necessitated by theoretical limitations and computational requirements, prevented a more complete treatment of entropy. The configurational entropies reflect the greater structural stability of the trimer helices compared to the monomer. The configurational entropies of the backbone and side-chain atoms increase with temperature in both trimer and monomer, but the increase is markedly less in the trimer helices than in the monomer. Pairs of glutamate and arginine residues involved in the salt bridges of the trimer helices evidence distinctly larger configurational entropies associated with correlation and much smaller decreases with temperature than the corresponding pairs of the monomer. The same trend is evident in pairs of residues involved in hydrophobic interactions.
The increase of configurational entropy and decrease in correlations of the trimer helices at the higher temperature while the salt-bridge occupancies increase and atom-positional RMSFs remain constant appear to pose a paradox. However, the increased number of possible salt bridges provided by formation of the trimer, i.e., the addition of interhelical interactions, implies that desolvation and increased random motion of the charged residues can increase occupation frequencies while pair correlations decrease. Thus, our study of ccβ-p, designed to fold into a stable coiled coil with a well-packed hydrophobic core and an optimal number of intra- and interhelical surface salt bridges, suggests a mechanism in which optimally designed salt-bridge networks accommodate stochastically the disorder of increased thermal motion to produce thermostability.
Materials and Methods
Simulation setup
The five MD simulations employed the GROMOS96 program modules and the 43A1 force field (van Gunsteren et al. 1996; Daura et al. 1998b; Hünenberger and van Gunsteren 1999). The X-ray structure of ccβ-p (Protein Data Bank entry 1s9z), displayed in Supplemental Figure S1, supplied the initial coordinates of the peptides in the three-stranded α-helical coiled-coil trimer configuration (Kammerer et al. 2004).
For each simulation, the initial configuration placed the solute at the center of a periodic truncated octahedral box, requiring that the minimum distance from any atom of the peptide to the box walls exceed 1.4 nm. A cubic periodic array of 216 pre-equilibrated water molecules, modeled according to SPC water (Berendsen et al. 1981), provided the initial configuration of the solvent in the box so that the distance between an oxygen atom of water and nonhydrogen atoms of the solute exceeded 0.23 nm.
Relaxation of the solute-solvent contacts, while positionally restraining the solute atoms using a harmonic potential with a force constant of 250 kJ mol−1 nm−2, entailed a steepest-descent energy minimization of the system. A second steepest-descent energy minimization of the system without restraints followed to eliminate any residual strain; the energy minimizations terminated when the energy change per step became <0.1 kJ mol−1.
Sampling from a Maxwellian distribution at 100°K provided the initial velocities for the MD simulations. Solvent and solute were independently weakly coupled to a temperature bath with a relaxation time of 0.1 ps (Berendsen et al. 1984). The systems were also coupled to a pressure bath at 1 atm with a relaxation time of 0.5 ps and an isothermal compressibility of 0.4575 × 10−3 (kJ mol−1 nm−3)−1 (Berendsen et al. 1984). The SHAKE algorithm constrained bond lengths with a geometric tolerance of 10−4 (Ryckaert et al. 1977), so that the leapfrog integration time step could be set to 0.002 ps. Treating the nonbonded interactions employed a triple-range method with cutoff radii 0.8 nm and 1.4 nm. Outside the outer cutoff radius, a reaction field with a relative dielectric permittivity of 66.6 (Glättli et al. 2002) approximated the electrostatic interactions. Within the inner cutoff radius, the evaluation used a charge-group pair list. Short-range interactions entailed updating the list at every time step; interactions between pairs separated by distances >0.8 nm and <1.4 nm entailed updating only every fifth time step.
Analysis
Registration of trajectory coordinates and energies at 0.5-ps intervals served the analysis. Analysis procedures comprised: (1) calculations of atom-positional root-mean-square differences between initial and trajectory coordinates and root-mean-square fluctuations between trajectory coordinates and their mean over the entire simulation after translational superposition of the centers of mass of the backbone N, Cα, and C atoms of all but the N- and C-terminal residues; (2) assignment of secondary structure at 10-ps intervals according to the method of Kabsch and Sander (Kabsch and Sander 1983) using the program PROCHECK (Laskowski et al. 1993); (3) determination for the complete trajectories of hydrogen bonds as defined by a minimum donor–hydrogen–acceptor angle of 135° and a maximum hydrogen–acceptor distance of 0.25 nm; (4) determination for the complete trajectories of salt bridges as defined by a maximum hydrogen–acceptor distance of 0.6 nm; and (5) estimation of configurational entropies.
Evaluation of configurational entropies uses the procedure of Schlitter (Schlitter 1993) as implemented by Schäfer and colleagues (Schäfer et al. 2000, 2001), which estimates an upper bound for the solute configurational entropy, S, of a nondiffusive system with two assumptions: (1) harmonicity of the individual eigenmodes and (2) negligible supralinear correlations among eigenmodes (Schäfer et al. 2000; Baron et al. 2006)
 |
where kB is Boltzmann's constant, T the absolute temperature, e Euler's number, 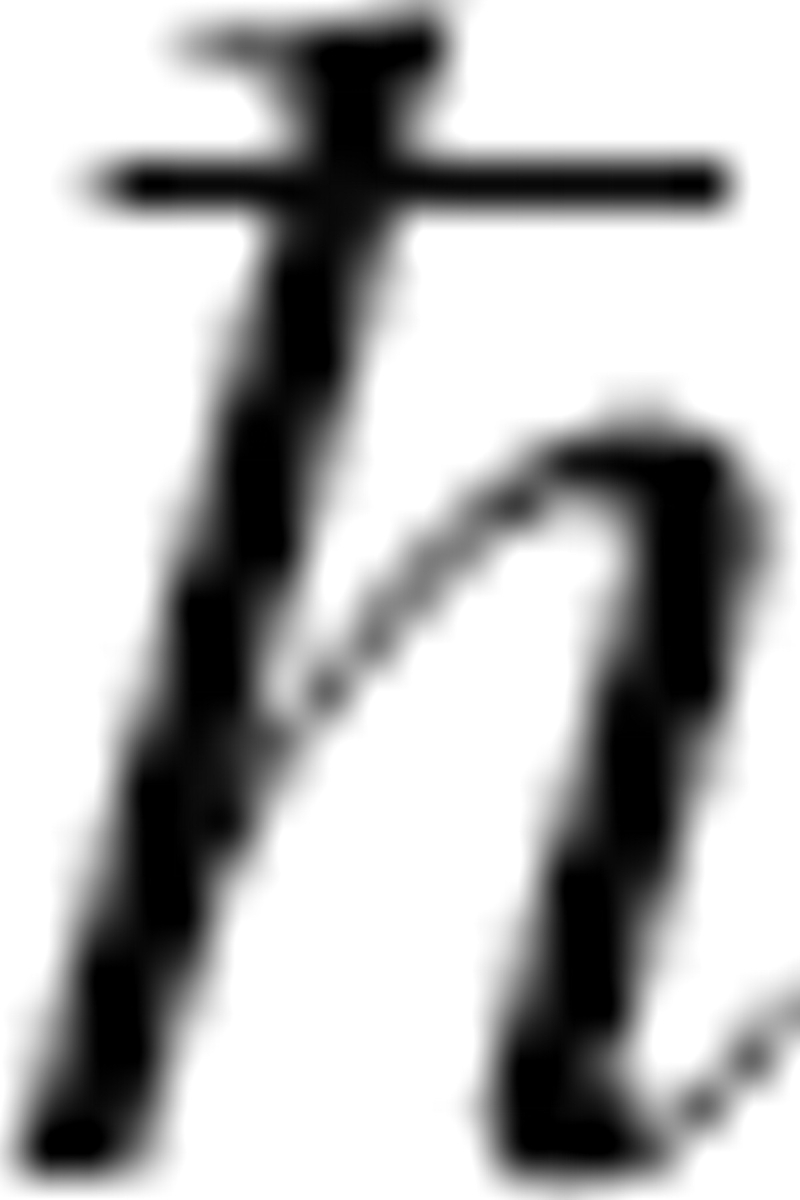 Planck's constant divided by 2π, and
Planck's constant divided by 2π, and  the mass-weighted covariance matrix of atom-configurational fluctuations in Cartesian space,
the mass-weighted covariance matrix of atom-configurational fluctuations in Cartesian space,
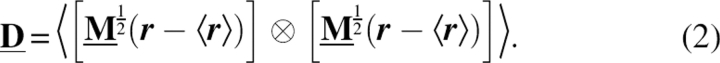 |
Solute configurations were sampled at 0.5-ps intervals, and entropies derived from the mass-weighted covariance matrix recalculated every nanosecond.  is the 3N-dimensional diagonal matrix containing the N masses of the subset of N atoms for which the entropy is calculated. r is the 3N-dimensional vector of Cartesian coordinates describing a solute configuration after subtracting the translational degrees of freedom relative to a reference configuration via translational superposition of the centers of mass. The subtraction thus yields the upper bound of the configurational entropy due to internal (“i”) and rotational (“r”) degrees of freedom, denoted by the superscript “ir.” The first configuration of the analyzed trajectory provided the reference configuration. For calculating the configurational entropies, the least-squares fit included the N, Cα, and C atoms of the backbone of all but the N- and C-terminal residues, consistent with the calculations of RMSD and RMSF. Also consistent with the latter calculations, the entropies of the complete backbone omitted contributions of oxygen and hydrogen atoms. The per-atom entropies of interest, derived by dividing by the number of heavy atoms (C, N, O), are little affected by this approximation, since the hydrogen atoms contribute little to the entropies. The configurational entropy due to correlated motion of two residues (i) and (j) is defined (Schäfer et al. 2001)
is the 3N-dimensional diagonal matrix containing the N masses of the subset of N atoms for which the entropy is calculated. r is the 3N-dimensional vector of Cartesian coordinates describing a solute configuration after subtracting the translational degrees of freedom relative to a reference configuration via translational superposition of the centers of mass. The subtraction thus yields the upper bound of the configurational entropy due to internal (“i”) and rotational (“r”) degrees of freedom, denoted by the superscript “ir.” The first configuration of the analyzed trajectory provided the reference configuration. For calculating the configurational entropies, the least-squares fit included the N, Cα, and C atoms of the backbone of all but the N- and C-terminal residues, consistent with the calculations of RMSD and RMSF. Also consistent with the latter calculations, the entropies of the complete backbone omitted contributions of oxygen and hydrogen atoms. The per-atom entropies of interest, derived by dividing by the number of heavy atoms (C, N, O), are little affected by this approximation, since the hydrogen atoms contribute little to the entropies. The configurational entropy due to correlated motion of two residues (i) and (j) is defined (Schäfer et al. 2001)
where the entropy Sir bb(i + j) denotes the total entropy of the atoms in the subset composed of (i) and (j).
Other plausible choices of atoms with which to perform the superposition had little effect on the calculated entropies, nor did removal of the rotational degrees of freedom via a least-squares fit, yielding only the contribution of the internal degrees of freedom. The contribution of rotation to the configurational entropies of all atoms constituted ∼1% of the sum. For the coiled coil, each helix of the trimer was fit independently (the same procedure used in computing RMSD and RMSF), neglecting the contribution to the entropy due to the relative motion of the helices. In contrast to the contributions of the individual helices, this latter contribution did not converge completely during the simulations and is, therefore, not treated in this analysis. Convergence of the configurational energies of the individual helices of monomer and trimer is shown in Supplemental Figure S6.
Validation
The quality of the simulated properties presented here will depend on (1) the quality of the theory or model, (2) the accuracy of the interatomic interaction function or force field used, (3) the degree of sampling, statistics, and convergence reached in the simulations, (4) the quality of the simulation software, and (5) how competently the simulation software is used (van Gunsteren and Mark 1998). The theory used is standard equilibrium statistical mechanics (calculation of entropy as discussed by Baron et al. 2006) and continuum electrostatics to evaluate long-range forces (Smith and van Gunsteren 1993; Tironi et al. 1997; Hünenberger et al. 2001). The GROMOS force field used here has been tested by comparison of simulated with measured average properties for a variety of systems (Oostenbrink et al. 2004), and the high temperature stability of salt bridges has been reproduced (de Bakker et al. 1999). A reduction of the structural stability of ankyrin proteins upon mutation of charged residues has been correctly predicted (Yu et al. 2006). Water sites in crystals of the protein BPTI were correctly simulated (Schiffer and van Gunsteren 1999). Structural changes in the protein α-lactalbumin upon a change of pH were simulated in agreement with NMR data (Smith and van Gunsteren 1993). The effects of pH (Gee and van Gunsteren 2006) or the presence of charged side chains (Glättli et al. 2005) upon the folding equilibria of β-polypeptides in methanol solution were found to be simulated in agreement with experimental NMR and CD data. Temperature and pH effects on the configurational stability of helix-forming polypeptides in aqueous solution were simulated and compared to experimental data (Missimer et al. 2005). These studies indicate that the GROMOS force field is able to predict conformational properties as a function of changes in the environment of a particular solute molecule. Regarding the third validation issue, the properties shown as a function of time (van Gunsteren et al. 1995) in the figures confirm that sufficient statistics can be obtained within 100 ns of simlulation for the relatively small peptides considered. Finally, we note that the GROMOS software (van Gunsteren et al. 1996; Scott et al. 1999; Christen et al. 2005) is widely used and hence thoroughly tested.
Electronic supplemental material
A supplemental document file contains six figures.
Acknowledgments
We thank Indira Chandrasekhar, Chris Oostenbrink, Christine Peter, Alice Glättli, Markus Christen, Mika Kastenholz, and Tim Heinz for assistance. We thank Philippe Hünenberger for critically reading the manuscript and making many useful suggestions. Financial support by the National Center of Competence in Research (NCCR) Structural Biology of the Swiss National Science Foundation (SNSF) is gratefully acknowledged.
Footnotes
Supplemental material: see www.proteinscience.org
Reprint requests to: John H. Missimer, Biomolecular Research, Structural Biology, Paul Scherrer Institut, CH-5232 Villigen PSI, Switzerland; e-mail: john.missimer@psi.ch; fax: 41-56-310-5288.
Abbreviations: MD, molecular dynamics; RMSD, root-mean-square deviation; RMSF, root-mean-square fluctuation; ccβ-p, de novo 17-residue peptide.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.062542907.
References
- Baron R., van Gunsteren, W.F., and Hünenberger, P.H. 2006. Estimating the configurational entropy from molecular dynamics simulations: Anharmonicity and correlation corrections to the quasi-harmonic approximation. Trends in Physical Chemistry (in press).
- Berendsen H.J.C., Postma, J.P.M., van Gunsteren, W.F., and Hermans, J. 1981. Interaction models for water in relation to protein hydration. In Intermolecular forces (ed. B. Pullman), pp. 331–342. Reidel, Dordrecht, Germany.
- Berendsen H.J.C., Postma, J.P.M., van Gunsteren, W.F., DiNola, A., and Haak, J.R. 1984. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81: 3684–3690. [Google Scholar]
- Cambillau C. and Claverie, J.M. 2000. Structural and genomic correlates of hyperthermostability. J. Biol. Chem. 275: 32383–32386. [DOI] [PubMed] [Google Scholar]
- Christen M., Hünenberger, P.H., Bakowies, D., Baron, R., Bürgi, R., Geerke, D.P., Heinz, T.N., Kastenholz, M.A., Kräutler, V., Oostenbrink, C., et al. 2005. The GROMOS software for biomolecular simulation: GROMOS05. J. Comput. Chem. 26: 1719–1751. [DOI] [PubMed] [Google Scholar]
- Daura X., Jaun, B., Seebach, D., van Gunsteren, W.F., and Mark, A.E. 1998a. Reversible peptide folding in solution by molecular dynamics simulation. J. Mol. Biol. 280: 925–932. [DOI] [PubMed] [Google Scholar]
- Daura X., Mark, A.E., and van Gunsteren, W.F. 1998b. Parametrization of aliphatic CHn united atoms of GROMOS96 force field. J. Comput. Chem. 19: 535–547. [Google Scholar]
- de Bakker P.I.W., Hünenberger, P.H., and McCammon, J.A. 1999. Molecular dynamics simulations of the hyperthermophilic protein sac7d from Sulfolobus acidocaldarius: Contribution of salt bridges to thermostability. J. Mol. Biol. 285: 1811–1830. [DOI] [PubMed] [Google Scholar]
- Dobson C.M. 2003. Protein folding and misfolding. Nature 426: 884–890. [DOI] [PubMed] [Google Scholar]
- Duan Y. and Kollman, P.A. 1998. Pathways to a protein folding intermediate observed in a 1-microsecond simulation in aqueous solution. Science 282: 740–744. [DOI] [PubMed] [Google Scholar]
- Gee P.J. and van Gunsteren, W.F. 2006. Terminal-group effects on the folding behaviour of selected beta-peptides. Proteins 63: 136–143. [DOI] [PubMed] [Google Scholar]
- Glättli A., Daura, X., and van Gunsteren, W.F. 2002. Derivation of an improved simple point charge model for liquid water: SPC/A and SPC/L. J. Chem. Phys. 116: 9811–9828. [Google Scholar]
- Glättli A., Daura, X., Bindschädler, P., Jaun, B., Mahajan, Y.R., Mathad, R.I., Rueping, M., Seebach, D., and van Gunsteren, W.F. 2005. On the influence of charged side chains on the folding-unfolding equilibrium of beta-peptides: A molecular dynamics simulation study. Chemistry 11: 7276–7293. [DOI] [PubMed] [Google Scholar]
- Hummer G., Garcia, A.E., and Garde, S. 2001. Helix nucleation kinetics from molecular simulations in explicit solvent. Proteins 42: 77–84. [PubMed] [Google Scholar]
- Hünenberger P.H. and van Gunsteren, W.F. 1999. Empirical classical force fields for molecular systems, Springer-Verlag Berlin.
- Hünenberger P.H., Börjesson, U., and Lins, R.D. 2001. Electrostatic interactions in biomolecular systems. Chimia (Aarau) 55: 861–866. [Google Scholar]
- Jaenicke R. and Bohm, G. 1998. The stability of proteins in extreme environments. Curr. Opin. Struct. Biol. 8: 738–748. [DOI] [PubMed] [Google Scholar]
- Kabsch W. and Sander, C. 1983. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22: 2577–2637. [DOI] [PubMed] [Google Scholar]
- Kammerer R.A. and Steinmetz, M.O. 2006. De novo design of a two-stranded coiled-coil switch peptide. J. Struct. Biol. 155: 146–153. [DOI] [PubMed] [Google Scholar]
- Kammerer R.A., Kostrewa, D., Zurdo, J., Detken, A., Garcia-Echeverria, C., Green, J.D., Müller, S.A., Meier, B.H., Winkler, F.K., Dobson, C.M., et al. 2004. Exploring amyloid formation by a de novo design. Proc. Natl. Acad. Sci. 101: 4435–4441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kammerer R.A., Kostrewa, D., Progias, P., Honnappa, S., Avila, D., Lustig, A., Winkler, F.K., Pieters, J., and Steinmetz, M.O. 2005. A conserved trimerization motif controls the topology of short coiled coils. Proc. Natl. Acad. Sci. 102: 13891–13896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karshikoff A. and Ladenstein, R. 2001. Ion pairs and the thermotolerance of proteins from hyperthermophiles: A “traffic rule” for hot roads. Trends Biochem. Sci. 26: 550–556. [DOI] [PubMed] [Google Scholar]
- Kumar S. and Nussinov, R. 2001. How do thermophilic proteins deal with heat? Cell. Mol. Life Sci. 58: 1216–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski R.A., MacArthur, M.W., Moss, D.S., and Thornton, J.M. 1993. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26: 283–291. [Google Scholar]
- Missimer J.H., Steinmetz, M.O., Jahnke, W., Winkler, F.K., van Gunsteren, W.F., and Daura, X. 2005. Molecular dynamics simulations of helical 16-residue C- and N-terminal peptide derivatives of GCN4-p1 in aqueous solution. Chem. Biodivers. 2: 1086–1104. [DOI] [PubMed] [Google Scholar]
- Oostenbrink C., Villa, A., Mark, A.E., and van Gunsteren, W.F. 2004. A biomolecular force field based on the free enthalpy of hydration and solvation: The GROMOS force-field parameter sets 53A5 and 53A6. J. Comput. Chem. 25: 1656–1676. [DOI] [PubMed] [Google Scholar]
- Ryckaert J.-P., Ciccotti, G., and Berendsen, H.J.C. 1977. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 23: 327–341. [Google Scholar]
- Sanchez-Ruiz J.M. and Makhatadze, G.I. 2001. To charge or not to charge? Trends Biotechnol. 19: 132–135. [DOI] [PubMed] [Google Scholar]
- Schäfer M., Bartels, C., and Karplus, M. 1998. Solution conformations and thermodynamics of structured peptides: Molecular dynamics simulation with an implicit solvation model. J. Mol. Biol. 284: 835–848. [DOI] [PubMed] [Google Scholar]
- Schäfer H., Mark, A.E., and van Gunsteren, W.F. 2000. Absolute entropies from molecular dynamics simulation trajectories. J. Chem. Phys. 113: 7809–7817. [Google Scholar]
- Schäfer H., Daura, X., Mark, A.E., and van Gunsteren, W.F. 2001. Entropy calculations on a reversibly folding peptide: Changes in solute free energy cannot explain folding behavior. Proteins 43: 45–56. [DOI] [PubMed] [Google Scholar]
- Schiffer C.A. and van Gunsteren, W.F. 1999. Accessibility and order of water sites in and around proteins: A crystallographic time-averaging study. Proteins 36: 501–511. [PubMed] [Google Scholar]
- Schlitter J. 1993. Estimation of absolute and relative entropies of macromolecules using the covariance-matrix. Chem. Phys. Lett. 215: 617–621. [Google Scholar]
- Scott W.R.P., Hünenberger, P.H., Tironi, I.G., Mark, A.E., Billeter, S.R., Fennen, J., Torda, A.E., Huber, T., Krüger, P., and van Gunsteren, W.F. 1999. The GROMOS biomolecular simulation package. J. Phys. Chem. A. 103: 3596–3607. [Google Scholar]
- Simmerling C., Strockbine, B., and Roitberg, A.E. 2002. All-atom structure prediction and folding simulations of a stable protein. J. Am. Chem. Soc. 124: 11258–11259. [DOI] [PubMed] [Google Scholar]
- Smith P.E. and van Gunsteren, W.F. 1993. Methods for the evaluation of long-range electrostatic forces in computer simulations of molecular systems. In Computer simulation of biomolecular systems, theoretical and experimental applications (eds. W.F. van Gunsteren et al.). Vol. Vol. 2, pp. 182–212. Escom Science Publishers, Leiden, The Netherlands. [Google Scholar]
- Smith L.J., Dobson, C.M., and van Gunsteren, W.F. 1999. Molecular dynamics simulations of human α-lactalbum. Changes to the structural and dynamical properties of the protein at low pH. Proteins 36: 77–86. [DOI] [PubMed] [Google Scholar]
- Snow C.D., Nguyen, H., Pande, V.S., and Gruebele, M. 2002. Absolute comparison of simulated and experimental protein-folding dynamics. Nature 420: 102–106. [DOI] [PubMed] [Google Scholar]
- Szilagyi A. and Zavodszky, P. 2000. Structural differences between mesophilic, moderately thermophilic and extremely thermophilic protein subunits: Results of a comprehensive survey. Struct. Fold. Des. 8: 493–504. [DOI] [PubMed] [Google Scholar]
- Takano M., Yamato, T., Higo, J., Suyama, A., and Nagayama, K. 1999. Molecular dynamics of a 15-residue poly(L-alanine) in water: Helix formation and energetics. J. Am. Chem. Soc. 121: 605–612. [Google Scholar]
- Thomas A.S. and Elcock, A.H. 2004. Molecular simulations suggest protein salt bridges are uniquely suited to life at high temperatures. J. Am. Chem. Soc. 126: 2208–2214. [DOI] [PubMed] [Google Scholar]
- Tironi I.G., Luty, B.A., and van Gunsteren, W.F. 1997. Space-time correlated reaction field: A stochastic dynamical approach to the dielectric continuum. J. Chem. Phys. 106: 6068–6075. [Google Scholar]
- van Gunsteren W.F. and Mark, A.E. 1998. Validation of molecular dynamics simulation. J. Chem. Phys. 108: 6109–6116. [Google Scholar]
- van Gunsteren W.F., Hünenberger, P.H., Mark, A.E., Smith, P.E., and Tironi, I.G. 1995. Computer simulation of protein motion. Comput. Phys. Commun. 91: 305–319. [Google Scholar]
- van Gunsteren W.F., Billeter, S.R., Eising, A.A., Hünenberger, P.H., Krüger, P., Mark, A.E., Scott, W.R.P., and Tironi, I.G. 1996. Biomolecular simulation: The GROMOS96 manual and user guide. Vdf Hochschulverlag AG an der ETH Zürich, Zürich, Switzerland.
- Yu H., Kohl, A., Binz, H.K., Plückthun, A., Grütter, M.G., and van Gunsteren, W.F. 2006. Molecular dynamics study of the stabilities of consensus designed ankyrin repeat proteins. Proteins 65: 285–295. [DOI] [PubMed] [Google Scholar]