

Abstract
In bacteria, the dehydration of 2-methylcitrate to yield 2-methylaconitate in the 2-methylcitric acid cycle is catalyzed by a cofactor-less (PrpD) enzyme or by an aconitase-like (AcnD) enzyme. Bacteria that use AcnD also require the function of the PrpF protein, whose function was previously unknown. To gain insights into the function of PrpF, the three-dimensional crystal structure of the PrpF protein from the bacterium Shewanella oneidensis was solved at 2.0 Å resolution. The protein fold of PrpF is strikingly similar to those of the non-PLP-dependent diaminopimelate epimerase from Haemophilus influenzae, a putative proline racemase from Brucella melitensis, and to a recently deposited structure of a hypothetical protein from Pseudomonas aeruginosa. Results from in vitro studies show that PrpF isomerizes trans-aconitate to cis-aconitate. It is proposed that PrpF catalysis of the cis–trans isomerization proceeds through a base-catalyzed proton abstraction coupled with a rotation about C2–C3 bond of 2-methylaconitate, and that residue Lys73 is critical for PrpF function. The newly identified function of PrpF as a non-PLP-dependent isomerase, together with the fact that PrpD-containing bacteria do not require PrpF, suggest that the isomer of 2-methylaconitate that serves as a substrate of aconitase must have the same stereochemistry as that synthesized by PrpD. From this, it follows that the 2-methylaconitate isomer generated by AcnD is not a substrate of aconitase, and that PrpF is required to generate the correct isomer. As a consequence, the isomerase activity of PrpF may now be viewed as an integral part of the 2-methylcitric acid cycle.
Keywords: cis–trans isomerases, non-PLP-dependent isomerases protein fold, propionate catabolism, 2-methylcitric acid cycle, 2-methylcitrate dehydratase, 2-methylaconitate isomerase, aconitase, Shewanella physiology
In nature, short-chain fatty acids (e.g., acetate, propionate, butyrate) are very abundant (>0.1 M) in environments such as soil and the human intestine, and are of great relevance to agriculture and human health and nutrition (Cummings et al. 1987; Cummings 1995; Buckel 1999). Not surprisingly, bacteria occupying these habitats have evolved enzymatic capabilities for the utilization of these compounds as sources of carbon and energy. Interestingly, one short-chain fatty acid, propionate, is also commonly used as a food preservative because it has broad negative effects on cell functions. These negative effects include cytosol acidification, dissipation of the proton motive force, and disruption of CoA homeostasis. In addition, in some cases, propionate leads to synthesis of 2-methylcitric acid (2-MC) which is a powerful inhibitor of aconitase and citrate synthase (Roe et al. 1998; Horswill et al. 2001; Roe et al. 2002; Ma et al. 2003; Wolfe 2005). This raises the question of how some bacteria are able to utilize propionate as the sole carbon source, whereas for others, propionate is bactericidal. The answer lies in the existence of a catabolic pathway that has evolved to degrade this compound.
Several propionate catabolic pathways have been described (Horswill and Escalante-Semerena 1997), but bioinformatics analysis of sequence genomes shows that the most widely distributed one is the 2-methylcitric acid cycle (2-MCC). The latter was first shown to occur in fungi (Tabuchi and Hara 1974), but the identity of the genes encoding the enzymes of the pathway were first established in the enterobacterium Salmonella enterica (Horswill and Escalante-Semerena 1997). Subsequent work and the bioinformatics analyses of available genome sequences established the widespread nature of this metabolic capability in prokaryotes (Textor et al. 1997; Russell et al. 1998; Bramer and Steinbuchel 2001; Bramer et al. 2002; Claes et al. 2002; Grimek and Escalante-Semerena 2004). Briefly, the 2-MCC is used to oxidize the Cα methylene of propionate to a keto group yielding pyruvate, which is used as a precursor for the synthesis of other metabolites, or it can be readily used to generate energy via oxidative phosphorylation.
In S. enterica, the sequence of reactions of the 2-MC pathway begins with the activation of propionate to propionyl–CoA by the AMP-forming propionyl–CoA synthetase (PrpE, EC 6.2.1.17), followed by the synthesis of 2-MC from propionyl–CoA and oxaloacetate by the 2-MC synthase (PrpC, EC 2.3.3.5); the dehydration of 2-MC to 2-methylaconitate by the cofactor-less 2-MC dehydratase (PrpD, 4.2.1.79); the rehydration of 2-methylaconitate to 2-methylisocitrate by aconitase (AcnB, 4.2.1.3), and ending with the cleavage of 2-methylisocitrate into pyruvate and succinate by the 2-methylisocitrate lyase (PrpB, 4.2.1.99) (Fig. 1).
Figure 1.

Schematic representation of the 2-methylcitric acid cycle with an emphasis on the chemical transformations catalyzed by either PrpD or AcnD/PrpF.
Some bacteria (e.g., Shewanella oneidensis, Vibrio cholerae, Burkholderia sacchari, Ralstonia eutropha CH34, and Neisseria meningitidis) have replaced prpD with a pair of genes (acnD, prpF) that encode an aconitase-like protein (AcnD), and a 397 amino acid protein of unknown function with a molecular weight of 41,660 (PrpF) (Horswill and Escalante-Semerena 2001; Grimek and Escalante-Semerena 2004). AcnD was previously shown to be a Fe-S cluster containing protein capable of dehydrating 2-MC into 2-methylaconitate (Grimek and Escalante-Semerena 2004). In the same studies, it was shown that PrpF was required for AcnD-dependent growth of a prpD mutant strain on propionate as the sole source of carbon and energy (Grimek and Escalante-Semerena 2004). Attempts to link PrpF to AcnD function were unfruitful. Bioinformatics analyses showed PrpF to be a protein widely distributed in nature, but with sequence similarity only to itself. Thus, a structure elucidation approach was taken to gain insights into the role of the PrpF protein in propionate catabolism.
In this paper we present the three-dimensional crystal structure of the PrpF protein from the Shewanella oneidensis strain MR-1. The structure is strikingly similar to those reported for diaminopimelate epimerase (DapF) from Haemophilus influenzae (Pillai et al. 2006), a putative proline racemase from Brucella melitensis (RCSB PDB code 1TM0), and to a recently deposited structure of a hypothetical protein from Pseudomonas aeruginosa (RCSB PDB code 2H9F). Evidence is presented to show that PrpF can catalyze the interconversion of cis- and trans-aconitate, suggesting that PrpF has cis–trans isomerase activity. Insights into the putative active site of the PrpF enzyme were obtained by cocrystallization of PrpF with trans-aconitate. The structure of this complex suggests that the cis–trans isomerization proceeds through a base-catalyzed proton abstraction coupled with a rotation about the C2–C3 bond.
Results and Discussion
Overview of tertiary and quaternary structure
Apo-PrpF crystallizes in space group P21 with cell dimensions a = 51.6 Å, b = 103.75 Å, c = 78.1 Å, and β = 104.3°, and contains two monomers per asymmetric unit. The structure was determined at 2.0 Å resolution by multiple wavelength anomalous dispersion (MAD) phasing from crystals of selenomethionine-labeled protein (Table 1). The two monomers in the asymmetric unit are related by a noncrystallographic twofold axis and form a molecular dimer. The two monomers which were refined independently are highly similar; the RMS difference between 309 α-carbon atoms is 0.21 Å. Given the similarity between the two monomers, all of the discussion of the structure of a single protein chain is based on that of subunit A.
Table 1.
Data collection and refinement statistics

Examination of the tertiary structure of PrpF (Fig. 2) immediately suggests that it consists of two structural domains that exhibit the same topology and are related by an approximate twofold rotation about their interface (rotation of ∼167°). Indeed, 90 structurally equivalent α-carbons superimpose with an RMS difference of 1.64 Å, which is remarkably high given that the sequence identity between the two domains is very low (12% between structurally equivalent residues whose α-carbons lie closer than 3.0 Å). There is a prominent cleft formed by the juxtaposition of the two domains. This constitutes the active site as will be discussed later.
Figure 2.
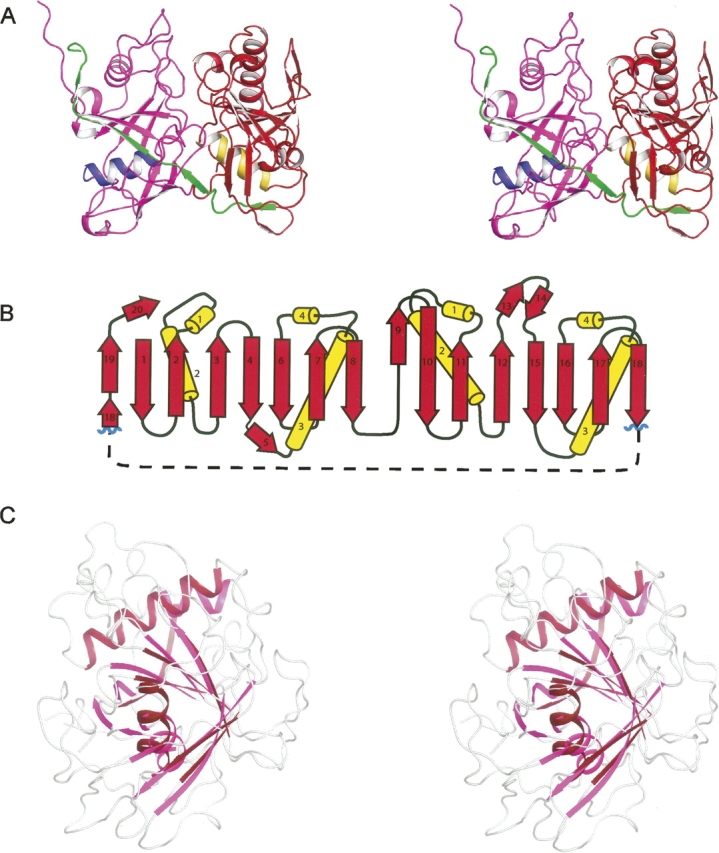
Structural representations and topology of apo PrpF. (A) A stereo ribbon representation of monomer A where the N- and C-terminal domains are colored in magenta and red, respectively. The central α-helix of the N- and C-terminal domains are depicted in blue and yellow, respectively, whereas the final β-strands that extend across both domains are colored in green. (B) The topology of the two domains. (C) A stereo superposition of the N- and C-terminal domains where the structurally similar secondary structural elements of the barrel are depicted in magenta and red for the N- and C-terminal domains, respectively. Figures 2–5 were prepared with the program PyMol (DeLano 2002).
Each domain is dominated by a central α-helix surrounded by eight mainly antiparallel β-strands to form a pronounced β-barrel. An additional α-helix is packed against the outside of the β-barrel, and interacts with strands β2, β3, β4, β20, and β9, β10, β11, β12, in the N- and C-terminal domains, respectively. A long β-strand extends from residues Glu372–Glu389 and topologically connects the two domains (β18 and β19). This β-strand is also the last strand in each barrel of both domains. A comparison of the topology of the two domains suggests that this protein arose from a gene duplication event. Furthermore, since the strand that completes the tertiary structure of the first domain is physically located at the C terminus and is almost continuous with the last strand of the second domain, this suggests that the duplicated gene was inserted within the primordial gene following the eighth β-strand (Fig. 2B).
The PrpF assembles to form a homodimer (Fig. 3) and buries 2412 Å2 of surface area per monomer, which represents 15% of each monomer's total surface area. In addition, ∼65% of the buried surface area is nonpolar which together with the large buried surface area, implies a tight binding interaction between subunits in the dimer. The homodimer interaction occurs primarily between the N-terminal domains, and accounts for 90% of the buried surface area. A striking feature of this interface is the interaction between the last two strands (β19 and β20) of monomer A with the twofold related strands (β19′ and β20′) of monomer B. This produces a very large β-sheet that encompasses both subunits of the dimer. The remaining 10% of buried surface area is located in an interaction between the N termini of helices α7 and α7′ in the C-terminal domains. These helixes are packed on the outside of the β-barrel and are directed toward each other across the molecular twofold axis.
Figure 3.

Ribbon representation of the PrpF homodimer.
Comparison of PrpF with diaminopimelate epimerase
The overall fold for PrpF has been observed before: first in diaminopimelate epimerase (Cirilli et al. 1998; Pillai et al. 2006), and subsequently in a phenazine biosynthesis-related protein (Grassick et al. 2004) and a putative proline racemase from Brucella melitensis (RCSB PDB code 1TM0). In addition, the structure of a protein of unknown function from Pseudomonas aeruginosa was deposited in the RCSB (2H9F), and appears closely related to PrpF. Together, these proteins define a superfamily that shares the same topology and domain duplication. In particular, they exhibit the unique insertion of the second domain just prior to the last strand of the first domain, such that it is likely that they arose from the same gene duplication event and thus share a common ancestor.
The superposition of the monomer A of PrpF with diaminopimelate epimerase (Pillai et al. 2006) is shown in Figure 4. Although the loops that connect the β-strands are structurally divergent, the central barrels of each domain are remarkably similar. The RMS difference between 162 structurally equivalent α-carbon atoms is 1.65 Å, whereas the sequence identity is only 22% (Lassmann and Sonnhammer 2005).
Figure 4.

Stereo overlay of apo PrpF and diaminopimelate epimerase (2GKJ). The β-strands for PrpF are depicted in light blue, whereas the helices and random coil are shown in white. In contrast, the entire molecule for diaminopimelate epimerase is colored in dark blue. The superposition was performed with the program ALIGN (Cohen 1997).
The active site for diaminopimelate epimerase has been defined by the location of conserved residues and through the structure of inhibitor complexes (Cirilli et al. 1998; Pillai et al. 2006). These studies show that the active site is located in the prominent cleft that lies between the two structural domains. The catalytic residues implicated in the epimerization mechanism reside at the base of the cleft and are situated at the ends of the central α-helices of each domain. In the case of diaminopimelate epimerase, the catalytic groups are provided by cysteine residues Cys73 and Cys217, which were proposed to function as the general acids/bases in the epimerization reaction. The structurally equivalent residues in PrpF are Cys107 and Met321.
At the onset of this structural and functional investigation of PrpF, the biological role of this protein was unknown. The discovery that the protein fold was similar to non-PLP-dependent amino acid epimerases and that there is one cysteine residue in the anticipated active site, suggested that PrpF might facilitate an isomerization reaction in the propionate utilization pathway. In particular, it was immediately hypothesized that its function might be to isomerize 2-methylaconitate. This is in contrast to the earlier speculation about its function (Grimek and Escalante-Semerena 2004).
Results from previously published in vivo studies of PrpF function indicated that this protein was involved in the dehydration of 2-methylcitrate to 2-methylaconitate (Grimek and Escalante-Semerena 2004). Although these authors considered the possibility that PrpF was an isomerase of an intermediate of propionate catabolism, the focus of the assignment was not on 2-methylaconitate. But as shown below, PrpF has the ability to isomerize trans-aconitate. Furthermore, the structure of its complex with trans-aconitate provides convincing evidence for this functionality.
Active site complex with trans-aconitate
Crystals of PrpF were grown in the presence of 9 mM trans-aconitate, and the structure was determined by molecular replacement starting from the apo structure described above. Overall there is little difference between the conformation of the polypeptide chain of the apo- and ligand-bound state of PrpF. The RMS difference between 369 α carbon atoms is only 0.29 Å, although one subunit of the homodimer in the complex shows several disordered sections in the ligand-bound crystal lattice. Even so, the electron density for the trans-aconitate is clearly defined in both active sites. As expected, based on the ligand-bound state of diaminopimelate epimerase, trans-aconitate binds in the prominent cleft formed by the junction of the two barrel domains and is directly opposite the N termini of the central α-helices (Fig. 5).
Figure 5.
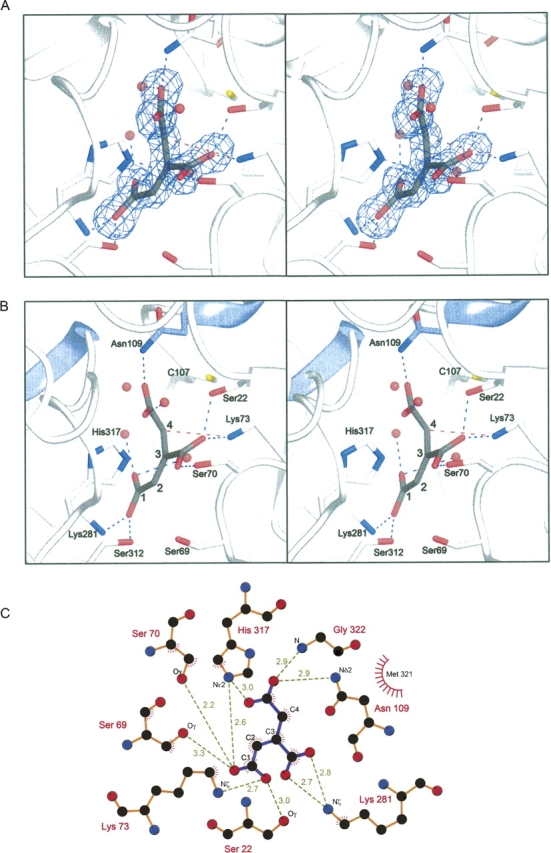
Details of the interaction of trans-aconitate with PrpF. (A) Stereoview of the electron density corresponding to trans-aconitate. The map, contoured at 3σ, was calculated from coefficients of the form F o–F c, where the ligand was omitted from the phase calculation and refinement. (B) Stereoview of the active site depicting the residues that interact with trans-aconitate. The N termini of the buried α-helices are colored in blue and show that the helices are directed toward C4 of the substrate. (C) Schematic representation of the interactions made by PrpF with trans-aconitate. Polar interactions and distances in Å are indicated as green dashed lines. Figure 5C was produced with the program LIGPLOT (Wallace et al. 1995).
The absence of conformational change between the structures of the apo and trans-aconitate complex stands in contrast to other proteins that exhibit the DapF fold. In the case of PhzF, there is a significant domain movement induced by ligand binding that causes closure of the active site cleft (Blankenfeldt et al. 2004). Examination of a surface representation of the trans-aconitate complex indicates that the ligand is not solvent exposed, which implies that domain movements must be necessary to allow the substrate to bind (figure not shown). In this instance, it would appear that the crystallization conditions favor the closed state.
The three carboxylates of trans-aconitate are coordinated by an extensive network of hydrogen bond donors as shown in Figure 5B and C. There are three charged residues in the coordination sphere (Lys73, Lys281, and His317) that serve to formally neutralize the three negative charges on the substrate. Lys73 and Lys281 form direct hydrogen bonds with trans-aconitate, whereas His317 lies within the coordination sphere of the carboxylate moieties on C3 and C4 of trans-aconitate. In addition, there are at least six more hydrogen bonds formed between the carboxylate oxygens and other polar residues and water molecules in the active site. The most extensive hydrogen-bonding network is found surrounding the carboxylate groups on C3 and C4 of trans-aconitate where this network completely satisfies the hydrogen bonding capacity of these polar atoms. This network of amino acid side chains is highly conserved across 85 sequences that can be identified as a PrpF. There are comparatively fewer hydrogen bonds between the protein atoms and the C1 carboxylate, which faces into a more open environment, where it interacts with water molecules.
The C4 carbon, that is implicated in the base catalyzed mechanism proposed below, lies 3.2 Å away from Lys73 and is ideally positioned for proton abstraction. Conversely, Cys107 lies at a somewhat greater distance from C4 (3.6 Å). The latter residue is formally equivalent to the catalytic base in diaminopimelate epimerase; however, its position in PrpF does not appear to be ideal for this function. Importantly, Lys73 is completely conserved across 85 full-length sequences that appear related to PrpF. A structural superposition of PrpF with other members of this superfamily reveals that Lys73 is in a structurally equivalent position with the catalytic glutamate, E45, of phenazine biosynthetic protein PhzF from Pseudomonas fluorescens (Blankenfeldt et al. 2004). Together, this suggests that this family of proteins can utilize a variety of residues as catalytic acids or bases, and that it is the position of the functional group relative to the substrate that is most important.
In order for Lys73 to function as a catalytic base it must be in the uncharged state during catalysis. Examination of the atoms surrounding Lys73 suggests that this is accomplished by placing the Nζ in a substantially hydrophobic environment. First, the amino group abuts the side chain of Phe91 and the Sγ and Cβ methylene group of Cys107. Second, it does not form any stereochemically sound hydrogen bonds to the amino acid residues that constitute the active site. Together this would suggest that this functional group would have a lower pKa than normally observed for a lysine side chain in solution.
Demonstration of aconitate isomerase activity
The structural similarity of PrpF with diaminopimelate epimerase and proline racemase prompted the performance of experiments aimed at determining whether PrpF was an isomerase. PrpF protein (200 μg) was incubated at 37°C in HEPES buffer (25 mM, pH 7.5) containing trans-aconitate (0.5 mM); samples were taken as a function of time, and the composition of the reaction mixture was analyzed by HPLC as described below. Under the assay conditions used, PrpF-dependent synthesis of cis-aconitate was calculated to occur at 4.8 ± 0.5 pmol min−1 μg−1 of PrpF protein (Fig. 6). The lack of a more robust isomerase activity associated with PrpF may be due to the use of aconitate rather than 2-methylaconitate, the true substrate of the enzyme.
Figure 6.

HPLC analysis of the formation of cis-aconitate over time by PrpF. Chromatograms of the conversion of trans- to cis-aconitate by PrpF monitored at 210 nm. Reaction time points and the pmol of cis-aconitate formed are labeled as is the 90-min no-protein control reaction.
Molecular mechanism for PrpF
Early biochemical and mechanistic studies on the aconitate isomerase from Pseudomonas putida suggested that the isomerization reaction proceeded via an allylic rearrangement (Fig. 7A; Klinman and Rose 1971a,b). This was based on isotope labeling and tritium exchange measurements. These studies suggested that the enzymatic reaction proceeded via a base-catalyzed mechanism that involved double-bond migration in a carbanion transition state. Furthermore, the tritium exchange reactions can be explained in terms of a polyprotic base, which implies the presence of a catalytic lysine residue. The isomerization of 2-methyl-trans-aconitate to 2-methyl-cis-aconitate catalyzed by PrpF cannot proceed by an allylic rearrangement because bond migration would yield a different product, 4-methyl aconitate. In addition, the active site of PrpF is inconsistent with an allylic mechanism, since it appears to contain only one catalytic acid/base, Lys73, where this lies adjacent to C4. It is expected that an enzyme that catalyzes an allylic rearrangement would require two catalytic acids/bases: one for proton abstraction at C2, and the other for the complementary reactions at C4.
Figure 7.

Established mechanism for aconitate isomerase from P. putida and a proposed mechanism for PrpF. (A) Shows the mechanism for aconitate isomerase that was established from isotope labeling and tritium exchange measurements (Klinman and Rose 1971a). (B) Shows the proposed mechanism for PrpF. In the latter case, the isomerization is proposed to occur through a single proton abstraction coupled to rotation about the C2–C3 bond.
The structure for PrpF therefore suggests that this enzyme utilizes a different mechanism from that established for aconitate isomerase (Fig. 7B). Given that there is only one base in the active site, the isomerization reaction must include a conformational rearrangement about the C2–C3 single bond in the ionic intermediate. The active site is consistent with this type of mechanism, since there are fewer protein ligand interactions with the C1 carboxylate and because there appears to be ample space to accommodate the cis-conformation. Indeed, when cis-aconitate is modeled into the active site with the assumption that the carboxylates associated with C4 maintain the same set of interactions, the C1 carboxylate adopts a position where it can interact with side chains of Ser69 and Ser312 with minor adjustments in their conformations.
Abstraction of a proton from the C4 carbon is a daunting chemical problem. The pKa of a proton alpha to a carboxyl moiety would normally be expected to be ∼34 (Richard et al. 2002); thus, it might seem unlikely that a lysine residue would be able to accomplish proton abstraction without a perturbation in the pKa. The same mechanistic problem confronts diaminopimelate epimerase, which has the same fold but utilizes a cysteine residue instead of lysine. Studies of the latter enzyme have suggested that the positive end of the helix dipoles contributed by both the N termini of the buried α-helices play an important role in lowering the apparent pKa of the α-carbon and stabilizing the negative intermediates (Pillai et al. 2006). Given the similar orientation of these α-helices in PrpF (Fig. 5B) it is likely that these structural features serve the same role across the entire family of proteins that share this fold.
Conclusions
The data reported here strongly support the argument that PrpF is responsible for the isomerization of 2-methylaconitate, the product of the dehydration of 2-methylcitrate by the AcnD enzyme. The fact that some bacteria use the PrpF/AcnD system and others use only the PrpD enzyme to catalyze the dehydration of 2-MC (Horswill and Escalante-Semerena 2001), predicts that 2-methylaconitate generated by AcnD and PrpD are distinct isomers. Therefore, on the basis of the newly identified PrpF function as an isomerase, and the fact that PrpD-containing bacteria do not require PrpF, it is hypothesized that the isomer of 2-methylaconitate that serves as the substrate of aconitase must have the stereochemistry of the PrpD product. It is also hypothesized that the 2-methylaconitate isomer generated by AcnD is not a substrate of aconitase, and that PrpF generates the 2-methylaconitate isomer that aconitase can use as substrate. If this idea were correct, it would make the isomerase activity of PrpF an integral part of the 2-MCC. Current work focuses on determining the stereochemistry of the PrpD and AcnD reaction product.
PrpF homologs catalyze similar isomerizations in other metabolic pathways
The widespread distribution of PrpF homologs in nature suggests that this protein fold is an effective template for the catalysis of isomerizations occurring via the same mechanism. One example is the isomerization of (R)-3-methylitaconate to 2,3-dimethylmaleate by the methylitaconate isomerase (Mii, EC 5.3.3.6) (Beatrix et al. 1994), a reaction that is part of the anaerobic nicotinate fermentation pathway in Eubacterium barkeri (Beatrix et al. 1994; Alhapel et al. 2006). The PrpF and Mii proteins are only 39% identical and 58% similar. Nevertheless, it is likely that their mechanism of catalysis is the same. Knowledge of the isomerase activity associated with PrpF provides a solid foundation for the analysis of the mechanism of catalysis of this widely distributed protein.
Materials and Methods
Culture media and chemicals
Cell cultures were grown in lysogenic broth (LB) (Bertani 1951, 2004) for overnight cultures and for expression of native protein. LB medium containing 1.5% (w/v) Bacto Agar (Difco) was used as solid medium. When added to the medium, antibiotic concentrations were as follows: ampicillin, 100 μg/mL and kanamycin, 50 μg/mL. All other chemicals used were purchased from Sigma unless otherwise stated.
Plasmid and strain construction
Restriction endonucleases NheI and NcoI were purchased from Promega, and BspHI was purchased from New England Biolabs. All cloning was done in Escherichia coli strain DH5α/F′ (New England Biolabs) unless otherwise stated. The S. oneidensis prpF gene was amplified from plasmid pPRP153 (S. oneidensis prpF+) (Grimek and Escalante-Semerena 2004). The reaction mixture contained TripleMaster polymerase (5 U, Eppendorf), 0.2 μM of each dNTP, 125 pmol each primer, and 0.1% (v/v) dimethylsulfoxide (DMSO). Approximately 200 ng of template DNA was used. PCR amplification reactions' conditions were: 95°C for 5 min, followed by 35 cycles of 95°C for 1 min, 55°C for 45 sec, 72°C for 90 sec, ending with 72°C for 10 min. Product bands were gel extracted using QIAquick gel extraction kit (Qiagen). All plasmids carrying prpF alleles were sequenced using two internal primers: 5′-GCC GCC GAA GTC CAA ATC GA-3′ and 5′-GCG TTA AAG CGA CCA ATG-3′. Sequencing reactions were prepared using nonradioactive BigDye protocols (ABI PRISM), purified using the CleanSEQ reaction cleanup procedure (Agencourt Biotechnology Corp.), and resolved at the UW-Madison Biotechnology Center.
Plasmid pPRP196
The wild-type prpF allele of S. oneidensis was amplified from plasmid pPRP153 using primers 5′-CCT TGG AGT TGC TAG CAT GAG TAA TAA AC-3′ and 5′-TGG GTT GAA CTC ATG ATA TTG GTC TAA GG-3′, cut with restriction enzymes NheI and BspHI and ligated with T4 DNA ligase (Fermentas) into a modified pET31b (Novagen) vector cut with NheI and NcoI. The pET31b vector was previously modified to contain an N-terminal His6 tag followed by a TEV protease cut site two amino acids upstream of the gene start. The N-terminal amino acid sequence of this construct is MSYYHHHHHHDYDIPTSELYFQGASM1S2 where the residues removed by the TEV protease are underlined.
Protein expression and purification
Native PrpF protein was overproduced using plasmid pPRP196 transformed into E. coli strain BL21(λDE3). A starter culture from a single colony was grown overnight at 37°C in LB medium supplemented with ampicillin. The following day, 10 mL of the starter culture was used to inoculate 1 L of LB + ampicillin medium in a 2-L shaker flask. Cultures were grown at 37°C until they reached an optical density (OD600) of ∼1.2. Cultures were transferred to a 16°C incubator and allowed to equilibrate for 30 min. After equilibration, prpF+ expression was induced by the addition of isopropyl-β-D-thiogalactopyranoside (IPTG) added to a final concentration of 1.0 mM. After a 20-h incubation period with IPTG under aerobic conditions, cells were harvested by centrifugation at 5000g, flash frozen in liquid nitrogen, and stored at −80°C until used. PrpF protein was purified from 60 g of cells thawed and resuspended in 420 mL of lysis buffer, which contained 2 mM 2-mercaptoethanol, 1 tablet/50 mL of Complete EDTA-free Inhibitor (Roche), and 100 mg lysozyme. This mixture was subjected to seven rounds of sonication (1 min each) separated by 5-min cooling. Cellular debris was removed by centrifugation at 40,000g for 30 min. The supernatant was loaded onto a 30-mL column of nickel-nitrilotriacetic acid-agarose (Qiagen) previously equilibrated with lysis buffer. The column was washed with lysis buffer until the A280 of the outflow reached background level. PrpF protein was eluted with a linear gradient of 10–300 mM imidazole in lysis buffer. Fractions containing PrpF protein, identified using SDS-PAGE (Laemmli 1970) and Coomassie Blue staining (Sasse 1991), were pooled and dialyzed against 2-amino-2-(hydroxymethyl)propane-1,3-diol hydrochloride (Tris-HCl) buffer (10 mM, pH 8.0 at 4°C) containing NaCl (400 mM), ethylenediaminetetraacetic acid (EDTA, 2 mM), tris(2-carboxyethyl)phosphine hydrochloride (TCEP, 0.5 mM), pH 8.0 at 4°C. The N-terminal hexahistidine (H6) tag was removed by treatment with recombinant tobacco etch virus (rTEV) protease (Shih et al. 2005). Remaining tagged PrpF protein enzyme and rTEV protease were removed from the mixture using nickel-nitrilotriacetic acid-agarose affinity chromatography. Tagless PrpF protein was concentrated to ∼9 mg/mL using a centriprep YM30 (Millipore) concentrator. Concentrated protein was dialyzed against Tris-HCl buffer (10 mM, pH 8.0 at 4°C) containing TCEP (0.5 mM). Yield was 15 mg of PrpF protein per gram of cell paste.
Preparation of selenomethionine-labeled PrpF protein
Cultures of E. coli strain BL21(λDE3) carrying plasmid pPRP196 were grown overnight at 37°C in M9 minimal medium (Atlas 1995) supplemented with ampicillin. The following day, 10 mL of the starter culture was used to inoculate 650 mL of M9 + ampicillin in a 2-L shaker flask. Cultures were grown at 37°C to an optical density (OD600) of ∼1.2. Cultures were cooled for 10 min in an ice bath, transferred to a 16°C incubator, and supplemented with 65 mg each of L-lysine, L-threonine, and L-phenyalanine, and 31 mg each of L-leucine, L-isoleucine, L-valine, and L-selenomethionine. After an additional 30 min of growth, expression of the prpF+ gene was induced with IPTG added to a final concentration of 1.0 mM. After a 20-h incubation period at 16°C, cells were harvested by centrifugation at 5000g and flash frozen in liquid nitrogen. Selenomethionine-labeled PrpF protein was purified as described above.
Crystallization and structural determination
A search for crystallization conditions was conducted at 4°C via the hanging-drop method of vapor diffusion utilizing an “in-house” designed sparse matrix screen composed of 144 conditions. The best crystals for 10 mM trans-aconitate plus 9 mg/mL enzyme were observed from hanging drop experiments with precipitant solutions of 19% methyl ether poly(ethyleneglycol) 5000 buffered with N-(2-hydroxyethyl)-piperazine-N′-2-ethanesulfonic acid (HEPES) buffer (100 mM, pH 7.5 at 25°C). Single crystals grew to 0.4 × 0.3 × 0.2 mm in 4 wk. Crystals were frozen by rapidly transferring the crystals into a solution containing 15% (v/v) ethyleneglycol and 85% mother liquor and then flash freezing into liquid nitrogen.
The best apo crystals were observed from hanging-drop experiments with precipitant solutions of 300–350 mM NaCl, 21%–24% methyl ether poly(ethylene glycol) 5000, buffered with 100 mM triethanolamine (pH 8.0 at 25°C). Large fractal plates grew to 0.8 × 0.8 × 0.3 mm after 3–5 d. Crystals were frozen by transferring into a solution containing 20% methyl ether poly(ethylene glycol) 5000, 300 mM NaCl, buffered with 100 mM triethanolamine (pH 8.0 at 25°C) and allowed to equilibrate for 1 h. After equilibration, the crystals were transferred stepwise into a solution containing 10% ethylene glycol, 20% methyl ether poly(ethylene glycol) 5000, 350 mM NaCl, buffered with 100 mM triethanolamine (pH 8.0 at 25°C).
X-ray data were collected from crystals of native and selenomethionine-substituted protein on a CCD detector at SBC Beamline 19-BM (Advanced Photon Source, Argonne National Laboratory). The X-ray data were processed and scaled with HKL2000 (Otwinowski and Minor 1997). Relevant X-ray data collection statistics are presented in Table 1.
The structure of S. oneidensis PrpF was solved via MAD phasing with crystals of the selenomethionine substituted protein. The software package SOLVE was utilized to determine the positions of 21 out of the 24 selenium atoms in the asymmetric unit and to generate initial protein phases (figure of merit = 0.55) (Terwilliger and Berendzen 1999). Solvent flattening with RESOLVE (figure of merit = 0.79) resulted in an interpretable electron density map calculated to 1.98 Å resolution (Terwilliger 2000). The initial map allowed for 454 of the 698 amino acid residues in the asymmetric unit to be modeled; the N terminus is disordered to residue Ala5 as well as between residues Ala186 and Leu201.
The incomplete structure obtained from MAD phasing was then used as the search model to solve both the native apo structure and the enzyme complexed with trans-aconitate via molecular replacement with the program EPMR (Kissinger et al. 1999). For the native apo structure, alternate cycles of manual model building and least-squares refinement with the programs COOT (Emsley and Cowtan 2004) and Refmac (Murshudov et al. 1997) reduced the R-factor to 15.6% for all X-ray data from 50–2.0 Å. Relevant refinement statistics are presented in Table 1. In this model there are two breaks in the polypeptide chain between Asp188 and Gly193, the N terminus is disordered to residue Ala5.
The complex of enzyme with trans-aconitate was refined with alternate cycles of manual model building and least-squares refinement with the programs COOT (Emsley and Cowtan 2004) and Refmac (Murshudov et al. 1997) and the final R-factor was reduced to 20.3% for all X-ray data from 50–1.57 Å. In this model there is one break in monomer A polypeptide chain for monomer A between Asp187 and Cys194. In the B model there are three chain breaks between Val204–Asn213, Asn247–Lys281, and Leu311–Lys315. In both monomers the N terminus is disordered to residue Ala5 and Phe6 for A and B, respectively. The Ramachandran plot as calculated by PROCHECK (Laskowski et al. 1993) has no residues in the disallowed regions, 92.7% in the most favored, 7% in the additionally allowed, and 0.3% in the generously allowed region. Refinement statistics are presented in Table 1.
Enzymatic reactions with trans-aconitate
Tagless PrpF protein was dialyzed against HEPES buffer (25 mM, pH 7.5 at 25°C) overnight at 4°C. PrpF protein (200 μg) was incubated in HEPES buffer (25 mM, pH 7.5 at 25°C) for 5 min in a 37°C water bath. The reaction was started by the addition of trans-aconitate (0.5 mM, pH 6.7). Samples (150 μL) were taken at 10, 45, and 90 min, were acidified with H2SO4 (5 mM final concentration), and filtered on a Spin-X centrifuge tube filter (Costar). The content of cis- and trans-aconitate in the samples was determined by isocratic high-pressure liquid chromatography using a Beckman/Coulter chromatograph equipped with an Aminex HPX-87H HPLC Organic Acid Analysis Column (BioRad). The column was equilibrated and developed isocratically with 5 mM H2SO4.
Data deposition
The atomic coordinates and structure factors for the apo-PrpF and trans-aconitate·PrpF complex have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/) with accession codes 2PVZ and 2PWO, respectively.
Acknowledgments
We thank Kirsten Dennison for constructing the modified pET vector utilized in construction of the overexpression plasmid for PrpF. This work was supported by funds from the NIH to I.R. (AR35186) and J.C.E.-S. (GM62203). Use of the Structural Biology BM19 beamline Argonne National Laboratory Advanced Photon Source was supported by the U.S. Department of Energy, Office of Energy Research, under Contract No. W-31-109-ENG-38.
Footnotes
Reprint requests to: Ivan Rayment, Department of Biochemistry, University of Wisconsin, 433 Babcock Drive, Madison, WI 53706, USA; e-mail: Ivan_Rayment@biochem.wisc.edu; fax: (608) 262-1319; or Jorge C. Escalante-Semerena, Department of Bacteriology, University of Wisconsin, 1550 Linden Drive, Madison, WI 53706, USA; e-mail: escalante@bact.wisc.edu.
Abbreviations: 2-MC, 2-methylcitrate; 2-MCC, 2-methylcitric acid cycle; AMP, adenosine monophosphate; DMSO, dimethylsulfoxide; dNTP, deoxynucleoside triphosphate; DTT, dithiothreitol; TCEP, tris(2-carboxyethyl)phosphine hydrochloride; HEPES, N-(2-hydroxyethyl)-piperazine-N′-2-ethanesulfonic acid; EDTA, ethylenediaminetetraacetic acid; RMS, root mean square; rTEV, recombinant tobacco etch virus; OD600, optical density measured at 600 nm; PEI, polyethyleneimine; MAD phasing, multiple wavelength anomalous dispersion phasing.
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.072801907.
References
- Alhapel A., Darley, D.J., Wagener, N., Eckel, E., Elsner, N., and Pierik, A.J. 2006. Molecular and functional analysis of nicotinate catabolism in Eubacterium barkeri . Proc. Natl. Acad. Sci. 103: 12341–12346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atlas R. 1995. Handbook of media for environmental microbiology. CRC Press, Boca Raton, FL.
- Beatrix B., Zelder, O., Linder, D., and Buckel, W. 1994. Cloning, sequencing and expression of the gene encoding the coenzyme B12-dependent 2-methyleneglutarate mutase from Clostridium barkeri in Escherichia coli . Eur. J. Biochem. 221: 101–109. [DOI] [PubMed] [Google Scholar]
- Bertani G. 1951. Studies on lysogenesis. I. The mode of phage liberation by lysogenic Escherichia coli . J. Bacteriol. 62: 293–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertani G. 2004. Lysogeny at mid-twentieth century: P1, P2, and other experimental systems. J. Bacteriol. 186: 595–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blankenfeldt W., Kuzin, A.P., Skarina, T., Korniyenko, Y., Tong, L., Bayer, P., Janning, P., Thomashow, L.S., and Mavrodi, D.V. 2004. Structure and function of the phenazine biosynthetic protein PhzF from Pseudomonas fluorescens . Proc. Natl. Acad. Sci. 101: 16431–16436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bramer C.O. and Steinbuchel, A. 2001. The methylcitric acid pathway in Ralstonia eutropha: New genes identified involved in propionate metabolism. Microbiology 147: 2203–2214. [DOI] [PubMed] [Google Scholar]
- Bramer C.O., Silva, L.F., Gomez, J.G., Priefert, H., and Steinbuchel, A. 2002. Identification of the 2-methylcitrate pathway involved in the catabolism of propionate in the polyhydroxyalkanoate-producing strain Burkholderia sacchari IPT101(T) and analysis of a mutant accumulating a copolyester with higher 3-hydroxyvalerate content. Appl. Environ. Microbiol. 68: 271–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckel W. 1999. Anaerobic energy metabolism. In Biology of the procaryotes (eds. J.W. Lengler, G. Drews, and H.G. Chlegel), pp. 278–326. Thieme, Stuttgart, Germany.
- Cirilli M., Zheng, R., Scapin, G., and Blanchard, J.S. 1998. Structural symmetry: The three-dimensional structure of Haemophilus influenzae diaminopimelate epimerase. Biochemistry 37: 16452–16458. [DOI] [PubMed] [Google Scholar]
- Claes W.A., Puhler, A., and Kalinowski, J. 2002. Identification of two prpDBC gene clusters in Corynebacterium glutamicum and their involvement in propionate degradation via the 2-methylcitrate cycle. J. Bacteriol. 184: 2728–2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen G.H. 1997. ALIGN: A program to superimpose protein coordinates, accounting for insertions and deletions. J. Appl. Crystallogr. 30: 1160–1161. [Google Scholar]
- Cummings J.H. 1995. Short chain fatty acids. In Human colonic bacteria: Role in nutrition, physiology and pathology (eds. G. Gibson and G. Macfarlane), pp. 101–105. CRC Press, London.
- Cummings J.H., Pomare, E.W., Branch, W.J., Naylor, C.P., and Macfarlane, G.T. 1987. Short chain fatty acids in human large intestine, portal, hepatic and venous blood. Gut 28: 1221–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLano W.L. 2002. The PyMOL molecular graphics system. DeLano Scientific, San Carlos, CA.
- Emsley P. and Cowtan, K. 2004. Coot: Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60: 2126–2132. [DOI] [PubMed] [Google Scholar]
- Grassick A., Sulzenbacher, G., Roig-Zamboni, V., Campanacci, V., Cambillau, C., and Bourne, Y. 2004. Crystal structure of E. coli yddE protein reveals a striking homology with diaminopimelate epimerase. Proteins 55: 764–767. [DOI] [PubMed] [Google Scholar]
- Grimek T.L. and Escalante-Semerena, J.C. 2004. The acnD genes of Shewenella oneidensis and Vibrio cholerae encode a new Fe/S-dependent 2-methylcitrate dehydratase enzyme that requires prpF function in vivo. J. Bacteriol. 186: 454–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horswill A.R. and Escalante-Semerena, J.C. 1997. Propionate catabolism in Salmonella typhimurium LT2: Two divergently transcribed units comprise the prp locus at 8.5 centisomes, prpR encodes a member of the sigma-54 family of activators, and the prpBCDE genes constitute an operon. J. Bacteriol. 179: 928–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horswill A.R. and Escalante-Semerena, J.C. 2001. In vitro conversion of propionate to pyruvate by Salmonella enterica enzymes: 2-Methylcitrate dehydratase (PrpD) and aconitase enzymes catalyze the conversion of 2-methylcitrate to 2-methylisocitrate. Biochemistry 40: 4703–4713. [DOI] [PubMed] [Google Scholar]
- Horswill A.R., Dudding, A.R., and Escalante-Semerena, J.C. 2001. Studies of propionate toxicity in Salmonella enterica identify 2-methylcitrate as a potent inhibitor of cell growth. J. Biol. Chem. 276: 19094–19101. [DOI] [PubMed] [Google Scholar]
- Kissinger C.R., Gehlhaar, D.K., and Fogel, D.B. 1999. Rapid automated molecular replacement by evolutionary search. Acta Crystallogr. D Biol. Crystallogr. 55: 484–491. [DOI] [PubMed] [Google Scholar]
- Klinman J.P. and Rose, I.A. 1971a. Mechanism of the aconitate isomerase reaction. Biochemistry 10: 2259–2266. [DOI] [PubMed] [Google Scholar]
- Klinman J.P. and Rose, I.A. 1971b. Purification and kinetic properties of aconitate isomerase from Pseudomonas putida . Biochemistry 10: 2253–2259. [DOI] [PubMed] [Google Scholar]
- Laemmli U.K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227: 680–685. [DOI] [PubMed] [Google Scholar]
- Laskowski R.A., MacArthur, M.W., Moss, D.S., and Thornton, J.M. 1993. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26: 283–291. [Google Scholar]
- Lassmann T. and Sonnhammer, E.L. 2005. Kalign—An accurate and fast multiple sequence alignment algorithm. BMC Bioinformatics doi: 10.1186/1471-2105-6-298. [DOI] [PMC free article] [PubMed]
- Ma Z., Gong, S., Richard, H., Tucker, D.L., Conway, T., and Foster, J.W. 2003. GadE (YhiE) activates glutamate decarboxylase-dependent acid resistance in Escherichia coli K-12. Mol. Microbiol. 49: 1309–1320. [DOI] [PubMed] [Google Scholar]
- Murshudov G.N., Vagin, A.A., and Dodson, E.J. 1997. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53: 240–255. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z. and Minor, W. 1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276: 307–326. [DOI] [PubMed] [Google Scholar]
- Pillai B., Cherney, M.M., Diaper, C.M., Sutherland, A., Blanchard, J.S., Vederas, J.C., and James, M.N. 2006. Structural insights into stereochemical inversion by diaminopimelate epimerase: An antibacterial drug target. Proc. Natl. Acad. Sci. 103: 8668–8673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard J.P., Williams, G., O'Donoghue, A.C., and Amyes, T.L. 2002. Formation and stability of enolates of acetamide and acetate anion: An Eigen plot for proton transfer at alpha-carbonyl carbon. J. Am. Chem. Soc. 124: 2957–2968. [DOI] [PubMed] [Google Scholar]
- Roe A.J., McLaggan, D., Davidson, I., O'Byrne, C., and Booth, I.R. 1998. Perturbation of anion balance during inhibition of growth of Escherichia coli by weak acids. J. Bacteriol. 180: 767–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roe A.J., O'Byrne, C., McLaggan, D., and Booth, I.R. 2002. Inhibition of Escherichia coli growth by acetic acid: A problem with methionine biosynthesis and homocysteine toxicity. Microbiol. 148: 2215–2222. [DOI] [PubMed] [Google Scholar]
- Russell R.J., Gerike, U., Danson, M.J., Hough, D.W., and Taylor, G.L. 1998. Structural adaptations of the cold-active citrate synthase from an Antarctic bacterium . Structure 6: 351–361. [DOI] [PubMed] [Google Scholar]
- Sasse J. 1991. Detection of proteins. In Current protocols in molecular biology (eds. F.A. Ausubel et al.). Wiley Interscience, New York.
- Shih Y.P., Wu, H.C., Hu, S.M., Wang, T.F., and Wang, A.H. 2005. Self-cleavage of fusion protein in vivo using TEV protease to yield native protein. Protein Sci. 14: 936–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabuchi T. and Hara, S. 1974. Production of 2-methylisocitric acid from N-paraffins by mutants of candida-lipolytica. Agric. Biol. Chem. 38: 1105–1106. [Google Scholar]
- Terwilliger T.C. 2000. Maximum-likelihood density modification. Acta Crystallogr. D Biol. Crystallogr. 56: 965–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terwilliger T.C. and Berendzen, J. 1999. Automated MAD and MIR structure solution. Acta Crystallogr. D55: 849–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Textor S., Wendisch, V.F., DeGraaf, A., Muller, U., Linder, M.I., Linder, D., and Buckel, W. 1997. Propionate oxidation in Escherichia coli: Evidence for operation of a methylcitrate cycle in bacteria. Arch. Microbiol. 168: 428–436. [DOI] [PubMed] [Google Scholar]
- Wallace A.C., Laskowski, R.A., and Thornton, J.M. 1995. LIGPLOT: A program to generate schematic diagrams of protein–ligand interactions. Protein Eng. 8: 127–134. [DOI] [PubMed] [Google Scholar]
- Wolfe A.J. 2005. The acetate switch. Microbiol. Mol. Biol. Rev. 69: 12–50. [DOI] [PMC free article] [PubMed] [Google Scholar]