

Abstract
Protein G-related albumin-binding (GA) modules occur on the surface of numerous Gram-positive bacterial pathogens and their presence may promote bacterial growth and virulence in mammalian hosts. We recently used phage display selection to evolve a GA domain, PSD-1 (phage selected domain-1), which tightly bound phylogenetically diverse albumins. With respect to PSD-1’s broad albumin binding specificity, it remained unclear how the evolved binding epitope compared to those of naturally occurring GA domains and whether PSD-1’s binding mode was the same for different albumins. We investigate these questions here using chemical shift perturbation measurements of PSD-1 with rabbit serum albumin (RSA) and human serum albumin (HSA) and put the results in the context of previous work on structure and dynamics of GA domains. Combined, these data provide insights into the requirements for broad binding specificity in GA-albumin interactions. Moreover, we note that using the phage-optimized PSD-1 protein significantly diminishes the effects of exchange broadening at the binding interface between GA modules and albumin, presumably through stabilization of a ligand-bound conformation. The employment of artificially evolved domains may be generally useful in NMR structural studies of other protein–protein complexes.
Keywords: albumin, protein G, host–pathogen interactions, NMR
Protein G is a multidomain protein residing on the surface of group C and group G streptococci that may play a role in bacterial growth (de Chateau et al. 1996), virulence, and evasion of the host's immune response system. It contains both IgG-Fc and albumin binding domains that are denoted here as GB and GA, respectively. At least 16 GA domains with relatively high sequence identity have been identified in numerous bacterial species (Johansson et al. 2002a). The presence of these domains may allow infecting bacteria to coat their surfaces with host proteins in mammalian plasma. In particular, the GA domain of the commensal bacteria Peptostreptococcus magnus appears to be exclusively associated with pathogenic strains isolated from patients with localized suppurative infections (de Chateau et al. 1996). Thus, the interaction between GA domains and albumin may play an important role in the pathogenesis of these organisms.
The 3D structures of two naturally occurring GA domains, ALB8–GA (Johansson et al. 1997), and G148–GA3 (Johansson et al. 2002a), have been determined by solution NMR. ALB8–GA is essentially specific for human serum albumin (HSA) with 1000-fold lower affinity for guinea pig serum albumin (GPSA), while G148–GA3 binds the phylogenetically distinct HSA and GPSA almost equally well. NMR dynamics studies have shown that G148–GA3 is considerably more flexible than ALB8–GA, and it has been suggested that this might be the reason for the broader species specificity of G148–GA3 (Johansson et al. 2002b).
Recently, we used offset recombinant-PCR to shuffle seven representatives of GA sequence space and scanned the phage-displayed domains for mutants that bound to both HSA and GPSA (Rozak et al. 2006). We found a strong preference for a single recombinant domain, PSD-1 (phage-selected domain-1), regardless of whether the phages were enriched for their abilities to bind one or both of the albumins. Compared with G148–GA3, PSD-1 exhibits fourfold tighter binding to HSA and 10-fold tighter binding to GPSA. Moreover, in contrast to the earlier studies with G148–GA3, this tight binding to diverse albumins is maintained even though PSD-1 has a relatively rigid backbone comparable to that of ALB8–GA, as determined from 15N relaxation studies (Rozak et al. 2006).
There are 17 amino acid differences between PSD-1 and G148–GA3 (Fig. 1A). Most of these residues are solvent accessible, yet only two seem to have direct interaction with the putative albumin contact surface based on a 2.7 Å X-ray structure of ALB8–GA with HSA (Lejon et al. 2004). Relative to G148–GA3, one residue difference represents the loss of a H-bond to albumin (G26S) while the other is a possible gain of a H-bond (S44A) (Fig. 1A). These interactions do not appear sufficient to explain the enhanced binding to albumin.
Figure 1.

(A) Alignment of PSD-1, streptococcal G148–GA3, and Finegoldia magna ALB8–GA albumin-binding domains. Numbering is for PSD-1 residues. Identical residues are in gray or black, corresponding to conservation across two or three sequences, respectively. Amino acids implicated at the albumin-binding interface according to the ALB8–GA/HSA X-ray structure are indicated with asterisks. (B) Sections from 15N HSQC spectra of albumins complexed with [15N]-PSD-1. Cross-peaks due to free PSD-1 are black and those due to albumin-complexed PSD-1 are red. (C) Histogram plots of chemical shift perturbation for PSD-1 with RSA (top) and HSA (bottom). The secondary structure regions (filled rectangles) and albumin binding epitope (filled circles) are indicated at the top of the figure. The epitope is based on corresponding GA module residues contacting albumin in the X-ray structure of the ALB8–GA/HSA complex (Lejon et al. 2004). Asterisks indicate positions for which Δδ could not be determined unambiguously due to overlapping or weak signals in the complex. Arrows indicate exchange broadened signals. (D) Ribbon representations of the uncomplexed PSD-1 NMR structure (He et al. 2006) are used to highlight RSA (top) and HSA (bottom) complexed PSD-1 residues with significant Δδ in red. Epitope residues for which Δδ could not be determined unambiguously are rendered in blue.
To better understand the reasons for PSD-1’s improved binding to diverse albumins, we report here the epitope map of PSD-1 with a range of albumins using chemical shift perturbation. In combination with earlier analysis of GA structure and dynamics, the results provide insight into the reasons for PSD-1’s broad specificity. The data offer a substantially clearer picture of the GA–albumin interface in solution than earlier NMR studies, and facilitate interpretation of the interactions of other GA domains with albumin. We also note that exchange broadening at the binding interface between GA modules and albumin is significantly diminished using the phage-optimized PSD-1 protein. The employment of artificially evolved domains may be useful in mitigating this effect in NMR structural studies of other protein–protein complexes.
Results and Discussion
Binding of PSD-1 to RSA
Rabbit serum albumin (RSA) was used in the initial experiments on epitope mapping of the PSD-1 albumin-binding module. This allowed for direct comparison with earlier efforts to map the RSA-binding epitopes of ALB8–GA and G148–GA3 (Johansson et al. 2002a). The binding of RSA to GA modules tends to be in the micromolar range, and signals due to free and bound 15N-labeled GA domains are in fast exchange on the NMR timescale. A similar PSD-1/RSA micromolar interaction makes assignment of resonances due to bound PSD-1 relatively straightforward. Experiments were carried out by titrating unlabeled RSA into a solution of 15N-labeled PSD-1 and recording 15N HSQC spectra at different PSD-1/RSA ratios. Spectra were acquired at 37°C and 42°C to facilitate assignment in cases where there was overlap. Shift perturbations (Fig. 1B) were plotted as a function of residue number, and the results are summarized in Figure 1C.
The PSD-1 residues with the largest changes in chemical shifts (Δδ > 0.2 ppm) on RSA binding are Y28, Y29, and L32 in the α2 helix, A36 in the α2–α3 loop, and G41, S44, and L45 in the α3 helix. Four residues (E2, A15, A16, and K18) had overlapping signals in the complex, and Δδ could not be determined unambiguously. Four other residues, G26 in the α1–α2 loop, K37 and T38 in the α2–α3 loop, and E40 at the N terminus of the α3-helix, were exchange broadened in the complex. Thus, the most perturbed resonances indicate a binding epitope that contains residues from the α1–α2 loop, α2 helix, α2–α3 loop, and the N-terminal half of the α3 helix (Fig. 1D). This is generally consistent with the crystallographic epitope for ALB8–GA with HSA (Lejon et al. 2004), although the X-ray structure also indicates extensive contacts along the entire length of the α3-helix.
Earlier work indicated that the free and RSA-bound states of G148–GA3 and ALB8–GA are also in fast exchange (Johansson et al. 2002a). The shift perturbations are considerably larger in the PSD-1/RSA complex (largest Δδ ∼0.5 ppm) than in previous studies (largest Δδ ∼0.15 ppm), presumably due in part to the use of a 1:2 GA/RSA complex here, whereas a 1:0.6 ratio was used in earlier studies. Saturation was reached in the titration of PSD-1 with RSA. There were no further changes in PSD-1 chemical shifts in going from a 1:1.5 ratio to a 1:2 ratio (see Materials and Methods). The backbone amide resonances of PSD-1 G26, K37, T38, and E40 and the resonances of the corresponding amides in ALB8–GA and G148–GA3 (Johansson et al. 2002a) are similarly exchange broadened in the RSA complex. Overall, however, more residues have detectable shifts in the epitope region of the PSD-1/RSA complex given that only four resonances are exchange broadened in the complex compared with nine in the ALB8–GA/RSA complex and 11 in the G148–GA3/RSA complex. Most of the exchange-broadened signals in the ALB8–GA/RSA complex are in the α1–α2 loop, α2–α3 loop, and N-terminal part of α3. In the G148–GA3/RSA complex, further exchange broadening is also observed for α2 helical resonances.
The extensive exchange broadening observed in these earlier studies made it very difficult to accurately define the albumin binding epitope. The shift perturbation results obtained with PSD-1 and RSA therefore provide a more complete picture of the epitope in solution. These results also highlight differences in GA–albumin interactions within this epitope, as evidenced by varying degrees of exchange broadening despite similar binding affinities. It is interesting to note that even in the X-ray structure of the ALB8–GA/HSA complex there is some suggestion of conformational disorder at the binding interface since the positions of the Cγ and Cδ carboxylate atoms of the E40 (residue 41 in PSD-1) side chain are not defined.
Binding of PSD-1 to HSA and GPSA
Epitope mapping to other albumins was also investigated. Binding of PSD-1 to both HSA and GPSA is tight (Kd ∼10−8 M) (Rozak et al. 2006) and exchange between the free and bound forms of PSD-1 is slow on the NMR timescale. HSQC spectra of [2H/15N]-PSD-1 complexed to HSA and GPSA indicated very similar patterns of shifts (Fig. 1B) and so we focused on obtaining assignments for the HSA-bound form of PSD-1. Backbone amide assignments were made for [2H/13C/15N]–labeled PSD-1 in the 76 kDa complex with HSA using TROSY triple resonance experiments (Pervushin et al. 1997). Of the 54 main-chain amides in PSD-1, 44 were assigned in the complex. The remaining 10 were either overlapped or did not give correlations with adequate signal to noise to provide unambiguous connectivities. A number of residues in the C-terminal half of the α3-helix could not be assigned unambiguously so the pattern of shift perturbation was not established for this region. The assignment of I49 was made based on canonical chemical shifts expected for the Cα and Cβ resonances of this amino acid type.
Overall, the chemical shift perturbations (Fig. 1C) are significantly larger for PSD-1/HSA than PSD-1/RSA. Twenty-seven residues have Δδ > 0.2 ppm in the PSD-1/HSA complex compared with seven residues with Δδ > 0.2 ppm in the PSD-1/RSA complex. Although the shift perturbation values are bigger in the HSA complex, the largest changes in chemical shifts map to essentially the same region of the RSA-complexed domain (Fig. 1D). Three of the four signals that are exchange broadened in PSD-1/RSA (G26, T38, E40) are observed in PSD-1/HSA and give large chemical shift perturbations on complex formation. For G26 and T38, these large shifts likely correspond with direct H-bond interactions between main-chain amide protons in the GA module and acidic residues in HSA, as seen for the equivalent residues in the X-ray structure of the ALB8–GA/HSA complex. It is also interesting to note that several of the relevant acidic residues are not present in RSA, and this may in part be responsible for its generally lower affinity for GA modules.
The most highly conserved GA family surface residues found within the albumin-binding epitope of PSD-1 are A36, K37, and T38 in the α2–α3 loop and E40 in the N terminus of the α3-helix. In addition, residues L45 and I49 in the α3-helix are apolar in all GA variants characterized to date (Rozak et al. 2006). Most of these highly conserved residues (A36, T38, E40, and L45) also have strongly perturbed main-chain amide signals in the PSD-1/HSA and RSA complexes. While G26 also shifts significantly on HSA binding, it is not highly conserved. As noted above, this residue interacts with albumin through a main-chain contact. Other surface residues such as Y28 and L32 in the α2-helix also display significant shifts in one or both of the complexes. Residues 28 and 32 are more variable across GA modules, and the identity of the residue at position 28 has been proposed to play a role in species specificity (Lejon et al. 2004; He et al. 2006). Indeed, Y28 is unusual in that it shifts more in the complex with RSA (Δδ 0.31 ppm) than with HSA (Δδ 0.03 ppm), perhaps reflecting less optimal interactions with its contact points on the surface of HSA. Earlier studies showed that when this position contains a Phe, as in ALB8–GA, an interaction with a hydrophobic pocket on HSA is detected in the crystal structure of the complex (Lejon et al. 2004). This hydrophobic pocket may be disrupted in RSA with a M329K mutation, which could contribute to the observed changes in shift perturbations.
Conclusions
The results presented here show that an artificially evolved GA module, PSD-1, uses essentially the same binding epitope to interact with phylogenetically diverse albumins. The data also facilitate interpretation of earlier shift perturbation results with ALB8–GA and G148–GA3, and suggest that all three GA domains have similar albumin-binding epitopes in solution. The binding interface is in general agreement with that determined crystallographically for the ALB8–GA/HSA complex. Our results provide further support to the hypothesis that PSD-1’s enhanced affinities for HSA and GPSA are due to structural reasons rather than any change in binding mode or flexibility. This is not to say that flexibility is unimportant; it may be important for G148–GA3’s affinity for diverse albumins, for example, but it is not a requirement for broad specificity in the particular case of PSD-1. In comparing the various GA structures available, we note that the NMR structure of free PSD-1 (He et al. 2006) most closely resembles the albumin-bound form of ALB8-GA (Lejon et al. 2004), indicating that the mutations made in generating PSD-1 stabilize a bound state conformation. The differences in backbone structure between free and bound GA domains are small with a displacement of the α3-helix of ∼1–1.5 Å (He et al. 2006), but these subtle effects appear to lead to changes in albumin affinity that can have a dramatic effect on species specificity.
Of further note, the considerable exchange-broadening effects observed for the naturally occurring GA domains ALB8–GA and G148–GA3 with RSA were mitigated by using the PSD-1 phage selected domain, even though the domain was originally selected for optimal binding to other related albumins (HSA and GPSA). These results suggest that artificially evolved domains may be generally useful in NMR structural studies of protein–protein binding epitopes. Often high-resolution NMR studies of complexes are confounded by exchange broadening such that signals due to key residues at the contact surface between two proteins are not observable. This can happen when binding is relatively weak (e.g., Parsons et al. 2005), but has also been observed in cases where the binding interaction is quite tight (e.g., Sari et al. 2007). One can experiment with temperature, pH, and salt conditions, for example, to alter the exchange kinetics, and this sometimes leads to improvements in spectra. An additional strategy that may be worthwhile is to optimize stability and binding through phage display selection. The results presented here indicate that this approach can lead to significantly less exchange broadening and improved NMR spectra, presumably through stabilization of a ligand-bound conformation.
Materials and Methods
Sample preparation
PSD-1 was cloned into a pG58 vector as described previously (Rozak et al. 2006). In this approach, the protein is expressed as a fusion with subtilisin propeptide and, subsequently, purified and cleaved on an immobilized subtilisin column (Ruan et al. 2004). Escherichia coli BL21(DE3) competent cells were transformed with the pG58/PSD-1 plasmid and grown at 37°C in 1 L of labeled minimal media containing 100 μg/mL of ampicillin. Three samples of PSD-1, [15N]-labeled, [2H/15N]-labeled, and [2H/13C/15N]-labeled, were prepared. For the [2H/15N]- and [2H/13C/15N]-labeled samples, the concentration of D2O in minimal media was increased gradually so that cells could adapt to the change in isotopic environment. The level of deuterium incorporation in these samples was estimated to be >90%. At an OD600 of ∼0.8, the cells were induced with 1 mM IPTG for 4 h. Cells were harvested by centrifugation, and the cell pellet was resuspended in buffer containing 100 mM potassium phosphate at pH 7.0, 30 mg/mL of DNaseI, and 0.1 mM PMSF, for sonication on ice. After centrifugation for 1 h at 100,000g, the cleared lysate was loaded onto a S189 column (5-mL bed volume) and purified using the method of Ruan et al. (2004). The eluent containing purified PSD-1 was dialyzed against 2 mM ammonium bicarbonate buffer (pH 7.0) at 4°C overnight and then lyophilized. NMR samples of PSD-1 were prepared at a concentration of 0.2–0.4 mM in 50 mM potassium phosphate buffer at pH 7.0 with 0.1 mM EDTA.
NMR spectroscopy
NMR spectra were acquired on a Bruker AVANCE-600 spectrometer equipped with a z-axis gradient triple resonance (1H/13C/15N) cryoprobe. Data were processed with nmrPipe (Delaglio et al. 1995) and analyzed using Sparky (T.D. Goddard and D.G. Kneller, University of California, San Francisco). Titrations of PSD-1 with RSA were carried out by adding a solution of [15N]-labeled PSD-1 to lyophilized RSA, followed by acquisition of HSQC spectra at 37°C and 42°C. Molar PSD-1/RSA ratios of 1:0.5, 1:1, 1:1.5, and 1:2 provided a series of spectra from which assignment of shift perturbations were made. HSQC spectra were also recorded at 37°C and 42°C on complexes of [2H/15N]-PSD-1 with HSA and GPSA using PSD-1/albumin ratios of 1:1, 1:1.5, and 1:2. Optimal spectra were obtained at a ratio of 1:2, and these conditions were used for triple-resonance data collection. TROSY (Pervushin et al. 1997) versions of the following experiments were recorded at 37°C and 42°C with deuterium decoupling to obtain backbone assignments in the [2H/13C/15N]–PSD-1–HSA 76-kDa complex: HNCA, HNCOCA, HNCACO, HNCO, CBCACONH, and HNCACB. Total chemical shift changes between free and bound states of PSD-1 were determined using Equation 1,
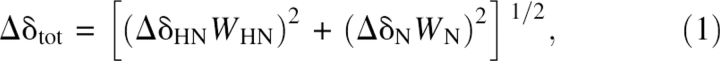 |
where the weighting factors were W HN = 1 and W N = 0.2 (Johansson et al. 2002a).
Acknowledgments
This work was supported by NIH Grants GM62154 and 1S10RR15744 and the W.M. Keck Foundation.
Footnotes
Reprint requests to: John Orban, Center for Advanced Research in Biotechnology, University of Maryland Biotechnology Institute, 9600 Gudelsky Drive, Rockville, MD 20850, USA; e-mail: orban@umbi.umd.edu; fax: (240) 314-6255.
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.072799507.
References
- de Chateau M., Holst, E., and Bjorck, L. 1996. Protein PAB, an albumin-binding bacterial surface protein promoting growth and virulence. J. Biol. Chem. 271: 26609–26615. [DOI] [PubMed] [Google Scholar]
- Delaglio F., Grzesiek, S., Vuister, G.W., Zhu, G., Pfeifer, J., and Bax, A. 1995. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6: 277–293. [DOI] [PubMed] [Google Scholar]
- He Y., Rozak, D.A., Sari, N., Chen, Y., Bryan, P., and Orban, J. 2006. Structure, dynamics, and stability variation in bacterial albumin binding modules: Implications for species specificity. Biochemistry 45: 10102–10109. [DOI] [PubMed] [Google Scholar]
- Johansson M.U., de Chateau, M., Wikstrom, M., Forsen, S., Drakenberg, T., and Bjorck, L. 1997. Solution structure of the albumin-binding GA module: A versatile bacterial protein domain. J. Mol. Biol. 266: 859–865. [DOI] [PubMed] [Google Scholar]
- Johansson M.U., Frick, I.M., Nilsson, H., Kraulis, P.J., Hober, S., Jonasson, P., Linhult, M., Nygren, P.A., Uhlen, M., Bjorck, L., et al. 2002a. Structure, specificity, and mode of interaction for bacterial albumin-binding modules. J. Biol. Chem. 277: 8114–8120. [DOI] [PubMed] [Google Scholar]
- Johansson M.U., Nilsson, H., Evenas, J., Forsen, S., Drakenberg, T., Bjorck, L., and Wikstrom, M. 2002b. Differences in backbone dynamics of two homologous bacterial albumin-binding modules: Implications for binding specificity and bacterial adaptation. J. Mol. Biol. 316: 1083–1099. [DOI] [PubMed] [Google Scholar]
- Lejon S., Frick, I.M., Bjorck, L., Wikstrom, M., and Svensson, S. 2004. Crystal structure and biological implications of a bacterial albumin binding module in complex with human serum albumin. J. Biol. Chem. 279: 42924–42928. [DOI] [PubMed] [Google Scholar]
- Parsons L.M., Liu, F., and Orban, J. 2005. HU-α binds to the putative double-stranded DNA mimic HI1450 from Haemophilus influenzae . Protein Sci. 14: 1684–1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pervushin K., Riek, R., Wider, G., and Wuthrich, K. 1997. Attenuated T2 relaxation by mutual cancellation of dipole–dipole coupling and chemical shift anisotropy indicates an avenue to NMR structures of very large biological macromolecules in solution. Proc. Natl. Acad. Sci. 94: 12366–12371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozak D.A., Alexander, P.A., He, Y., Chen, Y., Orban, J., and Bryan, P.N. 2006. Using offset recombinant polymerase chain reaction to identify functional determinants in a common family of bacterial albumin binding domains. Biochemistry 45: 3263–3271. [DOI] [PubMed] [Google Scholar]
- Ruan B., Fisher, K.E., Alexander, P.A., Doroshko, V., and Bryan, P.N. 2004. Engineering subtilisin into a fluoride-triggered processing protease useful for one-step protein purification. Biochemistry 43: 14539–14546. [DOI] [PubMed] [Google Scholar]
- Sari N., Ruan, B., Fisher, K.E., Alexander, P.A., Orban, J., and Bryan, P.N. 2007. Hydrogen–deuterium exchange in free and prodomain-complexed subtilisin. Biochemistry 46: 652–658. [DOI] [PubMed] [Google Scholar]