

Abstract
Glucose-1-phosphate uridylyltransferase, or UGPase, catalyzes the production of UDP-glucose from glucose-1-phosphate and UTP. Because of the biological role of UDP-glucose in glycogen synthesis and in the formation of glycolipids, glycoproteins, and proteoglycans, the enzyme is widespread in nature. Recently this laboratory reported the three-dimensional structure of UGPase from Escherichia coli. While the initial X-ray analysis revealed the overall fold of the enzyme, details concerning its active site geometry were limited because crystals of the protein complexed with either substrates or products could never be obtained. In an effort to more fully investigate the active site geometry of the enzyme, UGPase from Corynebacterium glutamicum was subsequently cloned and purified. Here we report the X-ray structure of UGPase crystallized in the presence of both magnesium and UDP-glucose. Residues involved in anchoring the ligand to the active site include the polypeptide chain backbone atoms of Ala 20, Gly 21, Gly 117, Gly 180, and Ala 214, and the side chains of Glu 36, Gln 112, Asp 143, Glu 201, and Lys 202. Two magnesium ions are observed coordinated to the UDP-glucose. An α- and a β-phosphoryl oxygen, three waters, and the side chain of Asp 142 ligate the first magnesium, whereas the second ion is coordinated by an α-phosphoryl oxygen and five waters. The position of the first magnesium is conserved in both the glucose-1-phosphate thymidylyltransferases and the cytidylyltransferases. The structure presented here provides further support for the role of the conserved magnesium ion in the catalytic mechanisms of the sugar-1-phosphate nucleotidylyltransferases.
Keywords: structure, crystallography, mechanism-enzymes
The importance of nucleotide-linked sugars in metabolism was recognized nearly 60 years ago by Luis F. Leloir and coworkers when they isolated UDP-glucose from yeast (Caputto et al. 1950). Indeed, Leloir's pioneering research efforts led to his receiving the Nobel Prize in Chemistry in 1970. Since these initial investigations, an enormous body of literature has accumulated with respect to the structure and function of nucleotide-linked sugars and the enzymes that are responsible for their synthesis. In general, there are two major biochemical roles for nucleotide-linked sugars: as intermediates in the formation of monosaccharides used in the production of complex carbohydrates and as glycosyl donors of these monosaccharides.
In recent years, our laboratory has been studying various enzymes that are involved in the biosynthesis of unusual di- and trideoxysugars found in the lipopolysaccharides of some Gram-negative bacteria (Schnaitman and Klena 1993) and in certain macrolide antibiotics (Trefzer et al. 1999). These sugars are produced via distinct biosynthetic pathways using dTDP-, CDP-, and GDP-activated sugars as the initiating ligands (Liu and Thorson 1994). The pathways for the synthesis of these unusual sugars typically begin with the attachment of α-D-glucose-1-phosphate or α-D-mannose-1-phosphate to an NMP moiety via a nucleotidylyltransferase and the concomitant loss of pyrophosphate. The three-dimensional structures of the thymidylyltransferases (Blankenfeldt et al. 2000; Barton et al. 2001, 2002; Zuccotti et al. 2001; Sivaraman et al. 2002) and the cytidylyltransferases (Koropatkin and Holden 2004; Koropatkin et al. 2005) have been well characterized.
To expand our understanding of these types of nucleotidylyltransferases, we recently solved the three-dimensional structure of glucose-1-phosphate uridylyltransferase (UGPase) from Escherichia coli, which catalyzes a similar reaction, namely, the production of UDP-glucose from UTP and glucose-1-phosphate (Scheme 1). Given the central role of UDP-glucose in galactose utilization (Holden et al. 2003), in glycogen synthesis (Alonso et al. 1995), and in the production of the carbohydrate moieties of glycolipids (Sandhoff et al. 1992), glycoproteins (Roth 1995; Verbert 1995), and proteoglycans (Silbert and Sugumaran 1995), the enzyme is ubiquitous in nature. Strikingly, the prokaryotic and eukaryotic forms of the enzyme are completely unrelated in amino acid sequence and three-dimensional structure (Flores-Diaz et al. 1997; Mollerach et al. 1998; Mollerach and Garcia 2000).
Scheme 1.

A ribbon representation of the E. coli UGPase tetramer in its unliganded form is depicted in Figure 1. Each subunit of the tetramer contains an eight-stranded mixed β-sheet with two additional layers of β-sheet and 10 α-helices. The tetramer displays 222 symmetry and can be described as a dimer of dimers. As labeled in Figure 1, Subunits 1 and 4 and 2 and 3 form the “tight” dimers with their C-terminal helices wrapping around one another. The overall fold of UGPase is similar to that observed for the glucose-1-phosphate thymidylyltransferases. Attempts to prepare crystals of the E. coli UGPase in the presence of its substrates, glucose-1-phosphate and UTP, or its product UDP-glucose were unsuccessful. Consequently, to more fully define the active site of the enzyme, we cloned, overexpressed, purified, and solved the three-dimensional structure of UGPase from Corynebacterium glutamicum complexed with UDP-glucose and magnesium to 2.0 Å resolution. Here we describe the detailed geometry of the UGPase active site and compare its mode of product binding to that observed for both the thymidylyltransferases and the cytidylyltransferases.
Figure 1.

Ribbon representation of the UGPase from E. coli. The structure was determined to 1.9 Å resolution in the absence of bound substrates, products, or analogs (Thoden and Holden 2007). The enzyme is a tetramer with 222 symmetry and can be aptly described as a dimer of dimers. The “tight” dimers are formed by Subunits 1 and 4 and 2 and 3.
Results and Discussion
The structure of UGPase complexed with UDP-glucose and magnesium was solved and refined to 2.0 Å resolution with an R-factor and R-free of 20.4% and 24.3%, respectively. The asymmetric unit contained a complete tetramer with molecular dimensions of ∼100 Å × 70 Å × 90 Å. In all of the subunits, the first nine to 11 residues were disordered, and several residues were missing at the C terminus as well. Also, there was a break in the electron density between Arg 89 and Asp 93 in Subunit 4. Not surprisingly, given that the amino acid sequence identity between the C. glutamicum and E. coli UGPases is 40%, the C. glutamicum UGPase quaternary structure is basically the same as that shown in Figure 1 for the E. coli enzyme. The total buried surface area for the “tight” dimer is ∼5600 Å2, whereas that for the “loose” dimer is 630 Å2. The four subunits of the C. glutamicum UGPase tetramer are similar such that their α-carbons superimpose with root-mean-square deviations of between 0.24 Å and 0.31 Å. Thus, for the sake of clarity, the following discussion refers only to Subunit 1 in the X-ray coordinate file unless otherwise noted.
Overall, the ϕ, ψ angles for the complete tetramer are good with 92.4%, 6.9%, 0.3%, and 0.4% lying in the core, allowed, generously allowed, and disallowed regions of the Ramachandran plot, respectively. The only significant outlier is Val 41 with dihedral angles of ϕ = 72.3° and ψ = −78.1°. This residue is positioned in a sharp reverse turn located in the random-coil region connecting β-strand 1 to the first α-helix of the subunit. It is also a valine in the E. coli UGPase and adopts the same strained dihedral angles. There are two cis prolines in the C. glutamicum UGPase at Pro 28, which is conserved in the E. coli enzyme, and Pro 147, which is a trans serine in the E. coli UGPase. Pro 147 resides in a surface loop connecting β-strand 5 to α-helix 6, whereas Pro 28 sits in an approximate Type III reverse turn formed by Leu 27 to Thr 30. Neither is close to the active site pocket.
A ribbon representation of Subunit 1 is presented in Figure 2A. The subunit has molecular dimensions of ∼50 Å × 50 Å × 60 Å and is dominated by a mixed β-sheet composed of nine strands. This β-sheet serves to cradle the UDP-glucose moiety. In addition to this large β-sheet, there is also a two-stranded antiparallel β-sheet, which forms another side of the active site pocket. The subunit also contains 11 major α-helical regions. As can be seen in Figure 2A, the subunit is roughly globular except for two regions, both of which form part of the “tight” dimer interface. The first of these is the helix–loop–helix motif delineated by Ser 80 to Leu 104, which curls into the active site of the second subunit of the tight dimer (Fig. 2B). As is discussed below, this loop was disordered in the E. coli enzyme structure, and it was postulated to become ordered upon substrate or product binding (Thoden and Holden 2007). The second region that extends the otherwise globular nature of the UGPase subunit is the C-terminal helix–loop–helix motif formed by Pro 270 to Ala 305. This motif wraps around its twofold-related partner from Subunit 4 as shown in Figure 2B.
Figure 2.
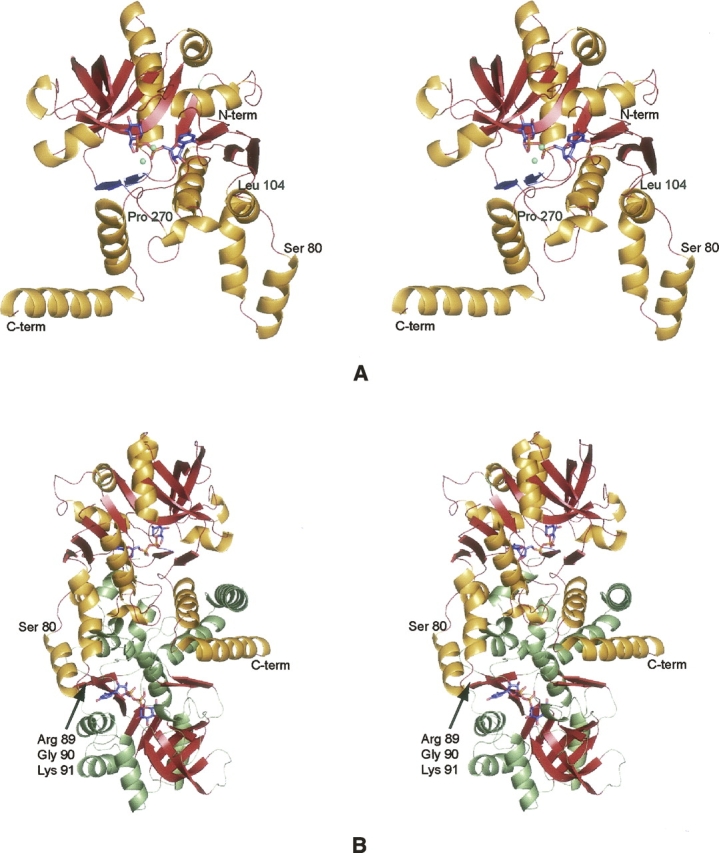
The structure of UGPase from C. glutamicum. (A) A stereoview of one subunit of the tetramer. The subunit structure is characterized by a nine-stranded mixed β-sheet as highlighted in red. There is a second two-stranded β-sheet as displayed in blue. The positions of the bound magnesium ions are indicated by the green spheres whereas the UDP-glucose ligand is depicted in a stick representation. (B) Stereoview of one of the “tight” dimers of the tetramer. Subunit 1 is depicted in red and gold whereas Subunit 2 is displayed in red and green. Note that the loop defined by Arg 89, Gly 90, and Lys 91 from Subunit 1 is located near the active site of Subunit 2.
Electron density corresponding to the (Mg2+)2UDP-glucose ligand in Subunit 1 is displayed in Figure 3A. Average B-values for these ligands in Subunits 1, 2, 3, and 4 were 32.3 Å2, 34.3 Å2, 35.8 Å2, and 34.7 Å2, respectively. The ribose of the nucleotide adopts the C2′-endo pucker, and the uracil ring is in the anti-conformation. Both magnesium ions are octahedrally coordinated by oxygen-containing ligands. One of the magnesium ions is ligated by an α- and a β-phosphoryl oxygen from the UDP-glucose, three water molecules, and the side chain of Asp 142. The specific metal:ligand distances, which range from 2.0 to 2.2 Å, are listed in Table 1. The second magnesium ion is coordinated by an α-phosphoryl oxygen and five waters with metal:ligand distances ranging from 2.0 to 2.3 Å. In the case of glucose-1-phosphate cytidylyltransferase, there is some indication in the kinetic data that a second Mg2+ ion may be required in addition to that forming the Mg2+-CTP complex (Koropatkin et al. 2005). Only one magnesium ion was observed, however, in the cytidylyltransferase/UDP-glucose structure (Koropatkin and Holden 2004).
Figure 3.
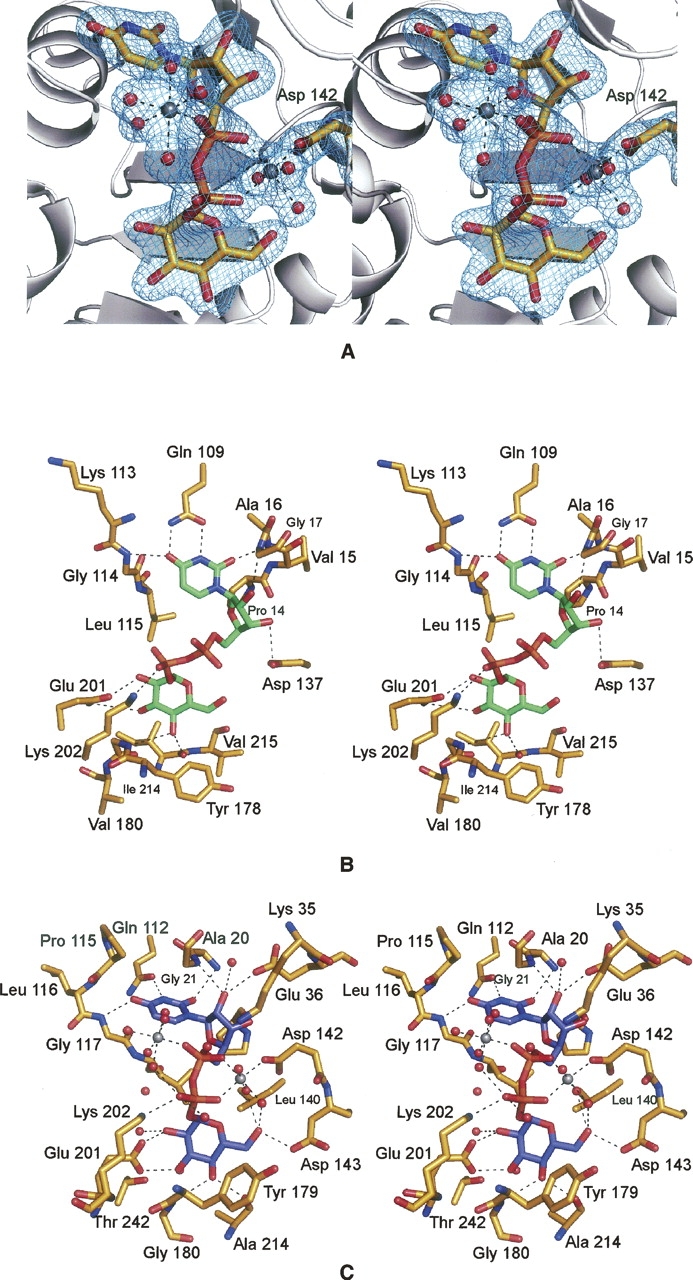
UGPase active sites. (A) Electron density corresponding to the two magnesium ions and UDP-glucose as observed in Subunit 1. The map was contoured at 4σ and was calculated with coefficients of the form (F O − F C), where F O and F C were the native and the calculated structure factor amplitudes, respectively. For the map calculation, the X-ray coordinates for the cations and the ligand were removed from the protein model. (B) Prediction of the UDP-glucose binding mode to E. coli UGPase. On the basis of the similarity between the E. coli forms of both UGPase and glucose-1-phosphate thymidylyltransferase, the manner in which UDP-glucose binds to UGPase was predicted as shown (Thoden and Holden 2007). UDP-glucose is displayed in green. Potential hydrogen bonds are indicated by the dashed lines. (C) Close-up view of the C. glutamicum UGPase active site. Those residues located within ∼3.2 Å of the UDP-glucose (highlighted in blue) are shown. Potential hydrogen bonds are depicted as dashed lines. The coordination geometries about the two magnesium ions, displayed in gray, are also indicated by dashed lines. Ordered water molecules are represented as red spheres.
Table 1.
Magnesium-ligand coordination distances observed in Subunit 1

In our previous study of the E. coli UGPase, we predicted the manner in which the product binds to the active site pocket as indicated in Figure 3B (Thoden and Holden 2007). This prediction was based on the structural similarities between UGPase and the well-characterized glucose-1-phosphate thymidylyltransferases. The quality of the modeling was limited, however, since there were no structures in the Protein Data Bank of a glucose-1-phosphate thymidylyltransferase complexed with both dTDP-glucose and magnesium. Thus we were unable to predict the location of the catalytically relevant cation when UDP-glucose is bound to UGPase, and this influenced our model of product binding within the active site cleft.
A close-up view of the C. glutamicum UGPase active site with bound UDP-glucose is presented in Figure 3C. The uracil ring of the product lies within hydrogen-bonding distance to the backbone nitrogens of Ala 20 and Gly 117 and the side chain carboxamide group of Gln 112. The ribose 2-hydroxyl group is situated within hydrogen-bonding distance to the side chain of Glu 36, a water molecule, and the backbone amide nitrogen of Gly 21. In our model of the E. coli enzyme, we postulated that Asp 137 would hydrogen-bond to the 3-hydroxyl group of the ribose (Fig. 3B). As indicated in Figure 3C, however, this aspartate (Asp 142) actually functions as a metal ion ligand. In the C. glutamicum enzyme, Lys 202 interacts with a β-phosphoryl oxygen of UDP-glucose, and Glu 201 hydrogen-bonds to both the 2′- and 3′-hydroxyl groups of the glycosyl moiety. The 4′-hydroxyl of the glycosyl group is anchored into the active site via interactions with the backbone nitrogen of Gly 180 and the carbonyl oxygen of Ala 214.
In the glucose-1-phosphate thymidylyltransferases, which are tetrameric, each active site is wholly contained within one subunit. This is in marked contrast to the hexameric glucose-1-phosphate cytidylyltransferases whereby the active sites are formed by residues from two subunits. One question that arises is whether, in the case of UGPase, the active site is contained within one subunit or formed by two monomers. As indicated in Figure 3C, all of the residues located within 3.2 Å of the product are contributed by one subunit. In the E. coli UGPase structure, solved in the absence of any substrates, products, or analogs, there was a disordered region between Glu 83 and Arg 88 that in one subunit was situated within ∼14 Å of the active site of the second subunit. It is this loop region where there is a 22-residue insertion in UGPase relative to the glucose-1-phosphate thymidylyltransferases. It was thus predicted in this first study of UGPase that this loop would become ordered upon substrate or product binding. Shown in Figure 4 is a superposition of the UGPases from E. coli and C. glutamicum near this loop region. As predicted, in the C. glutamicum UGPase with bound product, this loop is ordered, and both Arg 89 and Lys 91 from one subunit lie within ∼6 Å of the active site cavity of another subunit. It should be noted, however, that this loop is flexible and is disordered in Subunit 4.
Figure 4.

Superposition of the UGPases from E. coli and C. glutamicum. The major difference between the three-dimensional structures of the UGPases from E. coli and C. glutamicum is a loop region that becomes ordered upon product binding. Shown is a close-up view of this region. The UGPase from C. glutamicum is displayed in yellow and green to distinguish between Subunits 1 and 4, respectively. The light blue ribbon corresponds to the E. coli UGPase and as can be seen, the loop delineated by Glu 83 to Arg 88, indicated by the red asterisks, is missing. Note the manner in which the positively charged side chains of Arg 89 and Lys 91 from Subunit 1 project into the active site of Subunit 2 in the C. glutamicum enzyme. The bound magnesium ions are displayed as gray spheres.
To date, the catalytic mechanisms for both the glucose-1-phosphate thymidylyltransferases and the cytidylyltransferases have been well-characterized, and the reactions are known to proceed via an S N2 nucleophilic attack of the glucose-1-phosphate phosphoryl oxygen on the α-phosphorus of the nucleotide (Blankenfeldt et al. 2000; Barton et al. 2001; Zuccotti et al. 2001; Koropatkin et al. 2005). Both the thymidylyltransferases and cytidylyltransferases require divalent cations for activity (Zuccotti et al. 2001; Koropatkin et al. 2005). As discussed above, however, these ions were not observed in the crystal structures of the thymidylyltransferases solved in the presence of product (most likely because of crystallization conditions). However, they were observed in the structures of the thymidylyltransferase/dTDP complex (Sivaraman et al. 2002) and the cytidylyltransferase/UDP-glucose and cytidylyltransferase/UTP complexes (Koropatkin and Holden 2004; Koropatkin et al. 2005).
A superposition of the UGPase/UDP-glucose and the thymidylyltransferase/UDP-glucose models near the active site pockets is presented in Figure 5A. Overall, the active sites are remarkably similar. However, owing to the lack of magnesium ions in the thymidylyltransferase/UDP-glucose complex, the ligand is shifted in the active site away from Lys 26 and toward Lys 163. While magnesium is not necessary for product binding, enzymological analyses clearly implicate its importance in catalysis (Bernstein and Robbins 1965; Zuccotti et al. 2001). The only known structure of a glucose-1-phosphate thymidylyltransferase with a bound magnesium ion (in a physiologically relevant position) is that determined by Sivaraman et al. (2002). The structure was solved to 2.6 Å resolution in the presence of dTTP and Mg2+. Shown in Figure 5B is a superposition of it onto the UGPase structure. Note the nearly identical positions of the magnesium ions in the two enzyme models. In the thymidylyltransferase complex, the magnesium is coordinated by an α-phosphoryl oxygen from the nucleotide and the carboxylate groups of Asp 108 and Asp 223. Given the resolution of the X-ray data, it was not possible to completely define the coordination sphere for the cation, although was it thought to be octahedral and completed by two waters. The distances between the magnesium and the oxygens of Asp 223 are quite long, however, at ∼2.6 Å. In UGPase, a similar interaction is observed between the magnesium ion and Asp 142 (which is structurally homologous to Asp 108 in the thymidylyltransferase).
Figure 5.
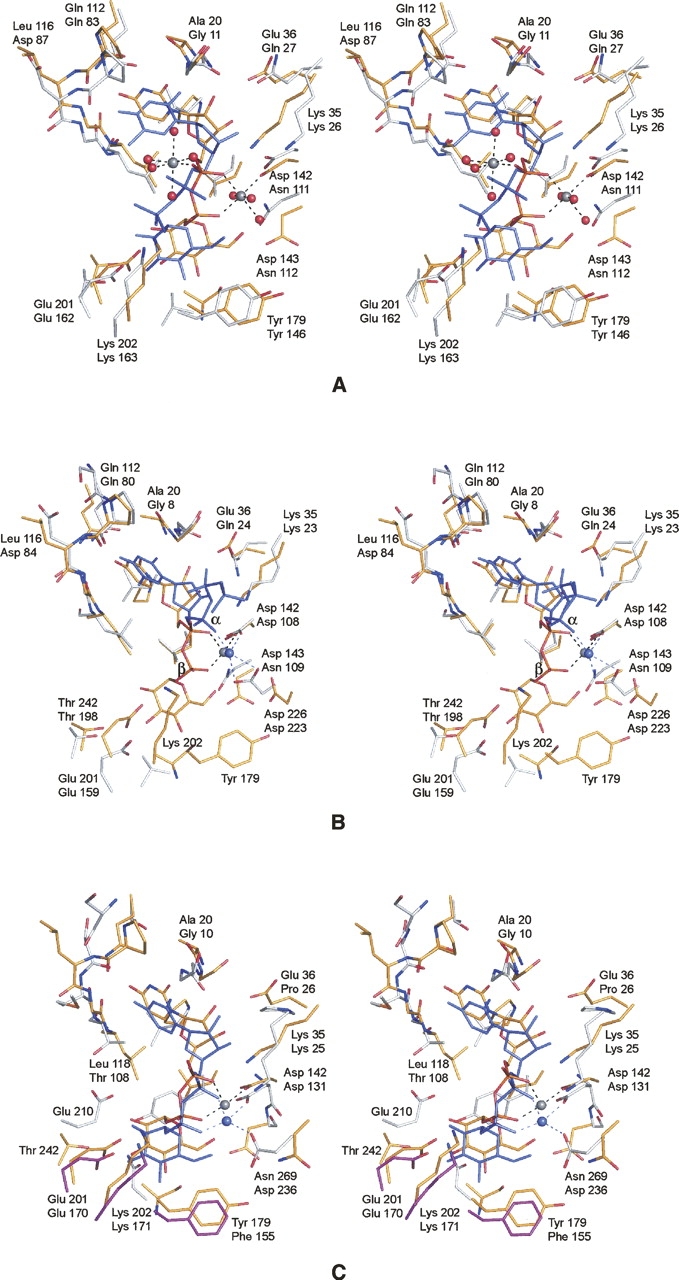
Comparison of UGPase with glucose-1-phosphate thymidylyltransferase and glucose-1-phosphate cytidylyltransferase. (A) Comparison of UGPase and the E. coli thymidylyltransferase complexed with UDP-glucose. X-ray coordinates for the thymidylyltransferase were obtained from the Protein Data Bank (accession code no. 1H5T). The protein atoms and the UDP-glucose ligand in the UGPase model are depicted in yellow bonds. The corresponding residues in the thymidylyltransferase are displayed in white bonds, whereas the UDP-glucose is depicted in blue. In each pair of labels, the top and bottom numbers refer to residues in UGPase and the thymidylyltransferase, respectively. (B) Comparison of the UGPase structure to the E. coli thymidylyltransferase complexed with magnesium and dTDP. The color coding is as described in A. X-ray coordinates were obtained from the Protein Data Bank (accession code no. 1MC3). Note the close structural correspondence of the bound magnesium ions. (C) Comparison of UGPase to glucose-1-phosphate cytidylyltransferase from Salmonella typhi. The color scheme is as described in A except for Glu 170, Lys 217, and Phe 155, which in the cytidylyltransferase are magenta to emphasize that they are contributed by a second subunit in the hexamer.
A comparison of UGPase with glucose-1-phosphate cytidylyltransferase from Salmonella typhi is presented in Figure 5C. The similarity between the two active sites is remarkable especially in light of the differences in their quaternary structures. Note that while Glu 201 (Glu 170), Lys 202 (Lys 217), and Tyr 179 (Phe 155) are located in similar positions in the active sites of these two enzymes, in the case of UGPase they are from the same subunit, whereas in the cytidylyltransferase, they are contributed by a second subunit. Like that observed in UGPase, the magnesium ion in the cytidylyltransferase assumes an octahedral coordination sphere, but in this case it is ligated by two waters (not shown for clarity), the carboxylate oxygens of Asp 131 and Asp 236, and an α- and a β-phosphoryl oxygen from the nucleotide. While Asp 131 structurally corresponds to Asp 142 in UGPase, Asp 236 is an asparagine in UGPase.
While initial reports on the glucose-1-phosphate nucleotidylyltransferases appeared in the literature well over 25 years ago, the last few years have witnessed a renewed interest in their structure and function. This has occurred, in part, because of their biological roles in the production of activated nucleotide-linked sugars, which are absolutely required for the synthesis of the unusual deoxysugars found in the lipopolysaccharides of some Gram-negative bacteria (Schnaitman and Klena 1993), in extracellular polysaccharides (Reeves 1994), and in certain macrolide antibiotics (Trefzer et al. 1999). By comparing the differences in dTTP binding in the thymidylyltransferase with UDP-glucose binding in UGPase (Fig. 5B), it can be speculated that when the substrates, glucose-1-phosphate and UTP, are bound in the active site of UGPase, the catalytically important magnesium ion is coordinated by a phosphoryl oxygen from the sugar-phosphate and an α-phosphoryl oxygen from UTP. As proposed by Sivaraman et al. (2002), the magnesium ion likely enhances the catalytic activity of UGPase by both charge neutralization and by properly orienting the phosphoryl oxygen of glucose-1-phosphate for nucleophilic attack on the α-phosphorus of UTP. Most likely all the bacterial glucose-1-phosphate nucleotidylyltransferases, including the cytidylyltransferases and the still structurally uncharacterized glucose-1-phosphate guanylyltransferases, catalyze their reactions in a similar manner.
Materials and Methods
Molecular cloning of the GalU gene
Genomic DNA from C. glutamicum (ATCC 13,032) was prepared by standard methods. The galU gene, without a 3′ stop codon to express a noncleavable C-terminally tagged protein, was PCR-amplified using primers that introduced a 5′ NdeI site and a 3′ XhoI site. The purified PCR product was A-tailed and ligated into the pGEM-T (Promega) vector for screening and sequencing. A GalU-pGEM-T vector construct of the correct sequence was then appropriately digested and ligated into a similarly digested pET31b(+) (Novagen) vector. The ligation mixture was used to transform E. coli DH5-α cells, which were plated onto LB Agar supplemented with 100 μg/mL ampicillin. Individual colonies were selected and cultured overnight, and the plasmid DNA was extracted with a Qiaprep spin miniprep kit (QIAGEN). Plasmids were tested for incorporation of the GalU gene by digesting with both NdeI and XhoI.
Protein expression and purification
The GalU-pET31 plasmid was used to transform Rosetta (DE3) E. coli cells (Novagen). The culture was grown at 37°C with shaking until an optical density of 0.7 was reached at 600 nm. The cultures were then induced with 1 mM IPTG and allowed to grow for an additional 5 h before harvesting by centrifugation at 5000g for 15 min. The cell paste was frozen in liquid nitrogen and stored at −80°C.
All protein purification steps were carried out at 4°C unless otherwise noted. The cells were disrupted by sonication on ice. The lysate was cleared by centrifugation, and GalU was purified using Ni-NTA resin (QIAGEN) according to the manufacturer's instructions. The GalU protein sample was dialyzed against 10 mM Tris-HCl and 200 mM NaCl at pH 8.0. Following dialysis, the sample was concentrated to 15 mg/mL based on an extinction coefficient of 6.4 (mg/mL)−1 · cm−1 as calculated with the program PROTEAN (DNAstar).
Enzymatic activity
Enzymatic activity was determined via HPLC chromatography. For each reaction, 0.1 mg of enzyme was added to a 1-mL solution composed of 2 mM UTP, 4 mM MgCl2, and 2 mM sugar-1-phosphate buffered at pH 8 with 25 mM HEPPS. The reactions were allowed to proceed for 30 min at ambient temperature, after which time the enzyme was removed by filtration. The reaction mixture was loaded onto a 1-mL Resource-Q column equilibrated in 10 mM ammonium bicarbonate (pH 8), and the reaction products were separated by a linear gradient of ammonium carbonate to a final concentration of 750 mM. Retention times for known samples of UTP, UDP, UMP, UDP-glucose, and UDP-galactose were determined to allow for comparisons with the reaction products. Both glucose-1-phosphate and galactose-1-phosphate were verified to be substrates for the enzyme.
Crystallization of GalU
Crystallization conditions were first surveyed by the hanging-drop method of vapor diffusion with a sparse matrix screen developed in the laboratory. Large crystals were subsequently grown at room temperature via the hanging-drop method of vapor diffusion. The protein samples contained 10 mM Mg2UDP-glucose, and the precipitant solutions were composed of 20%–23% poly(ethylene) glycol 3400, 200 mM MgCl2 buffered at pH 6.0 with 100 mM MES. The crystals grew to maximum dimensions of 0.8 mm × 0.3 mm × 0.2 mm in ∼2–3 wk. They belonged to the space group C2 with unit cell dimensions of a = 173.4 Å, b = 47.6 Å, c = 161.6 Å, β = 102.7°, and one tetramer in the asymmetric unit.
Structural analysis of GalU
All in-house X-ray data were measured at 4°C with a Bruker HISTAR area detector system. The X-ray source was CuKα radiation from a Rigaku RU200 X-ray generator operated at 50 kV and 90 mA. The X-ray data were processed and scaled with SAINT (Bruker AXS, Inc.). The final native data set used in the refinement of the UGPase/product model was collected at the Advanced Photon Source on the SBC-CAT beamline 19-ID. These data were processed and scaled with HKL3000 (Minor et al. 2006). Relevant X-ray data collection statistics are presented in Table 2.
Table 2.
X-ray data collection statistics

Crystals for in-house data collection were stabilized by harvesting into a synthetic solution composed of 25% poly(ethylene) glycol 3400, 200 mM NaCl, 150 mM MgCl2, 10 mM Mg2UDP-glucose, and 100 mM MES (pH 6.0). For synchrotron data collection, the harvested crystals were serially transferred to a cryo-protectant solution composed of 30% poly(ethylene) glycol 3400, 250 mM NaCl, 150 mM MgCl2, 10 mM Mg2UDP-glucose, 10% ethylene glycol, and 100 mM MES (pH 6.0).
The structure of UGPase was solved by single isomorphous replacement using a crystal soaked in 1 mM methylmercury acetate for 24 h. Six mercury-binding sites were identified with the program SOLVE (Terwilliger and Berendzen 1999), giving an overall figure-of-merit of 0.2 to 3.0 Å resolution. Solvent flattening and molecular averaging with RESOLVE (Terwilliger 2000) generated an interpretable electron density map. A preliminary model for one subunit was built on the basis of this averaged map. This model served as a search probe for molecular replacement with the software package PHASER (Storoni et al. 2004) against the high-resolution synchrotron X-ray data set. Alternate cycles of manual model building and least-squares refinement with TNT (Tronrud et al. 1987) reduced the R-factor to 20.4% for all measured X-ray data from 20.0 to 2.0 Å resolution. Relevant refinement statistics are given in Table 3.
Table 3.
Least-squares refinement statistics

Protein data bank deposition
X-ray coordinates have been deposited in the Protein Data Bank (2PA4).
Note added in proof
Since this paper was accepted, another manuscript appeared online describing the structure of UGPase from Sphingomonas elodea complexed with glucose-1-phosphate (Aragão et al. 2007). This structure, in combination with our results, provides a more detailed understanding of the active site geometry for the UGPases.
Acknowledgments
This research was supported by a grant from the NIH (DK47814 to H.M.H.). We thank Ivan Rayment, W.W. Cleland, and Paul Cook for helpful discussions. The assistance of Changsoo Chang during the X-ray data collection at the Structural Biology Center, Argonne National Laboratory is gratefully acknowledged. The Structural Biology Center is funded by the U.S. Department of Energy, Office of Biological and Environmental Research under contract DE-AC02-06CH11357.
Footnotes
Reprint requests to: Hazel M. Holden, Department of Biochemistry, University of Wisconsin, Madison, 433 Babcock Drive, Madison, WI 53706, USA; e-mail: Hazel_Holden@biochem.wisc.edu; fax: (608) 262-1319.
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.072864707.
References
- Alonso M.D., Lomako, J., Lomako, W.M., and Whelan, W.J. 1995. A new look at the biogenesis of glycogen. FASEB J. 9: 1126–1137. [DOI] [PubMed] [Google Scholar]
- Aragão D., Fialho, A.M., Marques, A.R., Mitchell, E.P., Sá-Correia, I., and Frazão, C. 2007. The complex of Sphingomonas elodea ATCC 31461 glucose-1-phosphate uridylyltransferase with glucose-1-phosphate reveals a novel quaternary structure, unique amongst NDP-sugar pyrophosphorylase members. J. Bacteriol. doi: 10.1128/JB.00277-07. [DOI] [PMC free article] [PubMed]
- Barton W.A., Lesniak, J., Biggins, J.B., Jeffrey, P.D., Jiang, J., Rajashankar, K.R., Thorson, J.S., and Nikolov, D.B. 2001. Structure, mechanism, and engineering of a nucleotidylyltransferase as a first step toward glycorandomization. Nat. Struct. Biol. 8: 545–551. [DOI] [PubMed] [Google Scholar]
- Barton W.A., Biggins, J.B., Jiang, J., Thorson, J.S., and Nikolov, D.B. 2002. Expanding pyrimidine diphosphosugar libraries via structure-based nucleotidylyltransferase engineering. Proc. Natl. Acad. Sci. 99: 13397–13402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein R.L. and Robbins, P.W. 1965. Control aspects of uridine 5′-diphosphate glucose and thymidine 5′-diphosphate glucose synthesis by microbial enzymes. J. Biol. Chem. 240: 391–397. [PubMed] [Google Scholar]
- Blankenfeldt W., Asuncion, M., Lam, J.S., and Naismith, J.H. 2000. The structural basis of the catalytic mechanism and regulation of glucose-1-phosphate thymidylyltransferase (RmlA). EMBO J. 19: 6652–6663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caputto R., Leloir, L.F., Cardini, C.E., and Paladini, A.C. 1950. Isolation of the coenzyme of the galactose phosphate-glucose phosphate transformation. J. Biol. Chem. 184: 333–350. [PubMed] [Google Scholar]
- Flores-Diaz M., Alape-Giron, A., Persson, B., Pollesello, P., Moos, M., von Eichel-Streiber, C., Thelestam, M., and Florin, I. 1997. Cellular UDP-glucose deficiency caused by a single point mutation in the UDP-glucose pyrophosphorylase gene. J. Biol. Chem. 272: 23784–23791. [DOI] [PubMed] [Google Scholar]
- Holden H.M., Rayment, I., and Thoden, J.B. 2003. Structure and function of enzymes of the Leloir pathway for galactose metabolism. J. Biol. Chem. 278: 43885–43888. [DOI] [PubMed] [Google Scholar]
- Koropatkin N.M. and Holden, H.M. 2004. Molecular structure of α-D-glucose-1-phosphate cytidylyltransferase from Salmonella typhi . J. Biol. Chem. 279: 44023–44029. [DOI] [PubMed] [Google Scholar]
- Koropatkin N.M., Cleland, W.W., and Holden, H.M. 2005. Kinetic and structural analysis of α-D-glucose-1-phosphate cytidylyltransferase from Salmonella typhi . J. Biol. Chem. 280: 10774–10780. [DOI] [PubMed] [Google Scholar]
- Liu H.W. and Thorson, J.S. 1994. Pathways and mechanisms in the biogenesis of novel deoxysugars by bacteria. Annu. Rev. Microbiol. 48: 223–256. [DOI] [PubMed] [Google Scholar]
- Minor W., Cymborowski, M., Otwinowski, Z., and Chruszcz, M. 2006. HKL-3000: The integration of data reduction and structure solution—From diffraction images to an initial model in minutes. Acta Crystallogr. D Biol. Crystallogr. 62: 859–866. [DOI] [PubMed] [Google Scholar]
- Mollerach M. and Garcia, E. 2000. The galU gene of Streptococcus pneumoniae that codes for a UDP-glucose pyrophosphorylase is highly polymorphic and suitable for molecular typing and phylogenetic studies. Gene 260: 77–86. [DOI] [PubMed] [Google Scholar]
- Mollerach M., Lopez, R., and Garcia, E. 1998. Characterization of the galU gene of Streptococcus pneumoniae encoding a uridine diphosphoglucose pyrophosphorylase: A gene essential for capsular polysaccharide biosynthesis. J. Exp. Med. 188: 2047–2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeves P.R. 1994. Bacterial cell wall. Elsevier Science, Oxford, UK.
- Roth J. 1995. Compartmentation of glycoprotein biosynthesis. In Glycoproteins (eds. J. Montreuil et al.), pp. 237–312. Elsevier, Amsterdam.
- Sandhoff K., van Echten, G., Schroder, M., Schnabel, D., and Suzuki, K. 1992. Metabolism of glycolipids: The role of glycolipid-binding proteins in the function and pathobiochemistry of lysosomes. Biochem. Soc. Trans. 20: 695–699. [DOI] [PubMed] [Google Scholar]
- Schnaitman C.A. and Klena, J.D. 1993. Genetics of lipopolysaccharide biosynthesis in enteric bacteria. Microbiol. Rev. 57: 655–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silbert J.E. and Sugumaran, G. 1995. Intracellular membranes in the synthesis, transport, and metabolism of proteoglycans. Biochim. Biophys. Acta 1241: 371–384. [DOI] [PubMed] [Google Scholar]
- Sivaraman J., Sauve, V., Matte, A., and Cygler, M. 2002. Crystal structure of Escherichia coli glucose-1-phosphate thymidylyltransferase (RffH) complexed with dTTP and Mg2+ . J. Biol. Chem. 277: 44214–44219. [DOI] [PubMed] [Google Scholar]
- Storoni L.C., McCoy, A.J., and Read, R.J. 2004. Likelihood-enhanced fast rotation functions. Acta Crystallogr. D Biol. Crystallogr. D60: 432–438. [DOI] [PubMed] [Google Scholar]
- Terwilliger T.C. 2000. Maximum-likelihood density modification. Acta Crystallogr. D Biol. Crystallogr. 56: 965–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terwilliger T.C. and Berendzen, J. 1999. Automated MAD and MIR structure solution. Acta Crystallogr. D Biol. Crystallogr. 55: 849–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thoden J.B. and Holden, H.M. 2007. The molecular architecture of glucose-1-phosphate uridylyltransferase. Protein Sci. 16: 432–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trefzer A., Salas, J.A., and Bechthold, A. 1999. Genes and enzymes involved in deoxysugar biosynthesis in bacteria. Nat. Prod. Rep. 16: 283–299. [DOI] [PubMed] [Google Scholar]
- Tronrud D.E., Ten Eyck, L.F., and Matthews, B.W. 1987. An efficient general-purpose least-squares refinement program for macromolecular structures. Acta Crystallogr. A 43: 489–501. [Google Scholar]
- Verbert A. 1995. From Glc3MangGlcNAc2-protein to Man5GlcNAc2-protein: Transfer ‘en bloc’. In Glycoproteins (eds. J. Montreuil et al.), pp. 145–152. Elsevier, Amsterdam.
- Zuccotti S., Zanardi, D., Rosano, C., Sturla, L., Tonetti, M., and Bolognesi, M. 2001. Kinetic and crystallographic analyses support a sequential-ordered bi bi catalytic mechanism for Escherichia coli glucose-1-phosphate thymidylyltransferase. J. Mol. Biol. 313: 831–843. [DOI] [PubMed] [Google Scholar]