

Abstract
Certain proteins of unicellular organisms are translated as precursor polypeptides containing inteins (intervening proteins), which are domains capable of performing protein splicing. These domains, in conjunction with a single residue following the intein, catalyze their own excision from the surrounding protein (extein) in a multistep reaction involving the cleavage of two intein–extein peptide bonds and the formation of a new peptide bond that ligates the two exteins to yield the mature protein. We report here the solution NMR structure of a 186-residue precursor of the KlbA intein from Methanococcus jannaschii, comprising the intein together with N- and C-extein segments of 7 and 11 residues, respectively. The intein is shown to adopt a single, well-defined globular domain, representing a HINT (Hedgehog/Intein)-type topology. Fourteen β-strands are arranged in a complex fold that includes four β-hairpins and an antiparallel β-ribbon, and there is one α-helix, which is packed against the β-ribbon, and one turn of 310-helix in the loop between the β-strands 8 and 9. The two extein segments show increased disorder, and form only minimal nonbonding contacts with the intein domain. Structure-based mutation experiments resulted in a proposal for functional roles of individual residues in the intein catalytic mechanism.
Keywords: protein splicing, inteins, NMR structure determination
Protein splicing is a method of protein self-processing that occurs in a variety of unicellular organisms and their viruses, including bacteria, archaea, fungi, and other single-cell eukaryotes. During protein splicing, an intervening sequence (intein) is seamlessly removed from a precursor protein while the flanking regions (exteins) are joined to form the mature extein protein (Noren et al. 2000; Paulus 2000). This process is catalyzed by combined action of the intein and the first C-extein residue, resulting in cleavage of the peptide bonds at the N and C termini of the intein followed by the formation of a new peptide bond between the N- and C-exteins. To contribute to a better understanding of the mechanism by which a single protein domain can catalyze such a complex process, we determined the NMR structure of a precursor of the Methanococcus jannaschii KlbA intein.
Experimental studies have elucidated a four-step mechanism of protein splicing (Noren et al. 2000; Paulus 2000), which requires the intein itself as well as the first C-extein residue (+1 residue), which is a key nucleophile (by convention, extein residues are numbered beginning with 1 at the splice junction and increasing outward from the splice junction. Residues in the C-extein are denoted by a plus sign, and residues in the N-extein are denoted by a minus sign). Splicing is initiated by an acyl shift reaction in which the N-terminal residue of the intein, a Cys or Ser, attacks the carbonyl carbon of the preceding peptide bond to create the first intermediate, a linear (thio)ester intermediate with the N-extein attached to the intein residue 1 side chain. The second step is a transesterification reaction in which the +1 residue, a Cys, Ser, or Thr, attacks the same carbonyl carbon to transfer the N-extein to its side chain, cleaving the N-terminal splice junction and creating a branched intermediate (“branched” indicating a polypeptide with two N termini). This intermediate is resolved in the third step, which comprises C-terminal cleavage by cyclization of the Asn residue at the C terminus of the intein to release the free intein with a C-terminal succinimide group and the exteins linked by a (thio)ester bond. These last intermediates are resolved by spontaneous reactions, namely an S–N or O–N acyl shift to form a native peptide bond between the exteins, and spontaneous hydrolysis of the succinimide group to yield the free intein with a C-terminal asparagine or isoasparagine residue. S–N and O–N acyl shift reactions occur rapidly under physiological conditions (Paulus 2000) and do not require enzymatic assistance.
To date, more than 375 intein sequences have been reported (http://www.neb.com/neb/inteins; Perler 2002). They share only minimal sequence similarity overall, but certain regions contain residues that are conserved in all or a large fraction of intein sequences (see Fig. 1, blocks A, B, F and G; Pietrokovski 1994, 1998; Perler et al. 1997). In addition to the protein-splicing domain, many inteins contain homing endonuclease domains, which are thought to be responsible for the spread of inteins by horizontal gene transfer (Gimble and Thorner 1992; Pietrokovski 2001); inteins lacking an endonuclease domain have been termed mini-inteins. Within the protein splicing domain, the most highly conserved residues, besides those at the splice junctions, are a threonine and a histidine in block B. The histidine is found in nearly all inteins identified to date, the exceptions being three inteins from three different archaea which occupy the same insertion sites in homologous extein genes, and are considered to be intein alleles. No studies have so far examined the functionality of the inteins that are devoid of the block B histidine. Previous structural investigations (Duan et al. 1997; Hall et al. 1997; Klabunde et al. 1998; Poland et al. 2000; Mizutani et al. 2002) revealed that a likely role of this histidine residue is to protonate the leaving group of the first reaction, i.e., the nitrogen of the N-terminal scissile bond. Structural studies have also suggested specific catalytic roles for other residues in N- and C-terminal cleavage, which differ significantly between different inteins (Ding et al. 2003; Sun et al. 2005).
Figure 1.
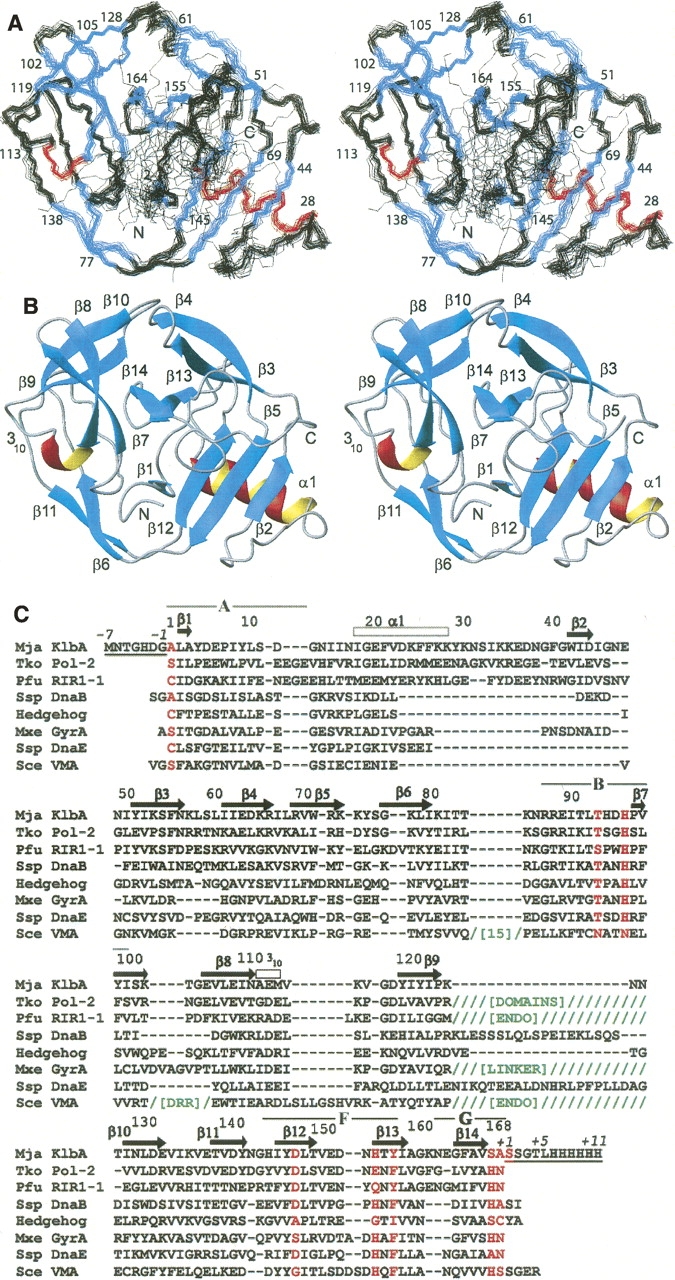
(A) Ensemble of 20 CYANA conformers representing the Mja KlbA intein precursor solution structure, superimposed for minimal RMSD of the backbone N, Cα, and C′ atom positions of residues −1 to +1. Residue numbers and the N and C termini of the protein are indicated. Helices are colored red, β-strands blue, and regions without regular secondary structure black. (B) Ribbon presentation of the Mja KlbA intein, with the same orientation as in A. Helices are colored red and β-strands cyan. Regions of regular secondary structure are labeled, and the N and C termini are indicated. The conformer with minimal RMSD to the mean coordinates of the ensemble in A is shown. (C) Amino acid sequence of the Mja KlbA intein precursor described in this study, and structure-based sequence alignment with the other inteins for which a three-dimensional structure is available. The residue numbers and regular secondary structure elements of the Mja KlbA intein precursor are indicated above the sequence. The residue numbering begins with 1 for the first residue of the intein and ends with 168 at the last residue of the intein. The seven extein residues at the N terminus (−7 to −1) and the 11 extein residues at the C terminus (+1 to +11) are underlined, and are numbered separately (italicized numbers). The splice junction residues, and other residues of interest that are discussed in the text, are colored red. All sequences include the amino acid replacements used to generate a construct for structure determination. In the multiple sequence alignment, hyphens (-) indicate that no residue is present at that position, while slashes (/) indicate the presence of multiple extra residues (insertions not shown due to space constraints). When entire additional domains are inserted, this is indicated by abbreviated domain names in green, where “ENDO” indicates an endonuclease domain, and “DRR” indicates a DNA-recognition region. The locations of the conserved intein sequence blocks A, B, F, and G (see text) are indicated above the sequence.
All inteins functioning by the standard mechanism require a Ser or Cys residue at position 1, and show no N-terminal cleavage if this residue is replaced by alanine or other nonconservative substitutions. A few inteins beginning with Ala were at first thought to be inactive, pseudo-inteins (Pietrokovski 1998), but as more examples were found, they were shown to be splicing-competent (Southworth et al. 2000; Yamamoto et al. 2001; Southworth and Perler 2002). Southworth et al. (2000) showed that one such intein, the KlbA intein from M. jannaschii (Mja), is active, and they proposed a splicing mechanism for Ala1 inteins. This variant mechanism is similar to the standard mechanism described above, but without the obligatory formation of the first linear (thio)ester intermediate; thus, the branched intermediate is formed directly from the precursor by nucleophilic attack of the +1 residue on the carbonyl carbon of the peptide bond preceding the intein.
Since the Mja KlbA intein has previously been studied in model systems (Southworth et al. 2000), we felt that structural studies of this intein would be helpful to provide further insight into the structure and function of inteins. For the structural studies, we employed a modified construct, in which the C-terminal Asn is replaced by Ala and the Cys +1 residue by Ser, which lacks splicing activity. This allowed us to study a stable precursor construct containing both an N- and a C-terminal extein segment. To avoid ambiguity in the interpretation, we chose to study a precursor polypeptide in which native extein residues are present, rather than a free intein or an intein with heterologous extein residues flanking the intein. We report here the NMR structure of this intein precursor in solution. The structural data suggested new mutational experiments, which helped to demonstrate the functional roles of several residues within and near the active site.
Results
The NMR structure of the KlbA intein precursor
The intein structure is composed mainly of β-strand regular secondary structures, which are arranged in a complex fold of the HINT (Hedgehog/Intein)-type (Fig. 1; Murzin et al. 1995). The molecule contains 16 regular secondary structures, i.e., 14 β-strands, one α-helix, and one turn of 310-helix. In the ensemble of 20 conformers resulting from the CYANA calculation the backbone atom positions of residues 1−168, corresponding to the intein itself, as well as the +1 C-extein residue, Ser +1, and the −1 N-extein residue, Gly −1, are well defined and superimpose with an RMSD value of 0.51 Å (Fig. 1A; Table 1). The remaining C-extein residues, Ser +2 to His +11, and the N-extein residues Met −7 to Asp −2 are flexibly disordered. For these disordered residues, only a small number of short-range NOEs are observed. Overall, the intein itself thus forms a stably folded globular domain, while the extein residues form flexibly extended “tails.” This may be due to the small size of the extein segments present in this precursor, which fail to fold in the absence of the remainder of the native exteins, but it may also reflect a requirement for some flexibility of the extein segments near the intein/extein junctions.
Table 1.
Input for the structure calculation and characterization of the bundle of 20 energy-minimized CYANA conformers representing the solution structure of the 186-residue Mja KlbA intein precursor

In sequential order, the intein fold (Fig. 1) begins with a short β-strand of residues 2−3 lying near the center of the protein. This is followed by an amphipathic α-helix of residues 18−28, which is exposed to solvent on one side, while the three phenylalanine residues in positions 21, 25, and 26, as well as other hydrophobic side chains, pack against a three-stranded antiparallel β-sheet formed by the strands β2, β5, and β12. A tight turn and a short segment of extended chain lead to the strand β2 (42−44), and a twisted antiparallel β-hairpin of strands β3 (51−56) and β4 (61−66). The strands β5 (69−73) and 6 (77−80) are separated only by a short coil region, and a 16-residue nonregular loop connects to the strands β7 (97−102) and β8 (105−110), which form another twisted antiparallel β-hairpin followed by a 310-turn (111−113). The next β-hairpin of β9 (119−123) and β10 (128−132) is oriented approximately at a right angle relative to β7 and β8. The strands β11 (138−141) and β12 (145−149) pair with the strands β6 and β5 to form a long, curving antiparallel β-ribbon that leads back to a position near the N terminus. From there, a loop connects to a final β-hairpin of the strands β13 (155−158) and β14 (164−167) located near β1 in the center of the disk-shaped molecule.
Relationship to other intein family members
A search of the Protein Data Bank with the DALI server (Holm and Sander 1993) allowed comparisons of the Mja KlbA intein structure to other inteins. The closest similarity was found to the two other archaeal inteins with known structures, the Thermococcus kodakaraensis Pol-2 intein (Tko Pol-2; Z-score 21.3; PDB codes 2CW7, 2CW8) (Matsumura et al. 2006) and the Pyrococcus furiosus RIR1–1 intein (Pfu RIR1–1; Z-score 19.5; PDB code 1DQ3) (Ichiyanagi et al. 2000). Neither of these proteins is a mini-intein, and in both the homing endonuclease domain and additional domains (the domains III and IV in Tko Pol-2; the Stirrup domain in Pfu RIR1–1) are inserted in positions corresponding to the end of the β9–β10 hairpin in the Mja KlbA intein. These insertions do not affect the intein fold itself, which is closely similar in the archaeal inteins, so that the Cα atoms can be superimposed with low RMSD values (1.9 Å for 164 aligned residues of KlbA vs. 2CW7; 2.2 Å for 164 aligned residues of KlbA vs. 1DQ3). All three archaeal inteins contain an α-helix in positions corresponding to the residues 18−28 in KlbA, which is a longer helix than is found in other HINT domains. Most intein structures solved to date actually contain only a 310-helix turn at the position of the α-helix in KlbA, which is an intriguing variation since Ile 18 in Mja KlbA is highly conserved and forms part of the core of the protein. The Synechocystis sp. PCC6803 DnaE intein (Ssp DnaE) (Sun et al. 2005) contains an α-helix of five residues in this location, but has no strand corresponding to β2 in KlbA. Overall, the archaeal inteins appear to have insertions of 10–20 residues at this position, relative to other inteins. A structure-based sequence alignment of Mja KlbA with Tko Pol-2 and Pfu RIR1–1 is included in Figure 1C. The alignment was generated based on the structurally corresponding positions identified by DALI, and highlights the positions of insertions and deletions within the HINT fold.
There is also significant structural similarity to other inteins: The Ssp DnaB intein (PDB code 1MI8) (Ding et al. 2003), Drosophila hedgehog autoprocessing domain (1AT0) (Hall et al. 1997), Mycobacterium xenopi GyrA mini-intein (1AM2) (Klabunde et al. 1998), Ssp DnaE intein (1ZDE, 1ZD7) (Sun et al. 2005), and Saccharomyces cerevisiae VMA intein (1VDE, 1EF0, 1JVA, 1LWS, 1LWT, 1GPP, 1DFA) (Duan et al. 1997; Hu et al. 2000; Poland et al. 2000; Mizutani et al. 2002; Moure et al. 2002; Werner et al. 2002); all have DALI Z-scores ranging from 9.0 to 17.1. As an illustration, the superpositions of the Mja KlbA intein with the Tko Pol-2 intein and with the Sce VMA intein are shown in Figure 2.
Figure 2.
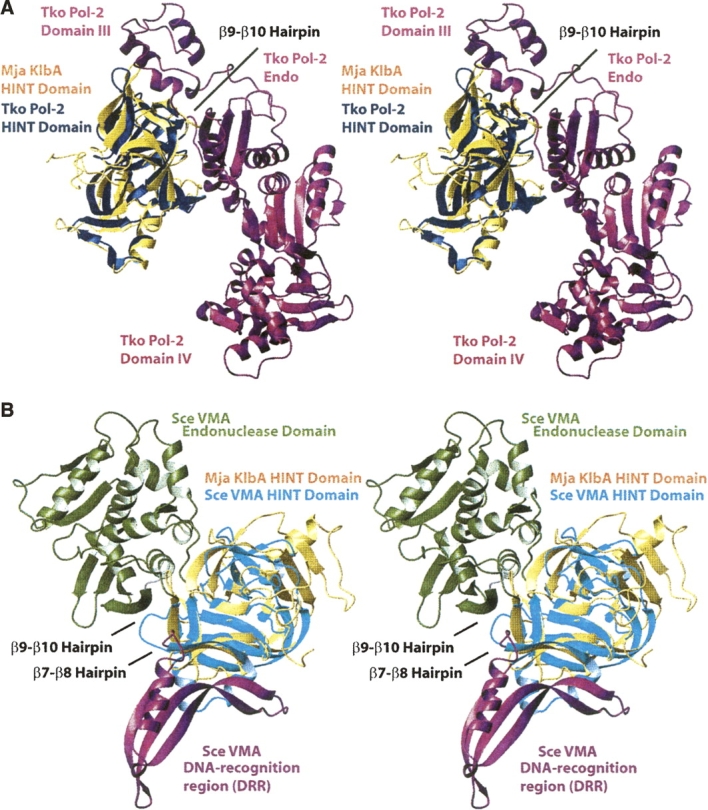
Superpositions of the Mja KlbA intein structure with two other inteins. In both panels, the Mja KlbA intein is shown in gold. The superpositions were generated by the DALI software (Holm and Sander 1993). (A) Protein splicing (HINT) domain of the Thermococcus kodakaraensis Pol-2 intein (PDB code 2CW7) (blue). The endonuclease domain and the additional domains III and IV of Tko Pol-2 are shown in magenta. The β9−β10 hairpin of Mja KlbA, which is the position at which the additional domains are inserted into the intein fold, is labeled. The aligned residues are: Mja KlbA intein residues 1−11, 12−36, 38−46, 49−125, 126−161, and 163−168, with Tko Pol-2 residues 1−11, 15−39, 40−48, 49−125, 496−531, and 532−537. In total, 164 Cα positions were superimposed, with an RMSD of 1.9 Å. (B) Protein splicing (HINT) domain of S. cerevisiae VMA intein (PDB code 1JVA) (cyan). The endonuclease domain (green) and the DNA-recognition region (DRR) (magenta) of Sce VMA are also shown. The β7−β8 and β9−β10 hairpins of Mja KlbA, corresponding to the points of insertion of these additional domains, are labeled. The aligned residues are: Mja KlbA intein residues 1−22, 49−55, 61−75, 78−85, 86−103, 104−114, 116−126, 127−133, 135−142, 144−151, 152−158, and 162−168, with Sce VMA residues 284−305, 307−313, 314−328, 329−336, 352−369, 437−447, 456−466, 698−704, 705−712, 713−720, 723−729, and 731−737. In total, 129 Cα positions were superimposed, with an RMSD of 2.5 Å.
Positions of conserved residues
As in other inteins, the N-terminal scissile bond (Gly −1−Ala 1) is found preceding the strand β1 near the center of the disk-shaped molecule. The conserved Thr and His residues of block B (Fig. 1C) are found within a type I β-turn formed by the residues 93−96, immediately N-terminal to the strand β7. The Thr 93 and His 96 side chains are hydrogen-bonded to each other, and are both near the backbone amide nitrogen of the residue Ala 1 (Fig. 3A). On the other side of the scissile bond and also in close proximity is Asp 147 on strand β12. The side chain of this residue extends into the active site close to the scissile bond, with a distance of 3.1 Å between its Oδ1 carboxyl oxygen and the carbonyl oxygen of Gly −1.
Figure 3.
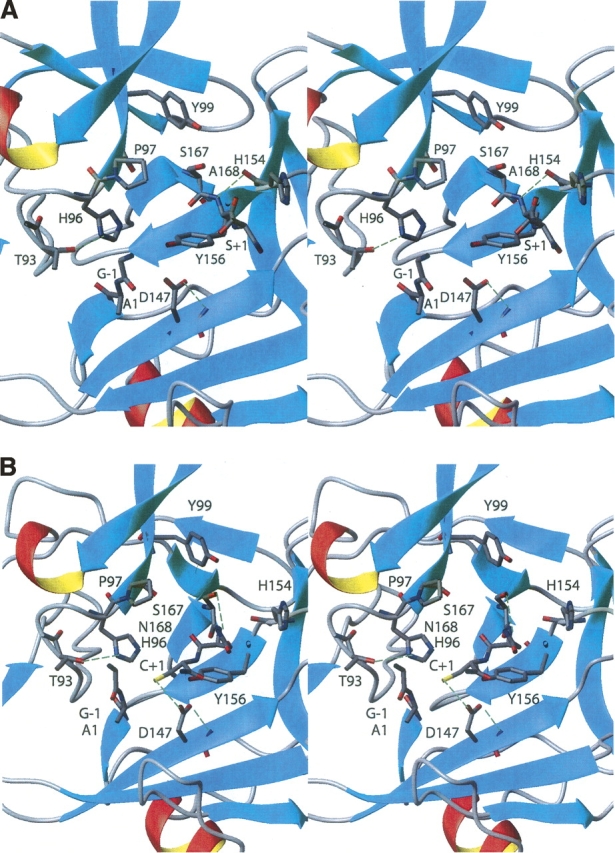
(A) Locations of the active-site residues in the NMR structure of the Mja KlbA intein. (B) Locations of the active-site residues in a computational model of a putative active conformation of the KlbA intein, which was derived from the NMR structure (see text). In both stereoviews the regular secondary structure elements are shown in a ribbon presentation, and the backbone and side chains of the scissile bonds G(−1)–A1 and A168–S(+1), and the active-site residues are shown in stick presentations, colored by atom types (C, gray; N, blue; O, red). The green dashed lines indicate hydrogen bonds.
While the backbone atoms of Ala 1 face block B (Fig. 1C), the side chain of Ala 1 is directed toward strand β12, and makes contact with a hydrophobic area formed by the side chain atoms of Ile 145 and the backbone atoms of Tyr 146 and Asp 147. This conformation of Ala 1 in the Mja KlbA intein is very similar to that observed in other intein precursors in which the Ser or Cys residues at position 1 have been replaced by Ala. Previous mutational studies of the KlbA intein had shown that if Ala 1 was replaced with Cys, the initial acyl shift typical of standard inteins could occur (Southworth et al. 2000). Consistent with these observations, a Cys 1 residue can readily be accommodated in the KlbA intein NMR structure, with its side chain directed toward strand β12. The positioning of the side chain in this hydrophobic pocket would allow the Cys Sγ atom to approach the −1 carbonyl carbon, in a conformation that would be favorable for the first acyl shift reaction.
The strand β14 ends at the penultimate residue of the intein, Ser 167, and the following three residues adopt an irregular extended conformation. The C-terminal residue of the intein, Ala 168 (which was inserted in the place of the Asn 168 in wild-type KlbA intein), is held close to the neighboring loop connecting the strands β12 and β13. This loop is contained in the conserved sequence block F (Fig. 1C). The C-terminal strand β14 is held near this loop by a hydrogen bond from Ala 168 HN to His 154 O (Fig. 3A). The side chains of the penultimate residue, Ser 167, and the catalytic residue, Ser +1, lie above the active site and are solvent-exposed. The side chain and the carbonyl group of Ala 168 are both directed toward a hydrophobic area formed by three side chains from the block F loop (Leu 148, Val 150, and Tyr 156). The C-terminal scissile bond between Ala 168 and Ser +1 lies near the top of the active site.
Backbone dynamics
It has been postulated that local mobility may be important in intein catalysis, considering that several subsequent reaction steps must occur within the same active site. We measured 15N R1 and R2 relaxation rates and heteronuclear NOE values to address this possibility. We find that the majority of the protein is well-ordered, with none of the active-site residues showing picosecond−nanosecond timescale motions of the backbone amide moieties (Fig. 4). Residues −5 to −3 and +3 to +6 show reduced heteronuclear NOE values (<0.6), lower R2 values, and slightly higher R1 values than the rest of the protein, indicating rapid motions, as is also indicated by a lack of long-range NOE crosspeaks, and increased structural disorder within the calculated bundle of 20 conformers (Fig. 1A). The residues of the active site, including block B and the splice junction residues (Fig. 1C), show no observable differences in relaxation properties from the rest of the protein. There is an indication of increased mobility in the loop connecting the strands β7 and β8, with the residues Lys 102, Thr 103, Gly 104, and Glu 105, which form an inverse type I β-turn. The Sce VMA intein is unique among the structures solved to date in that it contains a DNA-recognition region inserted at this position, in addition to the endonuclease domain insertion at the end of the hairpin corresponding to β9−β10 in Mja KlbA (Figs. 1C, 2B). The β9−β10 hairpin is a more common position for insertions, with endonuclease domains or linker regions occurring at this position in several other inteins as well (Figs. 1C, 2A).
Figure 4.

15N relaxation parameters of the Mja KlbA intein precursor recorded at 60.06 MHz 15N frequency. (A) Longitudinal relaxation rates R1 (sec−1). (B) Transverse relaxation rates R2 (sec−1). (C) Relative intensity, Irel, of 15N{1H}-NOEs. In panels A and B error bars were calculated from a least-squares fit of the data. In panel C, the error bars are given as half of the difference between two independent measurements. Residue numbers and the positions of the regular secondary structures are indicated at the bottom of panel C.
Site-directed mutagenesis
Biochemical and mutagenesis studies of inteins have established roles for certain residues in the catalytic mechanism (for further details, the reader is referred to recent reviews: Paulus 2001; Evans and Xu 2002; David et al. 2004; Saleh and Perler 2006; Muralidharan and Muir 2006; Perler 2006, and also to the structural studies described above). The three-dimensional structure of the Mja KlbA intein, combined with this previous biochemical work, suggested that several specific residues are likely participants in the catalytic mechanism. To check on these hypotheses, we constructed a series of mutants with designed single or double amino acid replacements. A survey of these mutations and their effects on splicing is given in Table 2.
Table 2.
Survey of the amino acid replacements introduced into the Mja KlbA intein and their effects on splicing

Block B histidine
The most highly conserved residues in intein sequences are those at the splice junctions and the Thr and His residues in block B (T93 and H96 in Mja KlbA). The mutations H96G, H96A, H96E, and H96R all abrogated N-terminal cleavage, confirming previous observations that His 96 is essential for the first step, i.e., the branched intermediate formation (Noren et al. 2000; Paulus 2000). These variant proteins also had strongly reduced amounts of C-terminal cleavage (Table 2). While His 96 is not hydrogen bonded to Ala 168 in the NMR structure, its presence may be important for correct alignment of the C-terminal strand, or in contributing to an oxyanion hole that would help to stabilize the transition state for Asn cyclization. Alternatively, structural changes on branched intermediate formation may stimulate Asn cyclization. Of the above variants, H96R had the least residual C-terminal cleavage (Table 2), presumably because the placement of the long Arg side chain into the small active site would require the largest displacements of other atom groups.
Splice junction residues
As expected, replacement of the C-terminal Asn residue in the variants N168A, N168D, N168E, and N168Q prevented C-terminal cleavage. Interestingly, these replacements also affected N-terminal cleavage, with <5% N-terminal cleavage for the N168D variant. The observation that the nature of the residue substituted for Asn 168 can affect N-terminal cleavage was unexpected, as this residue has not previously been noted to be involved in reactions at the N terminus. Single residue mutations of the +1 residue were also tested, and replacement of C +1 with Ser or Thr abrogated N-terminal cleavage, showing that an oxygen nucleophile cannot substitute for the wild-type sulfur nucleophile in this intein, which is likely due to the lower nucleophilicity of the hydroxyl moiety. The Cys +1 to Ser or Thr replacements also inhibited C-terminal cleavage (Table 2).
Block F residues
His 154 in Mja KlbA is found near the C-terminal splice junction (Fig. 3A), suggesting a possible role in assisting Asn cyclization, as has been demonstrated in the Ssp DnaB intein (Ding et al. 2003). However, the replacement H154A had no measurable effect on splicing (Table 2). Tyr 156 is highly conserved in inteins as either Phe or Tyr (Fig. 1C), and Figure 3A shows that it is centrally located in the active site. The replacement Y156F did not significantly inhibit splicing, but Y156A yielded equal amounts of precursor, N-terminal single cleavage product, and free intein, showing that the presence of an aromatic ring plays an important role in the correct alignment of the active site residues, especially with respect to C-terminal cleavage. Similar observations were made by Ding et al. (2003) regarding the Phe residue at this position in the Ssp DnaB intein. The double replacement H154A/Y156F also had no measurable effect on splicing, eliminating the possibility that these two residues might play redundant roles. These results demonstrate that in the Mja KlbA intein, assistance of Asn cyclization by the block F histidine is not necessary, and that Tyr 156 has a structural, but not catalytic, role in the splicing reactions.
An additional activating interaction was observed in the Ssp DnaE intein (Sun et al. 2005), which is a split intein and, like Mja KlbA, lacks a penultimate His. In the Ssp DnaE intein the residue following the block B His (Arg 73) extends across the active site and is hydrogen bonded to the Asn carbonyl oxygen, providing the activating interaction observed in other inteins from the side chain of the penultimate His residue. In the Mja KlbA intein, the corresponding residue is Pro 97, which cannot provide the same hydrogen bonding function. In the NMR structure we do not observe any hydrogen bonding partner of Ala 168, but we note that similar to Ssp DnaB intein, Asp 147 might participate in a water-mediated interaction, possibly jointly with the side chain of the penultimate residue Ser 167.
The NMR structure shows that the side chain of Asp 147 is located near the N-terminal splice junction (Fig. 3A), and thus suggests a second potential role for Asp 147 in activating branched intermediate formation by interacting with the carbonyl oxygen of Gly −1. To further investigate this indication from the NMR structure, we tested replacing Asp 147 with Ala or Glu. The D147A mutation yielded mainly intact precursor, with a small amount of C-terminal cleavage (Table 2), while the D147E replacement yielded predominantly C-terminal cleavage, with a small amount of intact precursor. We conclude that Asp 147 assists the reactions at both splice junctions, with the greatest effect in the branched intermediate formation. Since D147A completely blocks N-terminal cleavage, this residue must play an important role in activating branched intermediate formation; this might be by increasing the electrophilicity of the Gly −1 carbonyl carbon, by increasing the nucleophilicity of Cys +1, by contributing to the formation of an oxyanion hole, or by an as-yet undefined mechanism. Since the D147A mutation also significantly inhibits C-terminal cleavage, this residue must also play a role in Asn cyclization. Although Asp 147 is not hydrogen bonded to any partner on the C-terminal strand, and is in fact in closest proximity to the N-terminal splice junction, there remains the possibility that this residue may be involved in an indirect hydrogen bonding network with the C-terminal region that also includes one or several water molecules.
Since the D147E mutation also blocks N-terminal cleavage, but allows C-terminal cleavage, we hypothesize that a Glu residue at this position still provides the necessary electrostatic or hydrogen-bonding stabilization for the Asn cyclization occurring at the C terminus, but is bulky enough to inhibit the approach of the Cys +1 residue toward the N terminus for the branched intermediate formation, and thus cannot substitute for the wild-type Asp in the reactions at the N terminus.
In the structure of the Ssp DnaB intein (Ding et al. 2003), Asp 136 (corresponding to Asp 147 in Mja KlbA) hydrogen bonds to the Ala 154 carbonyl oxygen via a water molecule, but the D136A substitution did not significantly inhibit C-terminal cleavage. In contrast, a recent study by van Roey et al. (2007) showed that in the Mycobacterium tuberculosis RecA intein, Asp 422 assists both N- and C-terminal cleavage. The crystal structures of three different variants of the Mtu RecA intein showed that this residue can contact both Cys 1 and Asn 440 by adopting different side chain conformations, suggesting a structural mechanism by which this residue can assist reactions. Many intein sequences contain an Asp at this position, but Cys, Ser, Thr, Asn, Gln, and Glu are also common, and in some sequences nonpolar or aromatic residues are also observed. In those inteins that contain a polar residue, this residue may be involved in assisting the reactions at both the N- and C-terminal splice junctions.
Discussion
The N-terminal splice junction in inteins
The combination of the NMR structure of the Mja KlbA intein with previous biochemical studies (Southworth et al. 2000) and the mutational data of the preceding section allow us to present hypotheses regarding the roles of individual active-site residues in activating cleavage at the splice junctions. The residues Thr 93 and His 96 are hydrogen bonded to each other, and are located near the N-terminal scissile bond, close to the Ala 1 N–H moiety (Fig. 3A). His 96 likely catalyzes essential proton transfers during the branched intermediate formation, for example, by donating a proton to the amide group of Ala 1 to facilitate the breakdown of the intermediate chemical structure. The side chain of Thr 93 may help both to activate the His residue and to create an oxyanion hole with hydrogen-bonding potential to stabilize reaction intermediates.
The structure of the intein active site has some parallels with the active sites of cysteine proteases, which employ a Cys–His–Asn triad for catalysis. In some papain-like proteases, the Cys and His form an imidazolium−thiolate ion pair (Otto and Schirmeister 1997), which increases the nucleophilicity of the Cys side chain; the Asn residue is hydrogen bonded to the His and stabilizes its imidazolium form. The three intein residues shown to be important for N-terminal cleavage, Thr 93 (Southworth et al. 2000), His 96, and Cys +1, may constitute a similar catalytic triad, with the roles of these three residues being similar to the roles of the corresponding residues in protease active sites. In addition to assisting the breakdown of the intermediate chemical structure, the His may also help to activate the Cys residue and increase its nucleophilicity, and the role of Thr 93 may be analogous to that of the Asn of the catalytic triad in that it provides hydrogen-bonding stabilization that assists catalysis. A role of the block B His residue both in activating the nucleophilic Cys +1 and in protonating the departing amide group would be consistent with mutagenesis data on several inteins, which show that this residue is essential for catalysis. As in cysteine proteases, the replacement of either the Cys or the His in inteins leads to a complete loss of N-terminal cleavage, while replacement of the Thr slows catalysis but does not completely disrupt the reaction (Otto and Schirmeister 1997; Noren et al. 2000; Southworth et al. 2000).
In the Mja KlbA intein structure, the side chain of Ser +1, which replaces Cys +1, is directed away from the N-terminal area of the active site, and faces the solvent. The Ser +1 hydroxyl group is thus quite distant from the N-terminal scissile bond (8.1−13.4 Å between Ser +1 Oγ and Gly −1 C, with a mean value in the ensemble of Figure 1A of 10.9 Å). Thus, the +1 residue is not positioned for direct attack of the −1 carbonyl group, and the movement of the nucleophile toward the N terminus requires some structure rearrangement, as discussed further below.
The C-terminal splice junction in inteins
Unlike the cysteine proteases, in inteins the cleavage of the N-terminal scissile bond is followed by a second cleavage step, the cleavage of the C-terminal scissile bond, which is the peptide bond between Asn 168 and Cys +1 (Ala 168 and Ser +1 in the presently studied stable precursor). In the Mja KlbA intein structure, the penultimate residue Ser 167 is directed away from the active site, facing the solvent. The side chain of Ala 168 is directed toward a hydrophobic area on the block F loop formed by Leu 148, Val 150, and Tyr 156, while the C-terminal intein/extein bond between Ala 168 and Ser +1 is near the top of the active site in the orientation of Figure 3A. The Ser +1 side chain faces toward the N terminus, and is in a relatively solvent-exposed area of the active site. There are no obvious hydrogen-bonding partners for the Ala 168 carbonyl group that might be responsible for assisting Asn cyclization, although the side chains of His 96, Asp 147, Ser 167, and Tyr 99 are sufficiently close by to be involved in water-mediated activating interactions. Our mutagenesis data confirm that Asp 147 assists C-terminal cleavage, and that this assistance can also be provided by a Glu residue substituted for Asp at this position.
Currently, 18 crystal structures representing eight different inteins have been deposited in the Protein Data Bank. Twelve of these structures represent post-splicing forms, and thus information about the +1 and −1 residues is missing (Duan et al. 1997; Hu et al. 2000; Ichiyanagi et al. 2000; Moure et al. 2002; Sun et al. 2005; Matsumura et al. 2006; van Roey et al. 2007) (PDB codes 2CW7, 2CW8, 1DQ3, 1ZD7, 1VDE, 1LWS, 1LWT, 1DFA, 2IMZ, 2IN0, 2IN8, 2IN9). Two of the structures (Mxe GyrA, Sce VMA) (Klabunde et al. 1998; Werner et al. 2002) (PDB codes 1AM2, 1GPP) represent only the intein and one or two N-extein residues, so that the position of the +1 nucleophilic residue is unknown. Of the remaining four structures of intein precursors, three have separations of about 10 Å between the +1 nucleophile side chain and the carbonyl carbon atom of the N-terminal scissile bond (Poland et al. 2000; Ding et al. 2003; Sun et al. 2005) (PDB codes 1ZDE, 1MI8, 1EF0). The fourth structure, of a precursor of the Sce VMA intein, was trapped by a mutation of the penultimate Asn residue to Ser, rather than to Ala as in all other intein precursor structures (Mizutani et al. 2002) (PDB code 1JVA). It is thus possible that the Ala replacement causes a change in conformation at the C terminus in which the residues 167−168 (numeration of Mja KlbA intein) rearrange to allow the Ala side chain to make hydrophobic contacts with residues of block F.
In the following section, we discuss the hypothesis that the structure of the Sce VMA intein (Mizutani et al. 2002) represents a conformation that could also be adopted by the Mja KlbA intein during the approach of the +1 nucleophile to the N-scissile bond. A computational model of the wild-type Mja KlbA intein precursor was generated by replacing the residues Ala 168 and Ser +1 in the NMR structure with Asn and Cys, respectively, and adjusting the backbone torsion angles of the tripeptide segment Ser 167–Asn168–Cys +1 to values similar to those observed in the structure of the Sce VMA intein (Mizutani et al. 2002). Further small adjustments were made of the Cys +1 χ1 and χ2 angles, to minimize unfavorable van der Waals contacts, and of the backbone torsion angles of some of the proximal extein residues, including the Gly −1 ψ angle, to avoid steric clashes between the two exteins. After these interactive adjustments, the model was energy-minimized in a water shell using the Amber force field (Cornell et al. 1995) in the program OPALp (Luginbühl et al. 1996; Koradi et al. 2000). The resulting model (Fig. 3B) seems to represent a potentially functional active conformation, with a separation of 3.3 Å between the Cys +1 Sγ atom and the carbonyl carbon of the N-terminal scissile bond. This modeling approach shows that a plausible rearrangement of the backbone conformation near the C-terminal splice junction in the Mja KlbA intein enables the close approach of Cys+1 toward the N-terminal scissile bond.
We note also that our model shows the Asp 147 side chain hydrogen bonding to the Cys+1 Sγ atom (Fig. 3B), suggesting another possible mechanism by which Asp 147 might assist N-terminal cleavage. This hydrogen bonding assistance together with the presence of a positively charged block B His residue would favor the deprotonation of the Cys+1 side chain and help to accelerate the reaction. Similarly, in the Mtu RecA intein (van Roey et al. 2007), Asp 147 was shown to be capable of hydrogen bonding to the sulfur atom of the N-terminal Cys 1 residue, which suggests that it might assist linear intermediate formation in an analogous manner. We note, however, that the involvement of an Asp or other polar residue at this position may not be common to all inteins, while the block B His is essential for linear and branched intermediate formation in all inteins tested so far.
Basis of the alternate splicing mechanism in the Mja KlbA intein
A basis for the alternate reaction mechanism, by which the Mja KlbA intein can undergo a direct Cys +1−Gly −1 reaction and thus omit the first linear intermediate that has been shown to be necessary in all other native inteins tested (Noren et al. 2000; Paulus 2000; Southworth et al. 2000), is not directly apparent from the NMR structure. However, we observe a small difference in the width of the active site of the Mja KlbA intein when compared to those of some other inteins. In particular, the loop containing the block B residues is further apart from strand β12, with a distance between the Cα atoms of Thr 93 and Asp 147 of 11.7 Å, compared to 9.3 Å for Tko Pol-2 (Matsumura et al. 2006), 9.0 Å for Sce VMA (Mizutani et al. 2002), and 9.6 Å for Mxe GyrA (Klabunde et al. 1998). A widening of the active site could allow the +1 nucleophile to enter more deeply into the site and approach the −1 carbonyl group. In standard inteins, the first acyl shift reaction, which transfers the N-extein from the −1 carbonyl group to the Cys/Ser 1 side chain, would move the −1 carbonyl group further up in the active site into an area that is wider and presumably more accessible to the +1 nucleophile. This would also move the −1 carbonyl group away from the block B residues toward the strand β12, thus bringing the carbonyl group closer to the +1 nucleophile and lowering the barrier to reaction.
Overall, in the group of all available intein structures there seems to be a continuous variation of the active-site size, with similar distances between the Cα atoms of the residues corresponding to Thr 93 and Asp 147 in Pfu RIR1–1 (Ichiyanagi et al. 2000), Ssp DnaE (Sun et al. 2005), and Ssp DnaB (Ding et al. 2003) as in Mja KlbA (10.3 Å for Pfu RIR1–1; 10.2 Å for Ssp DnaE; 10.6 Å for Ssp DnaB). Furthermore, the replacement of Ala 1 with Gly in the Mja KlbA intein has been shown to prevent N-terminal cleavage (Southworth et al. 2000), suggesting that Ala 1 may be important in proper positioning of the scissile bond between the strand β12 and Cys +1, and that replacement of Ala 1 with Gly in fact allows the N-terminal splice junction to move too far toward the strands β12 and β5. The combination of all presently available data then leads us to suggest that the subset of inteins able to react by the presently discussed alternate mechanism is not necessarily limited to those in which an Ala residue occurs naturally at position 1.
Materials and Methods
Protein expression and purification for NMR analysis
The Mja KlbA intein precursor comprised the intein itself (168 residues) flanked by 7 N-extein residues and 11 C-extein residues. Of these, the seven N-extein residues (−1 to −7), as well as five of the 11 C-extein residues (+2 to +6) are wild type (i.e., derived from the Mja KlbA protein). The wild-type His +6 residue and the additional, nonnatural C-extein residues +7 to +11 constitute a 6×His tag for purification. The substitutions N168A and C+1S were introduced by site-directed mutagenesis using the QuikChange Kit as described by the manufacturer (Stratagene).
The Mja KlbA intein precursor (N168A, C+1S) was ligated into the plasmid pAII17 (Perler et al. 1992) and expressed in the E. coli strain T7 Express (New England BioLabs) along with pRIL (Stratagene). Cells were grown at 37°C and induced with 1 mM isopropyl β-D-thiogalactoside (IPTG) at an OD600 of 0.6–0.7, then grown for another 2–4 h at 37°C, 30 min at 30°C, and 20 h at 15°C. Cell pellets were lysed by sonication in 20 mM NaPO4, pH 7, with 0.5 M NaCl and 5 mM imidazole, and purified by affinity chromatography on Ni-NTA agarose (Qiagen) followed by ion-exchange chromatography on Toyopearl SuperQ-650M (Tosoh) and SP Sepharose Fast Flow (Amersham Pharmacia). Isotope labeling was accomplished by growing cultures in minimal medium containing either 1 g/L 15NH4Cl for a uniformly 15N-labeled sample, or 1 g/L 15NH4Cl and 4 g/L 13C6-D-glucose (Cambridge Isotope Laboratories) for a uniformly 15N,13C-labeled sample. Both procedures yielded ∼100 mg of pure protein from 1 L of culture. The pure protein was concentrated and exchanged by ultrafiltration with Pall Microsep 10K Omega concentrators (10-kDa molecular weight cutoff) into 20 mM sodium phosphate buffer, pH 5.3, with 100 mM NaCl and 2 mM NaN3. The final protein concentration in the 550 μl NMR samples was 2 mM.
NMR spectroscopy
NMR spectra were recorded at 308 K on Bruker Avance 600 and DRX 800 spectrometers equipped with TXI HCN z- or xyz-gradient probes. Resonance assignment was carried out using the following experiments (Sattler et al. 1999): 2D [15N,1H]-HSQC, 3D HNCA, 3D HNCO, 3D HNCACB, 3D CBCA(CO)NH, 3D HBHA(CO)NH, 3D 15N-resolved [1H,1H]-TOCSY (τm = 65 msec), 3D (H)CC(CO)NH-TOCSY, 3D HC(C)H-TOCSY, 3D 15N-resolved [1H,1H]-NOESY (τm = 60 msec), 3D 13C-resolved [1H,1H]-NOESY (τm = 60 msec). Assignment of aromatic side chain resonances was based on 3D 13C-resolved [1H,1H]-NOESY (Muhandiram et al. 1993), 2D [13C,1H]-TROSY (Pervushin et al. 1998), and 2D TOCSY-relayed ct-HMQC (Zerbe et al. 1996). Internal 2,2-dimethyl-2-silapentane-5-sulfonate (DSS) was used as a chemical shift reference for 1H, and 15N and 13C shifts were referenced indirectly to DSS (Wishart et al. 1995). The chemical shift lists (Johnson et al. 2007) have been deposited in the BioMagResBank under accession number 15,061.
15N relaxation data and 15N{1H}-NOE values were measured using 2D [15N,1H]-HSQC-based experiments (Farrow et al. 1994). The R1 relaxation rate was measured using 11 different time points: 20, 80 (2×), 150, 250, 350, 450, 600 (2×), 800, 1000 (2×), 1300, and 1600 msec. The R2 relaxation rate was measured using 12 different time points: 10 (2×), 20, 40, 60, 80, 120, 150, 180, 230, 280 (2×), 350, and 400 msec. Saturation in the 15N{1H}-NOE experiment was carried out with a series of 120° high-power 1H pulses separated by 5-msec delays (Markley et al. 1971) applied for a duration of 3.0 sec, during the interscan delay of 5.0 sec. Data processing and analysis were carried out with XWINNMR 3.5, TopSpin 1.3 (Bruker), XEASY (Bartels et al. 1995) and CARA (Keller 2005) (http://www.nmr.ch). Relaxation data were processed with NMRPipe (Delaglio et al. 1995) and analyzed with NMRView (Johnson 2004).
Three-dimensional structure determination
Structure determination was based on experimental NOE data obtained from 3D 15N-resolved [1H,1H]-NOESY, and two 3D 13C-resolved [1H,1H]-NOESY spectra optimized for the aliphatic and aromatic 13C regions. All NOESY spectra were recorded at 800-MHz 1H frequency with samples in 90% H2O/10% D2O and with mixing times of 60 msec. A stand-alone version of the program ATNOS/CANDID (Herrmann et al. 2002a,b) was employed in combination with CYANA version 1.0.3 for molecular dynamics in torsion-angle space (Güntert et al. 1997). The chemical shift lists from the resonance assignment (Johnson et al. 2007) and the three aforementioned NOESY spectra were used as input for ATNOS/CANDID. The standard ATNOS/CANDID/CYANA protocol (Herrmann et al. 2002a,b) comprising seven cycles of NOESY peak picking, NOE assignment, distance restraint generation, and structure calculation was employed. Supplemental dihedral angle restraints derived from 13Cα chemical shifts were employed throughout the calculation (Spera and Bax 1991; Luginbühl et al. 1995). The final set of unambiguous NOE assignments obtained in the last cycle led to 3254 meaningful distance restraints, i.e., on average 18 restraints/residue. The 20 structures with the lowest residual CYANA target function values obtained from cycle 7 were subjected to energy minimization in a shell of water molecules, using the program OPALp (Luginbühl et al. 1995; Koradi et al. 2000) with the Amber force field (Cornell et al. 1995). The quality of the final structures was assessed with PROCHECK (Morris et al. 1992; Laskowski et al. 1993). The atomic coordinates of the bundle of 20 conformers of Table 1 and Figure 1A (accession number 2JMZ) and of the conformer closest to their mean coordinates (Fig. 1B; accession number 2JNQ) have been deposited in the Brookhaven Protein Data Bank (http://www.rcsb.org/pdb/).
Protein expression and mutagenesis for protein splicing assays
A series of variant Mja KlbA intein precursor constructs, each of which included the designed replacements of one or two residues, were constructed in order to test the effects of individual residue positions on protein splicing, using site-directed mutagenesis with the QuikChange kit (Stratagene) as directed by the manufacturer. The constructs were expressed in Escherichia coli T7 Express cells along with pRIL (Stratagene) by induction with 0.4 mM IPTG either for 2 h at 37°C or overnight at 15°C. The protein splicing activity of the different mutant constructs was assessed by three different assays: (1) SDS-PAGE gel electrophoresis of soluble fractions of cell lysates. (2) SDS-PAGE of samples purified by nickel affinity chromatography over Ni-NTA resin (Qiagen) at pH 8.0, as described by the manufacturer. (3) Western blot analysis with anti-6×His antibody (Invitrogen), as described by the manufacturer. Soluble lysates or purified protein were boiled for 5 min in sample buffer with DTT (New England BioLabs), loaded onto 10%–20% SDS-PAGE gels (Invitrogen), and either stained with Coomassie Blue or transferred to nitrocellulose.
Identification of the resulting proteins was accomplished as follows. First, the intact precursor, the single-splice junction cleavage products, and the excised free intein were differentiated from each other by their mobility on the SDS-PAGE gel. The molecular weight of the intact precursor is 21.4 kDa, that of the free intein is 19.5 kDa, and those of the single splice junction cleavage products are 20.7 and 20.1 kDa, respectively. Further experiments were necessary to distinguish between the two possible single-splice junction cleavage products, since they have too closely similar molecular weights to be individually identified by SDS-PAGE. This was achieved considering that the C-terminal single-splice junction cleavage product has lost the C-terminal 6×His tag, while the N-terminal single-splice junction cleavage product retains it. Therefore, the single-splice junction cleavage products were further tested either by Western blotting of the SDS-PAGE gel with an anti-6×His antibody, by Ni-NTA column purification, or by both methods. The C-terminal single-splice junction cleavage product could then be unambiguously identified as a band near 20.5 kDa on the SDS-PAGE gel that showed no reaction with the anti-6×His antibody. The N-terminal single-splice junction cleavage product was identified either by its reaction with the anti-6×His antibody in the Western blot, by its binding affinity to the Ni-NTA column, or by both methods. (We note that if only Western blotting is used, ambiguity exists due to the fact that a band that reacts with the anti-6×His antibody might also contain some of the C-terminal single-splice junction cleavage product. However, only two of the variant proteins described in Table 2 were tested only by Western blotting, and such ambiguity exists only for one of these two, namely the H154A/Y156F variant.) The data reported in Table 2 represent the average of two to five independent experiments.
Acknowledgments
M.A.J. was supported by a fellowship from the Canadian Institutes of Health Research and by the Skaggs Institute for Chemical Biology. K.W. is the Cecil H. and Ida M. Green Professor of Structural Biology at TSRI and a member of the Skaggs Institute for Chemical Biology. F.B.P., M.W.S., and L.B. thank Don Comb for support and encouragement. We acknowledge the use of the high-performance computing facility at The Scripps Research Institute (TSRI).
The NMR analysis was performed by M.A.J., the preparation of protein for NMR analysis was performed by M.W.S., and the mutational analysis was performed by L.B. This is manuscript #18639 from TSRI.
Footnotes
Reprint requests to: Kurt Wüthrich, The Scripps Research Institute, Department of Molecular Biology and Skaggs Institute for Chemical Biology, 10550 North Torrey Pines Road, MB-44, La Jolla, CA 92037, USA; e-mail: wuthrich@scripps.edu; fax: (858) 784-8014.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.072816707.
References
- Bartels C., Xia, T.-H., Billeter, M., Güntert, P., and Wüthrich, K. 1995. The program XEASY for computer-supported NMR spectral analysis of biological macromolecules. J. Biomol. NMR 6: 1–10. [DOI] [PubMed] [Google Scholar]
- Cornell W.D., Cieplak, P., Bayly, C.I., Gould, I.R., Merz Jr, K.M., Ferguson, D.M., Spellmeyer, D.C., Fox, T., Caldwell, J.W., and Kollman, P.A. 1995. A second generation force field for the simulation of proteins, nucleic acids, and organic molecules. J. Am. Chem. Soc. 117: 5179–5197. [Google Scholar]
- David R., Richter, M.P.O., and Beck-Sickinger, A.G. 2004. Expressed protein ligation: Method and applications. Eur. J. Biochem. 271: 663–677. [DOI] [PubMed] [Google Scholar]
- Delaglio F., Grzesiek, S., Vuister, G.W., Zhu, G., Pfeifer, J., and Bax, A. 1995. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6: 277–293. [DOI] [PubMed] [Google Scholar]
- Ding Y., Xu, M.-Q., Ghosh, I., Chen, X., Ferrandon, S., Lesage, G., and Rao, Z. 2003. Crystal structure of a mini-intein reveals a conserved catalytic module involved in side chain cyclization of asparagine during protein splicing. J. Biol. Chem. 278: 39133–39142. [DOI] [PubMed] [Google Scholar]
- Duan X., Gimble, F.S., and Quiocho, F.A. 1997. Crystal structure of PI-SceI, a homing endonuclease with protein splicing activity. Cell 89: 555–564. [DOI] [PubMed] [Google Scholar]
- Evans T.C. and Xu, M.-Q. 2002. Mechanistic and kinetic considerations of protein splicing. Chem. Rev. 102: 4869–4883. [DOI] [PubMed] [Google Scholar]
- Farrow N.A., Muhandiram, R., Singer, A.U., Pascal, S.M., Kay, C.M., Gish, G., Shoelson, S.E., Pawson, T., Forman-Kay, J.D., and Kay, L.E. 1994. Backbone dynamics of a free and a phosphopeptide-complexed Src homology 2 domain studied by 15N NMR relaxation. Biochemistry 33: 5984–6003. [DOI] [PubMed] [Google Scholar]
- Gimble F.S. and Thorner, J. 1992. Homing of a DNA endonuclease gene by meiotic gene conversion in Saccharomyces cerevisiae . Nature 357: 301–306. [DOI] [PubMed] [Google Scholar]
- Güntert P., Mumenthaler, C., and Wüthrich, K. 1997. Torsion angle dynamics for NMR structure calculation with the new program DYANA. J. Mol. Biol. 273: 283–298. [DOI] [PubMed] [Google Scholar]
- Hall T.M.T., Porter, J.A., Young, K.E., Koonin, E.V., Beachy, P.A., and Leahy, D.J. 1997. Crystal structure of a hedgehog autoprocessing domain: Homology between hedgehog and self-splicing proteins. Cell 91: 85–97. [DOI] [PubMed] [Google Scholar]
- Herrmann T., Güntert, P., and Wüthrich, K. 2002a. Protein NMR structure determination with automated NOE-identification in the NOESY spectra using the new software ATNOS. J. Biomol. NMR 24: 171–189. [DOI] [PubMed] [Google Scholar]
- Herrmann T., Güntert, P., and Wüthrich, K. 2002b. Protein NMR structure determination with automated NOE assignment using the new software CANDID and the torsion angle dynamics algorithm DYANA. J. Mol. Biol. 319: 209–227. [DOI] [PubMed] [Google Scholar]
- Holm L. and Sander, C. 1993. Protein structure comparison by alignment of distance matrices. J. Mol. Biol. 233: 123–138. [DOI] [PubMed] [Google Scholar]
- Hu D., Crist, M., Duan, X., Quiocho, F.A., and Gimble, F.S. 2000. Probing the structure of the PI–SceI–DNA complex by affinity cleavage and affinity photocross-linking. J. Biol. Chem. 275: 2705–2712. [DOI] [PubMed] [Google Scholar]
- Ichiyanagi K., Ishino, Y., Ariyoshi, M., Komori, K., and Morizawa, K. 2000. Crystal structure of an archaeal intein-encoded homing endonuclease PI-PfuI. J. Mol. Biol. 300: 889–901. [DOI] [PubMed] [Google Scholar]
- Johnson B.A. 2004. Using NMRView to visualize and analyze the NMR spectra of macromolecules. Methods Mol. Biol. 278: 313–352. [DOI] [PubMed] [Google Scholar]
- Johnson M.A., Southworth, M.W., Perler, F.B., and Wüthrich, K. 2007. NMR assignment of a KlbA intein precursor from Methanococcus jannaschii . Biomol. NMR Assign. in press. [DOI] [PubMed]
- Keller R.L.J. 2005. “Optimizing the process of NMR spectrum analysis and computer aided resonance assignment.” ETH Zürich #15947, Zürich, Switzerland. Thesis.
- Klabunde T., Sharma, S., Telenti, A., Jacobs Jr, W.R., and Sacchettini, J.C. 1998. Crystal structure of GyrA intein from Mycobacterium xenopi reveals structural basis of protein splicing. Nat. Struct. Biol. 5: 31–36. [DOI] [PubMed] [Google Scholar]
- Koradi R., Billeter, M., and Güntert, P. 2000. Point-centered domain decomposition for parallel molecular dynamics simulation. Comput. Phys. Commun. 124: 139–147. [Google Scholar]
- Laskowski R.A., MacArthur, M.W., Moss, D.S., and Thornton, J.M. 1993. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26: 283–291. [Google Scholar]
- Luginbühl P., Szyperski, T., and Wüthrich, K. 1995. Statistical basis for the use of 13Cα chemical shifts in protein structure determination. J. Magn. Reson. B 109: 229–233. [Google Scholar]
- Luginbühl P., Güntert, P., Billeter, M., and Wüthrich, K. 1996. The new program OPAL for molecular dynamics simulations and energy refinements of biological macromolecules. J. Biomol. NMR 8: 136–146. [DOI] [PubMed] [Google Scholar]
- Markley J.L., Horsley, W.J., and Klein, M.P. 1971. Spin-lattice relaxation measurements in slowly relaxing complex spectra. J. Chem. Phys. 55: 3604–3605. [Google Scholar]
- Matsumura H., Takahashi, H., Inoue, T., Yamamoto, T., Hashimoto, H., Nishioka, M., Fujiwara, S., Takagi, M., Imanaka, T., and Kai, Y. 2006. Crystal structure of intein homing endonuclease II encoded in DNA polymerase gene from hyperthermophilic archaeon Thermococcus kodakaraensis strain KOD1. Proteins 63: 711–715. [DOI] [PubMed] [Google Scholar]
- Mizutani R., Nogami, S., Kawasaki, M., Ohya, Y., Anraku, Y., and Satow, Y. 2002. Protein-splicing reaction via a thiazolidine intermediate: Crystal structure of the VMA1-derived endonuclease bearing the N and C-terminal propeptides. J. Mol. Biol. 316: 919–929. [DOI] [PubMed] [Google Scholar]
- Morris A.L., MacArthur, M.W., Hutchinson, E.G., and Thornton, J.M. 1992. Stereochemical quality of protein structure coordinates. Proteins 12: 345–364. [DOI] [PubMed] [Google Scholar]
- Moure C.M., Gimble, F.S., and Quiocho, F.A. 2002. Crystal structure of the intein homing endonuclease PI-SceI bound to its recognition sequence. Nat. Struct. Biol. 9: 764–770. [DOI] [PubMed] [Google Scholar]
- Muhandiram D.R., Farrow, N.A., Xu, G.-Y., Smallcombe, S.H., and Kay, L.E. 1993. A gradient 13C NOESY-HSQC experiment for recording NOESY spectra of 13C-labeled proteins dissolved in H2O. J. Magn. Reson. B 102: 317–321. [Google Scholar]
- Muralidharan V. and Muir, T.W. 2006. Protein ligation: An enabling technology for the biophysical analysis of proteins. Nat. Methods 3: 429–438. [DOI] [PubMed] [Google Scholar]
- Murzin A.G., Brenner, S.E., Hubbard, T., and Chothia, C. 1995. SCOP: A structural classification of proteins database for the investigation of sequences and structures. J. Mol. Biol. 247: 536–540. [DOI] [PubMed] [Google Scholar]
- Noren C.J., Wang, J., and Perler, F.B. 2000. Dissecting the chemistry of protein splicing and its applications. Angew. Chem. Int. Ed. Engl. 39: 450–466. [PubMed] [Google Scholar]
- Otto H.-H. and Schirmeister, T. 1997. Cysteine proteases and their inhibitors. Chem. Rev. 97: 133–171. [DOI] [PubMed] [Google Scholar]
- Paulus H. 2000. Protein splicing and related forms of protein autoprocessing. Annu. Rev. Biochem. 69: 447–496. [DOI] [PubMed] [Google Scholar]
- Paulus H. 2001. Inteins as enzymes. Bioorg. Chem. 29: 119–129. [DOI] [PubMed] [Google Scholar]
- Perler F.B. 2002. InBase: The intein database. Nucleic Acids Res. 30: 383–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perler F.B. 2006. Protein splicing mechanisms and applications. IUBMB Life 57: 469–476. [DOI] [PubMed] [Google Scholar]
- Perler F.B., Comb, D.G., Jack, W.E., Moran, L.S., Qiang, B., Kucera, R.B., Benner, J., Slatko, B.E., Nwankwo, D.O., Hempstead, S.K., et al. 1992. Intervening sequences in an Archaea DNA polymerase gene. Proc. Natl. Acad. Sci. 89: 5577–5581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perler F.B., Olsen, G.J., and Adam, E. 1997. Compilation and analysis of intein sequences. Nucleic Acids Res. 25: 1087–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pervushin K., Riek, R., Wider, G., and Wüthrich, K. 1998. Transverse relaxation-optimized spectroscopy (TROSY) for NMR studies of aromatic spin systems in 13C-labeled proteins. J. Am. Chem. Soc. 120: 6394–6400. [Google Scholar]
- Pietrokovski S. 1994. Conserved sequence features of inteins (protein introns) and their use in identifying new inteins and related proteins. Protein Sci. 3: 2340–2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietrokovski S. 1998. Modular organization of inteins and C-terminal autocatalytic domains. Protein Sci. 7: 64–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietrokovski S. 2001. Intein spread and extinction in evolution. Trends Genet. 17: 465–472. [DOI] [PubMed] [Google Scholar]
- Poland B.W., Xu, M.-Q., and Quiocho, F.A. 2000. Structural insights into the protein splicing mechanism of PI-SceI. J. Biol. Chem. 275: 16408–16413. [DOI] [PubMed] [Google Scholar]
- Saleh L. and Perler, F.B. 2006. Protein splicing in cis and in trans . Chem. Rec. 6: 183–193. [DOI] [PubMed] [Google Scholar]
- Sattler M., Schleucher, J., and Griesinger, C. 1999. Heteronuclear multidimensional NMR experiments for the structure determination of proteins in solution employing pulsed field gradients. Prog. NMR Spectrosc. 34: 93–158. [Google Scholar]
- Southworth M.W. and Perler, F.B. 2002. Protein splicing of the Deinococcus radiodurans strain R1 Snf2 intein. J. Bacteriol. 184: 6387–6388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southworth M.W., Benner, J., and Perler, F.B. 2000. An alternative protein splicing mechanism for inteins lacking an N-terminal nucleophile. EMBO J. 19: 5019–5026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spera S. and Bax, A. 1991. Empirical correlation between protein backbone conformation and Cα and Cβ 13C nuclear magnetic resonance chemical shifts. J. Am. Chem. Soc. 113: 5490–5492. [Google Scholar]
- Sun P., Ye, S., Ferrandon, S., Evans, T.C., Xu, M.-Q., and Rao, Z. 2005. Crystal structures of an intein from the split dnaE gene of Synechocystis sp. PCC6803 reveal the catalytic model without the penultimate histidine and the mechanism of zinc ion inhibition of protein splicing. J. Mol. Biol. 353: 1093–1105. [DOI] [PubMed] [Google Scholar]
- van Roey P., Pereira, B., Li, Z., Hiraga, K., Belfort, M., and Derbyshire, V. 2007. Crystallographic and mutational studies of the Mycobacterium tuberculosis recA mini-inteins suggest a pivotal role for a highly conserved aspartate residue. J. Mol. Biol. 367: 162–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner E., Wende, W., Pingoud, A., and Heinemann, U. 2002. High resolution crystal structure of domain I of the Saccharomyces cerevisiae homing endonuclease PI-SceI. Nucleic Acids Res. 30: 3962–3971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wishart D.S., Bigam, C.G., Yao, J., Abildgaard, F., Dyson, H.J., Oldfield, E., Markley, J.L., and Sykes, B.D. 1995. 1H, 13C and 15N chemical shift referencing in biomolecular NMR. J. Biomol. NMR 6: 135–140. [DOI] [PubMed] [Google Scholar]
- Yamamoto K., Low, B., Rutherford, S.A., Rajagopalan, M., and Madiraju, M.V. 2001. The Mycobacterium avium-intracellulare complex dnaB locus and protein intein splicing. Biochem. Biophys. Res. Commun. 280: 898–903. [DOI] [PubMed] [Google Scholar]
- Zerbe O., Szyperski, T., Ottiger, M., and Wüthrich, K. 1996. Three-dimensional 1H-TOCSY-relayed ct-[13C,1H]-HMQC for aromatic spin system identification in uniformly 13C-labeled proteins. J. Biomol. NMR 7: 99–106. [DOI] [PubMed] [Google Scholar]