

Abstract
Recent insights into the molecular and cellular mechanisms underlying cancer development have revealed that immune cells functionally regulate epithelial cancer development and progression. Moreover, accumulated clinical and experimental data indicate that the outcome of an immune response toward an evolving breast neoplasm is largely determined by the type of immune response elicited. Acute tumor-directed immune responses involving cytolytic T lymphocytes appear to protect against tumor development, whereas immune responses involving chronic activation of humoral immunity, infiltration by Th2 cells, and protumor-polarized innate inflammatory cells result in the promotion of tumor development and disease progression. Herein we review this body of literature and summarize important new findings revealing the paradoxical role of innate and adaptive leukocytes as regulators of breast carcinogenesis.
Introduction
Breast cancer is the most frequent malignant tumor of women in North America [1]. Standard treatment modalities have improved the overall outlook and quality of life for women with breast cancer; however, the fact that 40% still succumb to disease highlights the need for new therapeutic approaches and identification of new therapeutic targets. While genetic and epigenetic changes in genes that regulate mammary epithelial cell proliferation, survival, polarity and/or differentiation are probable 'initiators' of breast carcinogenesis, several lines of evidence indicate that stromal cell responses in premalignant mammary tissue may 'promote' progression to cancer and/or the metastatic capability of malignant mammary epithelial cells. Cellular components of tumor stroma include (myo)fibroblasts, vascular cells, infiltrating leukocytes and specialized mesenchymal support cells unique to each tissue microenvironment. A growing body of evidence has recently implicated tumor-infiltrating leukocytes as causal players in cancer development [2-8].
The present review focuses on the paradoxical roles of innate and adaptive leukocytes as regulators of breast carcinogenesis, and highlights recent experimental data indicating that therapeutically targeting these diverse immune cell types by either neutralizing and/or bolstering their specific bioactivities may provide a therapeutic advantage to patients with breast cancer.
Leukocytes and carcinoma development
Leukocytes represent a diverse assortment of immune cells composed of both innate (myeloid) and adaptive (lymphoid) lineages. Innate immune cells, including macrophages, granulocytes, mast cells, dendritic cells (DCs), and natural killer (NK) cells, represent the first line of defense against pathogens and foreign agents. When tissue homeostasis is perturbed, tissue-resident macrophages and mast cells locally secrete soluble factors such as cytokines, chemokines, bioactive mediators, and matrix-remodeling proteins that recruit additional leukocytes from the circulation into damaged tissue (that is, inflammation) [3,9,10]. Recruited innate immune cells can directly eliminate pathogenic agents in situ. At the same time, DCs take up foreign antigens (including tumor antigens) and migrate to lymphoid organs, where they present their antigens to adaptive immune cells. Upon recognition of foreign antigen presented by DCs or other professional antigen-presenting cells, adaptive immune cells, such as T lymphocytes or B lymphocytes, undergo clonal expansion in order to mount an 'adaptive' response targeted against the foreign agent [11,12]. Acute activation of innate immunity therefore sets the stage for activation of more sophisticated, antigenically committed, adaptive immune responses. Once foreign agents have been eliminated, inflammation resolves and tissue homeostasis is restored.
The inflammatory responses necessary for enabling an immune reaction may, however, also set the stage for promoting neoplastic disease. As early as 1863, Virchow first postulated that cancer originates at sites of chronic inflammation, in part based on his hypothesis that some classes of irritants causing inflammation also enhance cell proliferation [13]. When tissues are injured or are exposed to chemical irritants, damaged cells are removed by the induction of cell death pathways, while cell proliferation is enhanced to facilitate tissue regeneration in an attempt to re-establish tissue homeostasis. Proliferation and inflammation resolve only after insulting agents are removed or tissue repair is completed. In contrast, when insulting agents persist over time, sustained cycles of cell proliferation and death in environments rich in inflammatory cells and their bioactive products may increase neoplastic risk and foster tumor progression [3]. While sporadic or inherited genetic mutations in critical genes regulating cell cycle, programmed cell death, differentiation and adhesion may represent initiating events in tumorigenesis ('initiation'), chronic inflammation favors selection of additional features in initiated cells that may promote their full malignant transition ('promotion').
Historically, leukocytes found in and around developing tumors were thought to represent an attempt by the host to eradicate transformed neoplastic cells. Certain leukocytes, such as cytotoxic T lymphocytes (CTLs) and NK cells, undeniably play a vital function in constraining tumor development [14], and as such it has been postulated that many more neoplasms arise than those that eventually develop to fully malignant disease. Epidemiologic data support this contention, as evidenced by the increased incidence of viral-associated cancers [15], including human papillomavirus-related cervical and squamous carcinoma, herpesvirus-8-associated Kaposi's sarcoma and Epstein–Barr virus-related non-Hodgkin's lymphoma in immunocompromised individuals [15-19]. Similar to viral-associated cancers, there are data revealing an increased incidence of carcinogen-associated cancers in immune-compromised populations, including melanoma and lung adenocarcinoma [17,20]. Where carcinogen exposure and pathogen exposure are not thought to be etiologic factors, however, immune-compromised women exhibit reduced relative risk for common epithelial cancers, including breast adenocarcinoma [17,20-23]. Together, these epidemiological studies indicate that the overall risk for, and development of, (breast) cancer may, in part, be regulated by the immune status of the individual.
Adaptive immunity and carcinoma development: a role for B lymphocytes
A growing body of literature has emerged demonstrating that leukocytes functionally contribute to the development of most human solid tumors. Leukocytic infiltrates into the neoplastic stroma increase, paralleling breast tumorigenesis (Figures 1 and 2). Profiles of these immune cells vary but, in general, include both lymphoid and myeloid lineages.
Figure 1.

Development of human breast carcinoma is characterized by abundant infiltration of immune cells. Representative sections of normal, premalignant, and malignant human breast tissue stained with hematoxylin and eosin (H&E) (upper panels), and following immunodetection of CD45 (leukocyte common antigen, brown staining). DCIS, ductal carcinoma in situ.
Figure 2.
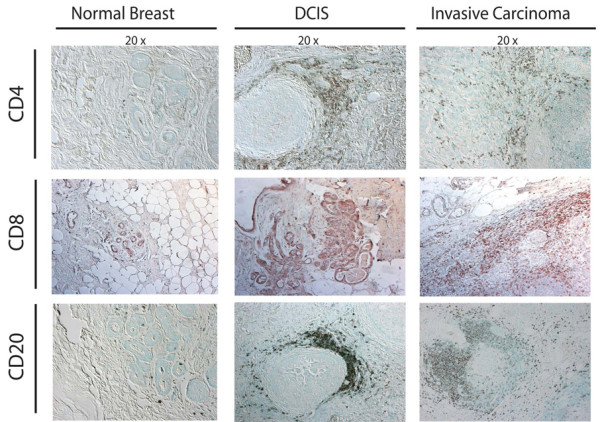
Development of human breast carcinoma is characterized by abundant infiltration of lymphocytes into neoplastic tissue. Representative sections of normal, premalignant, and malignant breast assessed for the presence of CD4+ (upper panels) and CD8+ (middle panels) T lymphocytes (brown staining in panels), as well as CD20+ B lymphocytes (red staining in lower panels), demonstrating the extent of immune-cell infiltration into premalignant and malignant stroma. DCIS, ductal carcinoma in situ.
B lymphocytes are recognized as contributing to anticancer immune responses via their secretion of antigen-specific immunoglobulins. While it is clear that acute activation of B cells may play a role in eradicating early neoplastic cells, or may participate in spontaneous regression of tumors through classical and well-studied immunoglobulin-mediated mechanisms, recent data also indicate that chronic activation of B cells may paradoxically play a role in potentiating carcinoma development (Figure 3).
Figure 3.
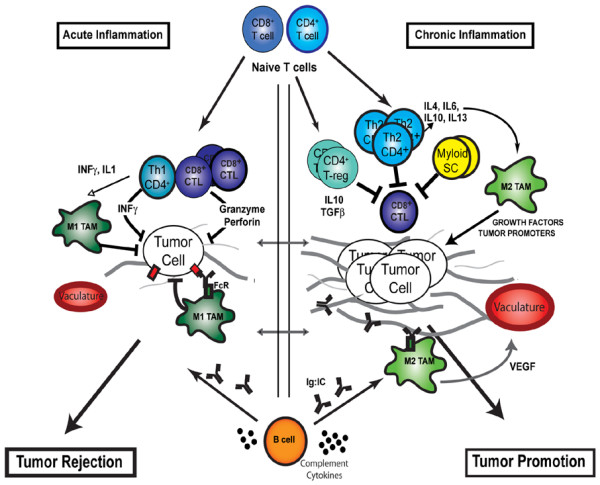
Contrasting roles of adaptive leukocytes during cancer development. During acute inflammatory responses (left panel), Th1 CD4+ and CD8+ T cells directly regulate tumor cell cytotoxicity, while indirectly polarizing innate immune cells toward tumor suppression (such as M1 polarization of tumor-associated macrophages [TAMs]). B-cell-derived factors (immunoglobulins and complement) facilitate recruitment of innate leukocytes and targeted destruction of neoplastic cells. During chronic inflammation, however (right panel), myeloid suppressor cells, Th2 CD4+ T cells and regulatory T (T-reg) cells function in combination to both repress CD8+ cytotoxicity and to induce protumoral polarization of innate immune response (such as M2 polarization of TAMs) via cytokine secretion (IL-4, IL-13, IL-10, IL-6 and transforming growth factor beta (TGFβ)). Chronically activated B cells promote accumulation of innate cells in the neoplastic stroma by immunoglobulin and cytokine production. When polarized, as during chronic inflammation, these innate immune cells in turn provide a rich proangiogenic and protumoral microenvironment. CTL, cytotoxic T lymphocyte; FcR, Fc receptor; INF, interferon; SC, suppressor cells; VEGF, vascular endothelial growth factor.
B-cell precursors mature within bone marrow, where somatic recombination of immunoglobulin genes results in expression of a diverse array of B-cell receptors. Mature antigen-committed B cells migrate to secondary lymphoid organs (lymph nodes or spleen, predominantly). Upon antigen recognition by B-cell receptors, the B lymphocytes become activated and undergo clonal expansion, resulting in their enhanced capacity to recognize foreign antigens [24]. Acute activation of B-lymphocyte responses (to foreign antigens or tissue damage) can also result in rapid induction of several soluble mediators, including diverse immunoglobulin subtypes, B-cell-derived cytokines such as IL-6 and activation of complement cascades, which together trigger recruitment of innate immune cells from the circulation. In this manner, acutely activated B cells orchestrate phagocytic or cytotoxic destruction of immunoglobulin-complexed antigens (pathogens or damaged cells) by innate immune cells. Such acute B-cell responses are critical for protecting tissues from pathogens and nonself-antigens. Chronic activation of B cells can be deleterious, however, as evidenced by their association with several pathologic disease states (rheumatoid arthritis and other autoimmune diseases) and some cancer types [25].
During breast carcinogenesis, mature B cells (including naive cells and activated cells) can be found in secondary lymphoid tissues as well as in tumor-associated stroma (Figure 2). As compared with healthy patients without evidence of cancer, the sentinel (draining) lymph nodes of breast cancer patients contain enriched populations of proliferating and affinity matured (IgG+) B lymphocytes [26]. Moreover, data from retrospective studies examining the percentages of B cells present in sentinel and auxiliary lymph nodes of breast cancer patients reveal that their presence and/or maturation (IgG+) correlates with increases in disease stage (stage I versus stage II) and in total tumor burden [27,28]. Urdiales-Viedma and colleagues utilized immunohistochemical detection of IgA, IgG and IgM in axillary lymph nodes from 50 unselected ductal breast carcinomas, and found that lymph nodes with IgG+ lymphoid follicles and/or metastatic lymph nodes with IgM+ lymphoid cells were statistically related to breast tumors of high histologic grade and more than three lymph node metastases [29].
Similarly, B cells present in breast tumor-associated stroma may also play a role in disease progression. Several studies have reported that infiltrating B lymphocytes represent the predominant lymphocytic population (in excess of T lymphocytes) in premalignant breast tissue, including hyperplasia and early ductal carcinoma in situ (DCIS) [30,31] (Figure 2). Approximately 20% of invasive breast cancers contain high numbers of B cells; when present, these cells can comprise up to 60% of the neoplasia-associated lymphocyte population [32]. These observations are not specific to breast cancer – 70% of solid tumors contain elevated populations of B cells [33]. Although the presence of chronically activated B cells in breast cancer patients is clear, how these cells might be effecting disease progression has not been established. In DCIS and invasive carcinoma, tumor-associated B cells are typically found in perivascular locales clustering in aggregates with T cells, forming ectopic follicles [32,34,35]. These follicles contain B cells interdigitated around CD21+ follicular DCs, thus identifying them as authentic ectopic follicles. Formation of ectopic follicles containing mature plasma cells indicates that the presence of B cells in neoplastic mammary tissue is the result of chronic activation rather than nonspecific chemoattraction. Extra-nodal B-cell proliferation and ectopic follicle formation have also been described in several autoimmune diseases (rheumatoid arthritis, multiple sclerosis, Sjogren's disease and Graves disease), where they are thought to underlie the disease pathogenesis [36-39].
How might B lymphocytes regulate carcinoma development? A vast literature exists describing the occurrence of (auto)-antibodies in either the serum of cancer patients or in interstitial antibody deposition in tumors [40]. An early presence of autoantibodies (in particular, antinuclear antibodies and smooth muscle antibodies) in serum of cancer patients is well known to correlate with an unfavorable prognosis [41]. Approximately 50% of breast cancer patients contain circulating immunoglobulins that specifically react with tumor-derived antigens – autoantibodies against ErbB2/HER2/neu are present in 20% of patients with ErbB2-positive breast cancer, making it the most common breast cancer 'autoantigen' [42]. Paradoxically, the presence of specific autoantibodies in serum and/or at tumor sites correlates with poor patient survival [40,41,43] – perhaps indicating that immunoglobulins resulting from chronic B-cell activation in response to tumor-specific antigens might promote disease progression (Figure 3).
Despite the presence of antitumor antibodies in greater than one-half of all breast cancer patients, there are only few reports of spontaneous tumor regression (presumed to be immunologic) in the absence of therapy [44,45]. Several factors may influence the efficiency of antitumor antibodies in inducing tumor regression/destruction, including the immunoglobulin concentration, HLA expression, tumor tolerance/immune suppression, and impaired cytotoxic T-cell activity. Whether individuals with progressing tumors harbor a higher antigen load that thus triggers enhanced immunoglobulin production, or whether an increased presence of serum or interstitial immunoglobulins predisposes patients to development of more advanced or recurrent cancers, therefore requires further study. While the answer is not clear, the combined implication of the data is that B lymphocytes do play a role in human carcinoma development, therefore necessitating a mechanistic evaluation of their role and specificity to determine whether they represent tractable targets for anticancer therapy.
Adaptive immunity and carcinoma development: a role for T lymphocytes
Histochemical detection of T lymphocytes in archival human carcinoma tissues has revealed that they also are commonly associated with developing carcinomas (Figure 2); however, the prognostic significance of T-cell infiltrations during breast carcinogenesis is subject to great debate. While B cells appear to represent the predominant lymphocyte during early breast cancer progression [34], infiltrating T lymphocytes (both CD4+ and CD8+) are more extensive in higher grade DCIS and invasive carcinomas [46].
The extent of T-cell infiltration into invasive breast carcinomas has been reported to range from 1% to 45% of the total cellular mass [47]. In rapidly proliferating tumors, the presence of T lymphocytes (by histopathological determination) at tumor sites is a good prognostic indicator when compared with nonimmunogenic tumors, and correlates with auxillary lymph node negativity, a smaller tumor diameter, a lower histological grade and recurrence-free survival [48] – therefore supporting an overall role for T cells in immune surveillance. The exact composition of T lymphocyte infiltration varies greatly, however, and may profoundly affect disease progression and overall patient survival.
During breast carcinogenesis, the presence of metastatic mammary epithelial cells in sentinel lymph nodes draining the primary tumor represents the strongest prognostic indicator for disease progression and overall patient outcome [49-51]. While it is unclear whether the presence of CD8+ CTLs alone provides any prognostic information, the presence of high percentages of CD4+ T-helper cells at primary tumor sites positively correlates with disease progression, including metastatic spread to sentinel lymph nodes and increased primary tumor size [47,52]. Perhaps more significant is the ratio of CD4+ to CD8+ cells, where primary tumors with ratios greater than one correlate with lymph node metastasis and reduced patient survival [47,52]. Similar results have been reported for colorectal carcinoma [53], renal carcinoma, esophageal carcinoma, and small-cell lung carcinoma [54].
Why are CD8+ CTL-mediated responses not more effective in eradicating or minimizing cancer occurrence and how might CD4+ T cells be involved in enhancing breast cancer progression? One plausible mechanism may have to do with the 'polarity' of the CD4+ T-helper-cell response at primary tumor sites and/or their distant metastases (Figure 3). CD4+ T-helper cells are activated in response to soluble factors and can be classified generally into two categories as either Th1 cells or Th2 cells (Figure 4) [55]. Following an activating stimulus, CD4+ T-helper cells that are Th1-polarized secrete IFNγ, transforming growth factor beta, TNFα and IL-2 [56]. These cytokines collaborate with the cytotoxic/cell killing functions of CD8+ T cells [57] and can induce upregulation of antigen processing (in the proteasome), can induce expression of MHC class I and II molecules, and can induce other antigen display cofactors in neoplastic cells. Th1 CD4+ T-helper cells also enhance antitumor immune responses by secretion of INFγ, which in turn induces activation of macrophage cytotoxic activity [58]. In contrast Th2-polarized CD4+ T-helper cells express IL-4, IL-5, IL-6, IL-10 and IL-13, which induce T-cell anergy and loss of T-cell-mediated cytotoxicity while also enhancing humoral immunity (B-cell function) [59]. Taken together, Th1 responses are thought to be beneficial toward antitumor immunity [60-63] whereas Th2 responses may downregulate cell-mediated antitumor immunity [63-67] and enhance protumor humoral responses [68,69].
Figure 4.
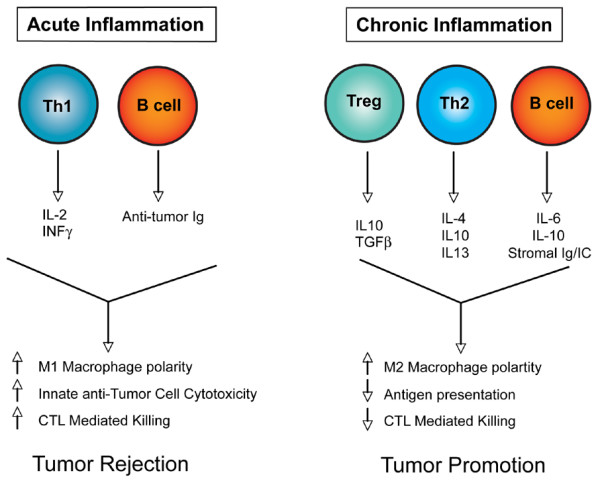
Model depicting the consequences of acute inflammation versus chronic inflammation. During acute antitumor inflammatory responses (left panel), Th1-polarized T cells secrete antitumor cytokines (IL-2 and INFγ, for example), which in combination with antitumor-directed B-cell-derived factors (such as immunoglobulins (Igs)) activate tumor inhibitory responses in recruited innate immune cells and cytotoxic T lymophocytes (CTLs) that together favor tumor rejection. In contrast, chronic activation of immune response (right panel) without resolution (of damage) often results in accumulation of regulatory T (Treg) cells, Th2 cells, and activated B cells, which in turn secrete progrowth factors (IL-4, IL-6, IL-10, IL-13, transforming growth factor beta (TGFβ) and immunoglobulins, for example) that enhance protumor responses in innate immune cells and inactivate CTL cytotoxicity, thus favoring tumor promotion.
Role for regulatory T lymphocytes
In addition to T-helper-cell polarity, developing neoplasms can escape CD8+ T-cell cytotoxicity by promoting expansion of regulatory T cells (Treg). Treg cells normally function to protect tissue from autoimmune disease by suppressing self-reactive cells. Typically characterized by expression of CD4, CD25 and FOXP3, Treg cells can account for approximately 5–10% of all T lymphocytes in healthy tissues.
In breast cancers the percentage of Treg cells, as assessed by FOXP3 positivity, increases in parallel with the disease stage, from normal to DCIS and from DCIS to invasive carcinoma [70]. In patients with invasive carcinoma the presence of high numbers of FOXP3+ T cells predicts worse relapse-free survival and decreased overall patient survival [70], and may indicate that the presence of Treg cells promotes tumor progression by inhibiting immunosuppression. The mechanisms underlying immunosuppression are not clear; however, CD4+CD25highFOXP3+ Treg cells retain the ability to actively inhibit CD4+CD25- T cells, CD8+ T cells, DCs, NK cells, and B cells in a cell-to-cell contact and dose-dependent manner [71-75]. The mechanisms by which developing tumors might stimulate proliferation and differentiation of Treg lymphocytes are still not well known, but production of prostaglandin E2 by tumor cells and of CCL22 by tumor-associated macrophages may act as chemotactic and differentiation agents for Treg cells [76-79].
Acquired immunity and carcinoma development
It is well established that chronic infiltration of tissue by some innate immune cell types (for example, immature monocytes, macrophages, mast cells or neutrophils) contributes to epithelial cancer development [3,8,80]. Many studies have reported that inflammatory leukocytes promote cancer development due to their enormous plasticity and their capacity to produce a myriad of cytokines, chemokines, metalloserine and metallocysteine proteases, reactive oxygen species, histamine and other bioactive mediators [3,8,80]. Several physiological processes necessary for tumor development, such as enhanced cell survival, tissue remodeling, angiogenesis and suppression of antitumor adaptive immune responses, are thus regulated by leukocytic infiltrates in neoplastic environments. This regulation is exemplified by positive correlations between numbers of innate immune cells (macrophages, mast cells and neutrophils) infiltrating human tumors with the number of blood vessels [81,82], and by experimental findings in mouse models where attenuating innate immune cell infiltration of premalignant tissue reduces angiogenesis and limits tumor development [83-90].
The earliest reports revealing a potential tumor-enhancing effect of adaptive immunity found that passive transfer of tumor-specific antibodies enhanced in vivo outgrowth of transplanted tumor cells or chemically induced tumors [91-93]. More recently, the availability of de novo carcinogenesis mouse models for the study organ specific cancer development has allowed experimentalist to revisit these analysis. Their experiments have revealed that the tumor enhancing roles of humoral immunity are more involved in innate immune cell responses, as opposed to direct regulation of neoplastic cell survival. For example, active immunization of mice harboring a mutant ras oncogene resulted in activation of humoral immune responses and enhanced papilloma formation upon chemical promotion [94,95].
Studies by Barbera-Guillem and colleagues reported that 'antitumor' humoral immune responses potentate in vivo growth and invasion of injected murine and human tumor cell lines via recruitment and activation of protumor granulocytes and macrophages [94,96,97]. Once in the neoplastic microenvironment, deposited immunoglobulin may mediate recruitment of innate immune cells via activation of complement cascades or engagement with Fc receptors expressed on resident or recruited innate immune cells that trigger various cellular responses, including phagocytosis, antigen presentation, secretion of proinflammatory mediators and antibody-dependent cytotoxicity [98,99]. During breast cancer development, immunoglobulin deposition in neoplastic stroma is known to increase the bioavailability of vascular endothelial growth factor (VEGF) by binding to Fc receptors on tumor-associated macrophages, thus triggering release of VEGF into the interstitium [96]. Moreover, the presence of endocytosed immunoglobulin in macrophages in breast carcinoma tissues corresponds with local extracellular VEGF protein levels and local angiogenic vascular buds [96].
The significance of tumor-associated macrophages has been experimentally addressed in a mouse model of mammary carcinogenesis; that is, mouse mammary tumor virus–polyomavirus middle T antigen transgenic mice [86]. In this model of mammary carcinogenesis, infiltration of macrophages into premalignant mammary tissue is associated with tumor progression [100]. Failure to recruit macrophages into neoplastic tissue does not alter the hallmarks of premalignancy, but instead significantly delays development of invasive carcinomas and results in reduced pulmonary metastases [86]. Metastatic potential is restored by transgenic expression of colony-stimulating factor (CSF)-1 in the mammary epithelium of CSF-1-deficient/polyomavirus middle T antigen mice [86]. Macrophage stimulation of mammary tumor progression is at least in part due to their ability to enhance angiogenesis via regulating VEGF levels and bioavailability [101]. These experimental data combined with the positive correlation between CSF-1 levels, macrophage recruitment and poor prognosis in human cancers [102] support the notion that macrophages and/or their products play a major role in facilitating late-stage metastatic progression of tumors [86]. Other cells of myeloid lineage have also been reported to contribute to tumor development – NK cells can play a role in protection against experimental tumor growth, in part by producing mediators with antiangiogenic properties [103,104]. These studies have together induced a paradigm shift regarding the role of immune cells during malignant progression. Whereas the historical viewpoint suggested that host immunity is protective with regards to cancer, it is now clear that certain subsets of chronically activated innate immune cells promote growth and/or facilitate survival of neoplastic cells.
In addition to providing a survival advantage to evolving neoplastic cells through their release of protumor mediators, is there evidence that recruited innate immune cells regulate any antitumor bioactivities of T cells present in premalignant/malignant mammary tissue? Neoplastic microenvironments favor polarized chronic protumorigenic inflammatory states, as opposed to those representing acute antitumor immune responses [8,105]. Clinical data indicate that the 'immune status' of healthy individuals is distinct as compared with those harboring malignant tumors, where T lymphocytes in the later population are found to be functionally impaired [106]. A subset of innate immune cells (for example, myeloid suppressor CD11b+Gr-1+ cells) accumulate in the peripheral blood of cancer patients [107,108], as well as in tumors and lymphoid organs [105,108,109]. Myeloid suppressor cells are known to induce T-lymphocyte dysfunction by direct cell–cell contact and by production of immunosuppressive mediators, and thus actively inhibit antitumor adaptive immunity [108,109]. Myeloid suppressor cells can also directly promote tumor growth by contributing to tumor-associated angiogenesis [110]. In addition, malignant lesions attract regulatory T cells that can suppress effector functions of CTLs [105]. Immune states such as these disable tumor-killing CD8+ CTL responses and enable states of immune privilege that foster escape from antitumor immunity while simultaneously exploiting activated innate immune cells that enhance cancer development. The degree to which similar mechanisms are at play during breast carcinogenesis is currently being investigated.
Cytokines derived from activated humoral immunity and/or Th2 T lymphocytes also actively regulate chemoattraction and polarization of tumor-associated leukocytes, especially macrophages. Chronically activated B cells (typically in germinal centers or in ectopic follicles) can produce granulocyte–macrophage-CSF, TNFα, IL-6, and IL-10 [111]. These cytokines, in combination with Th2 cytokines such as IL-4, IL-13, and IL-10, are potent effectors of innate immune cell polarization. Protumor M2 polarization in macrophages is induced by the Th2/humoral cytokines IL-4 and IL-10, while simultaneously repressing antitumor M1 macrophage polarization [112]. Granulocyte–macrophage-CSF, IL-6 and IL-10 secreted by activated B cells suppress macrophage cytotoxic activity [112], while IL-10 inhibits both antigen presentation by macrophages as well as monocyte differentiation into DC lineages [113]. Taken together, factors derived from chronically activated lymphocytes sculpt innate immune cell responses towards tumor tolerance and promotion of disease progression.
Conclusion
During the past decade, insights have been gained regarding mechanisms underlying the dynamic interplay between immune cells and tumor progression. The accumulated data indicate that the outcome of an immune response toward a tumor is largely determined by the type of immune response elicited (Figure 3). A tumor-directed immune response involving cytolytic CD8+ T cells, Th1 cells and NK cells appears to protect against tumor development and progression. If, on the other hand, the immune response involves B cells and activation of humoral immunity, and/or a Th2 polarized response the probable outcome is promotion of tumor development and progression. This balance between a protective cytotoxic response and a harmful humoral or Th2 response can be regulated systemically by the general immune status of the individual, as well as locally by myeloid suppressor cells and Treg cells, and thus offers clinicians attractive targets for anticancer immune-based therapies.
Abbreviations
CSF = colony-stimulating factor; CTL = cytotoxic T lymphocyte; DC = dendritic cell; DCIS = ductal carcinoma in situ; IFN = interferon; IL = interleukin; NK = natural killer; Th cells = T-helper cells; TNF = tumor necrosis factor; Treg cells = regulatory T cells; VEGF = vascular endothelial growth factor.
Competing interests
The authors declare that they have no competing interests.
Note
This article is part of a review series on Inflammation and breast cancer, edited by Mina J Bissell and Jeffrey W Pollard.
Other articles in the series can be found online at http://breast-cancer-research.com/articles/review-series.asp?series=bcr_Inflammation
Acknowledgments
Acknowledgements
The authors would like to acknowledge all the scientists who made contributions to the areas of research reviewed here that were not cited due to space constraints. The authors acknowledge support from a Postdoctoral Training Grant NCI T32 CA09043 (DDN), and grants from the National Institutes of Health, Sandler Program in Basic Sciences, the National Technology Center for Networks and Pathways, and a Department of Defense Era of Hope Scholar Award (LMC).
References
- American Cancer Society . Cancer Facts & Figures 2007. Alanta, GA: American Cancer Society; 2007. [Google Scholar]
- Balkwill F, Mantovani A. Inflammation and cancer: back to Virchow? Lancet. 2001;357:539–545. doi: 10.1016/S0140-6736(00)04046-0. [DOI] [PubMed] [Google Scholar]
- Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–867. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meylan E, Tschopp J, Karin M. Intracellular pattern recognition receptors in the host response. Nature. 2006;442:39–44. doi: 10.1038/nature04946. [DOI] [PubMed] [Google Scholar]
- Karin M. Nuclear factor-kappaB in cancer development and progression. Nature. 2006;441:431–436. doi: 10.1038/nature04870. [DOI] [PubMed] [Google Scholar]
- Karin M, Greten FR. NF-kappaB: linking inflammation and immunity to cancer development and progression. Nat Rev Immunol. 2005;5:749–759. doi: 10.1038/nri1703. [DOI] [PubMed] [Google Scholar]
- de Visser KE, Eichten A, Coussens LM. Paradoxical roles of the immune system during cancer development. Nat Rev Cancer. 2006;6:24–37. doi: 10.1038/nrc1782. [DOI] [PubMed] [Google Scholar]
- Balkwill F, Charles KA, Mantovani A. Smoldering and polarized inflammation in the initiation and promotion of malignant disease. Cancer Cell. 2005;7:211–217. doi: 10.1016/j.ccr.2005.02.013. [DOI] [PubMed] [Google Scholar]
- de Visser KE, Coussens LM. The inflammatory tumor microenvironment and its impact on cancer development. Contrib Microbiol. 2006;13:118–137. doi: 10.1159/000092969. [DOI] [PubMed] [Google Scholar]
- Coussens LM, Werb Z. Inflammatory cells and cancer: think different! J Exp Med. 2001;193:F23–F26. doi: 10.1084/jem.193.6.F23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osada T, Clay TM, Woo CY, Morse MA, Lyerly HK. Dendritic cell-based immunotherapy. Int Rev Immunol. 2006;25:377–413. doi: 10.1080/08830180600992456. [DOI] [PubMed] [Google Scholar]
- Ghiringhelli F, Apetoh L, Housseau F, Kroemer G, Zitvogel L. Links between innate and cognate tumor immunity. Curr Opin Immunol. 2007;19:224–231. doi: 10.1016/j.coi.2007.02.003. [DOI] [PubMed] [Google Scholar]
- David H. Rudolf Virchow and modern aspects of tumor pathology. Pathol Res Pract. 1988;183:356–364. doi: 10.1016/S0344-0338(88)80138-9. [DOI] [PubMed] [Google Scholar]
- Dunn GP, Old LJ, Schreiber RD. The immunobiology of cancer immunosurveillance and immunoediting. Immunity. 2004;21:137–148. doi: 10.1016/j.immuni.2004.07.017. [DOI] [PubMed] [Google Scholar]
- Boshoff C, Weiss R. AIDS-related malignancies. Nat Rev Cancer. 2002;2:373–382. doi: 10.1038/nrc797. [DOI] [PubMed] [Google Scholar]
- Penn I. Posttransplant malignancies. Transplant Proc. 1999;31:1260–1262. doi: 10.1016/S0041-1345(98)01987-3. [DOI] [PubMed] [Google Scholar]
- Euvrard S, Kanitakis J, Claudy A. Skin cancers after organ transplantation. N Engl J Med. 2003;348:1681–1691. doi: 10.1056/NEJMra022137. [DOI] [PubMed] [Google Scholar]
- Birkeland SA, Storm HH, Lamm LU, Barlow L, Blohme I, Forsberg B, Eklund B, Fjeldborg O, Friedberg M, Frodin L, et al. Cancer risk after renal transplantation in the Nordic countries, 1964–1986. Int J Cancer. 1995;60:183–189. doi: 10.1002/ijc.2910600209. [DOI] [PubMed] [Google Scholar]
- Fung JJ, Jain A, Kwak EJ, Kusne S, Dvorchik I, Eghtesad B. De novo malignancies after liver transplantation: a major cause of late death. Liver Transpl. 2001;7(11 Suppl 1):S109–S118. doi: 10.1053/jlts.2001.28645. [DOI] [PubMed] [Google Scholar]
- Pham SM, Kormos RL, Landreneau RJ, Kawai A, Gonzalez-Cancel I, Hardesty RL, Hattler BG, Griffith BP. Solid tumors after heart transplantation: lethality of lung cancer. Ann Thorac Surg. 1995;60:1623–1626. doi: 10.1016/0003-4975(95)00120-4. [DOI] [PubMed] [Google Scholar]
- Stewart T, Tsai SC, Grayson H, Henderson R, Opelz G. Incidence of de-novo breast cancer in women chronically immunosuppressed after organ transplantation. Lancet. 1995;346:796–798. doi: 10.1016/S0140-6736(95)91618-0. [DOI] [PubMed] [Google Scholar]
- Gallagher B, Wang Z, Schymura MJ, Kahn A, Fordyce EJ. Cancer incidence in New York State acquired immunodeficiency syndrome patients. Am J Epidemiol. 2001;154:544–556. doi: 10.1093/aje/154.6.544. [DOI] [PubMed] [Google Scholar]
- Oluwole SF, Ali AO, Shafaee Z, DePaz HA. Breast cancer in women with HIV/AIDS: report of five cases with a review of the literature. J Surg Oncol. 2005;89:23–27. doi: 10.1002/jso.20171. [DOI] [PubMed] [Google Scholar]
- McHeyzer-Williams MG. B cells as effectors. Curr Opin Immunol. 2003;15:354–361. doi: 10.1016/S0952-7915(03)00046-3. [DOI] [PubMed] [Google Scholar]
- Youinou P, Jamin C, Saraux A. B-cell: a logical target for treatment of rheumatoid arthritis. Clin Exp Rheumatol. 2007;25:318–328. [PubMed] [Google Scholar]
- Whitford PGW, Cambell AM. Flow cytometric analysis of tumour infiltrating lymphocyte activation and tumour cell MHC class I and II expression in breast cancer patients. Cancer Lett. 1992;61:157–164. doi: 10.1016/0304-3835(92)90174-T. [DOI] [PubMed] [Google Scholar]
- Morton BA, Ramey WG, Paderon H, Miller RE. Monoclonal antibody-defined phenotypes of regional lymph node and peripheral blood lymphocyte subpopulations in early breast cancer. Cancer Res. 1986;46(4 Pt 2):2121–2126. [PubMed] [Google Scholar]
- Wernicke M. Quantitative morphologic assessment of immunoreactivity in regional lymph nodes of patients with carcinoma of the breast. Surg Gynecol Obstet. 1975;140:919–924. [PubMed] [Google Scholar]
- Urdiales-Viedma M, Nogales-Fernandez F, Martos-Padilla S, Sanchez-Cantalejo E. Correlation of histologic grade and lymph node status with some histopathologic discriminants in breast cancer. Tumori. 1986;72:43–51. doi: 10.1177/030089168607200107. [DOI] [PubMed] [Google Scholar]
- Shimokawara I, Imamura M, Yamanaka N, Ishii Y, Kikuchi K. Identification of lymphocyte subpopulations in human breast cancer tissue and its significance: an immunoperoxidase study with anti-human T- and B-cell sera. Cancer. 1982;49:1456–1464. doi: 10.1002/1097-0142(19820401)49:7<1456::AID-CNCR2820490724>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- Lee AH, Happerfield LC, Bobrow LG, Millis RR. Angiogenesis and inflammation in ductal carcinoma in situ of the breast. J Pathol. 1997;181:200–206. doi: 10.1002/(SICI)1096-9896(199702)181:2<200::AID-PATH726>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- Coronella JA, Telleman P, Kingsbury GA, Truong TD, Hays S, Junghans RP. Evidence for an antigen-driven humoral immune response in medullary ductal breast cancer. Cancer Res. 2001;61:7889–7899. [PubMed] [Google Scholar]
- Punt CJ, Barbuto JA, Zhang H, Grimes WJ, Hatch KD, Hersh EM. Anti-tumor antibody produced by human tumor-infiltrating and peripheral blood B lymphocytes. Cancer Immunol Immunother. 1994;38:225–232. doi: 10.1007/BF01533513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coronella-Wood JA, Hersh EM. Naturally occurring B-cell responses to breast cancer. Cancer Immunol Immunother. 2003;52:715–738. doi: 10.1007/s00262-003-0409-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang BL, Springer GF, Kaufman MW. Concurrent immunohistochemical staining of tumor-infiltrating lymphocytes and carcinoma-associated T (Thomsen–Friedenreich)/Tn antigens in human breast carcinoma. J Histochem Cytochem. 1996;44:187–191. doi: 10.1177/44.2.8609376. [DOI] [PubMed] [Google Scholar]
- Jaume JC, Portolano S, Prummel MF, McLachlan SM, Rapoport B. Molecular cloning and characterization of genes for antibodies generated by orbital tissue-infiltrating B-cells in Graves' ophthalmopathy. J Clin Endocrinol Metab. 1994;78:348–352. doi: 10.1210/jc.78.2.348. [DOI] [PubMed] [Google Scholar]
- Stott DI, Hiepe F, Hummel M, Steinhauser G, Berek C. Antigen-driven clonal proliferation of B cells within the target tissue of an autoimmune disease. The salivary glands of patients with Sjogren's syndrome. J Clin Invest. 1998;102:938–946. doi: 10.1172/JCI3234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombo M, Dono M, Gazzola P, Roncella S, Valetto A, Chiorazzi N, Mancardi GL, Ferrarini M. Accumulation of clonally related B lymphocytes in the cerebrospinal fluid of multiple sclerosis patients. J Immunol. 2000;164:2782–2789. doi: 10.4049/jimmunol.164.5.2782. [DOI] [PubMed] [Google Scholar]
- Kim HJ, Krenn V, Steinhauser G, Berek C. Plasma cell development in synovial germinal centers in patients with rheumatoid and reactive arthritis. J Immunol. 1999;162:3053–3062. [PubMed] [Google Scholar]
- Tomer Y, Sherer Y, Shoenfeld Y. Autoantibodies, autoimmunity and cancer [review] Oncol Rep. 1998;5:753–761. doi: 10.3892/or.5.3.753. [DOI] [PubMed] [Google Scholar]
- Tan EM, Shi FD. Relative paradigms between autoantibodies in lupus and autoantibodies in cancer. Clin Exp Immunol. 2003;134:169–177. doi: 10.1046/j.1365-2249.2003.02259.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Disis ML, Pupa SM, Gralow JR, Dittadi R, Menard S, Cheever MA. High-titer HER-2/neu protein-specific antibody can be detected in patients with early-stage breast cancer. J Clin Oncol. 1997;15:3363–3367. doi: 10.1200/JCO.1997.15.11.3363. [DOI] [PubMed] [Google Scholar]
- Fernandez Madrid F. Autoantibodies in breast cancer sera: candidate biomarkers and reporters of tumorigenesis. Cancer Lett. 2005;230:187–198. doi: 10.1016/j.canlet.2004.12.017. [DOI] [PubMed] [Google Scholar]
- Sheikh KM, Quismorio FP, Friou GJ, Lee YT. Ductular carcinoma of the breast: serum antibodies to tumor-associated antigens. Cancer. 1979;44:2083–2089. doi: 10.1002/1097-0142(197912)44:6<2083::AID-CNCR2820440619>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- Lee YT, Sheikh KM, Quismorio FP, Jr, Friou GJ. Circulating anti-tumor and autoantibodies in breast carcinoma: relationship to stage and prognosis. Breast Cancer Res Treat. 1985;6:57–65. doi: 10.1007/BF01806011. [DOI] [PubMed] [Google Scholar]
- Wong PY, Staren ED, Tereshkova N, Braun DP. Functional analysis of tumor-infiltrating leukocytes in breast cancer patients. J Surg Res. 1998;76:95–103. doi: 10.1006/jsre.1998.5301. [DOI] [PubMed] [Google Scholar]
- Chin Y, Janseens J, Vandepitte J, Vandenbrande J, Opdebeek L, Raus J. Phenotypic analysis of tumor-infiltrating lymphocytes from human breast cancer. Anticancer Res. 1992;12:1463–1466. [PubMed] [Google Scholar]
- Aaltomaa S, Lipponen P, Eskelinen M, Kosma VM, Marin S, Alhava E, Syrjanen K. Lymphocyte infiltrates as a prognostic variable in female breast cancer. Eur J Cancer. 1992;28A:859–864. doi: 10.1016/0959-8049(92)90134-N. [DOI] [PubMed] [Google Scholar]
- Veronesi U, Paganelli G, Viale G, Luini A, Zurrida S, Galimberti V, Intra M, Veronesi P, Robertson C, Maisonneuve P, et al. A randomized comparison of sentinel-node biopsy with routine axillary dissection in breast cancer. N Engl J Med. 2003;349:546–553. doi: 10.1056/NEJMoa012782. [DOI] [PubMed] [Google Scholar]
- Page DL, Jensen RA, Simpson JF. Routinely available indicators of prognosis in breast cancer. Breast Cancer Res Treat. 1998;51:195–208. doi: 10.1023/A:1006122716137. [DOI] [PubMed] [Google Scholar]
- Badwe RA, Thorat MA, Parmar VV. Sentinel-node biopsy in breast cancer. N Engl J Med. 2003;349:1968–1971. doi: 10.1056/NEJM200311133492017. author reply 1968–1971. [DOI] [PubMed] [Google Scholar]
- Kohrt HE, Nouri N, Nowels K, Johnson D, Holmes S, Lee PP. Profile of immune cells in axillary lymph nodes predicts disease-free survival in breast cancer. PLoS Med. 2005;2:e284. doi: 10.1371/journal.pmed.0020284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pages C, Tosolini M, Camus M, Berger A, Wind P, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science. 2006;313:1960–1964. doi: 10.1126/science.1129139. [DOI] [PubMed] [Google Scholar]
- Naito Y, Saito K, Shiiba K, Ohuchi A, Saigenji K, Nagura H, Ohtani H. CD8+ T cells infiltrated within cancer cell nests as a prognostic factor in human colorectal cancer. Cancer Res. 1998;58:3491–3494. [PubMed] [Google Scholar]
- Moss RB, Moll T, El-Kalay M, Kohne C, Soo Hoo W, Encinas J, Carlo DJ. Th1/Th2 cells in inflammatory disease states: therapeutic implications. Expert Opin Biol Ther. 2004;4:1887–1896. doi: 10.1517/14712598.4.12.1887. [DOI] [PubMed] [Google Scholar]
- Munk ME, Emoto M. Functions of T-cell subsets and cytokines in mycobacterial infections. Eur Respir J Suppl. 1995;20:668s–675s. [PubMed] [Google Scholar]
- Romagnani S. The Th1/Th2 paradigm. Immunol Today. 1997;18:263–266. doi: 10.1016/S0167-5699(97)80019-9. [DOI] [PubMed] [Google Scholar]
- Stout RD, Bottomly K. Antigen-specific activation of effector macrophages by IFN-gamma producing (TH1) T cell clones. Failure of IL-4-producing (TH2) T cell clones to activate effector function in macrophages. J Immunol. 1989;142:760–765. [PubMed] [Google Scholar]
- Parker DC. T cell-dependent B cell activation. Annu Rev Immunol. 1993;11:331–360. doi: 10.1146/annurev.iy.11.040193.001555. [DOI] [PubMed] [Google Scholar]
- Kacha AK, Fallarino F, Markiewicz MA, Gajewski TF. Cutting edge: spontaneous rejection of poorly immunogenic P1.HTR tumors by Stat6-deficient mice. J Immunol. 2000;165:6024–6028. doi: 10.4049/jimmunol.165.11.6024. [DOI] [PubMed] [Google Scholar]
- Fallarino F, Gajewski TF. Cutting edge: differentiation of antitumor CTL in vivo requires host expression of Stat1. J Immunol. 1999;163:4109–4113. [PubMed] [Google Scholar]
- Lowes MA, Bishop GA, Crotty K, Barnetson RS, Halliday GM. T helper 1 cytokine mRNA is increased in spontaneously regressing primary melanomas. J Invest Dermatol. 1997;108:914–919. doi: 10.1111/1523-1747.ep12292705. [DOI] [PubMed] [Google Scholar]
- Tsung K, Meko JB, Peplinski GR, Tsung YL, Norton JA. IL-12 induces T helper 1-directed antitumor response. J Immunol. 1997;158:3359–3365. [PubMed] [Google Scholar]
- Hu HM, Urba WJ, Fox BA. Gene-modified tumor vaccine with therapeutic potential shifts tumor-specific T cell response from a type 2 to a type 1 cytokine profile. J Immunol. 1998;161:3033–3041. [PubMed] [Google Scholar]
- Pellegrini P, Berghella AM, Del Beato T, Cicia S, Adorno D, Casciani CU. Disregulation in TH1 and TH2 subsets of CD4+ T cells in peripheral blood of colorectal cancer patients and involvement in cancer establishment and progression. Cancer Immunol Immunother. 1996;42:1–8. doi: 10.1007/s002620050244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostrand-Rosenberg S, Grusby MJ, Clements VK. Cutting edge: STAT6-deficient mice have enhanced tumor immunity to primary and metastatic mammary carcinoma. J Immunol. 2000;165:6015–6019. doi: 10.4049/jimmunol.165.11.6015. [DOI] [PubMed] [Google Scholar]
- Kobayashi M, Kobayashi H, Pollard RB, Suzuki F. A pathogenic role of Th2 cells and their cytokine products on the pulmonary metastasis of murine B16 melanoma. J Immunol. 1998;160:5869–5873. [PubMed] [Google Scholar]
- Tan TT, Coussens LM. Humoral immunity, inflammation and cancer. Curr Opin Immunol. 2007;19:209–216. doi: 10.1016/j.coi.2007.01.001. [DOI] [PubMed] [Google Scholar]
- Johansson M, Tan T, de Visser KE, Coussens LM. Immune cells as anti-cancer therapeutic targets and tools. J Cell Biochem. 2007;101:918–926. doi: 10.1002/jcb.21230. [DOI] [PubMed] [Google Scholar]
- Bates GJ, Fox SB, Han C, Leek RD, Garcia JF, Harris AL, Banham AH. Quantification of regulatory T cells enables the identification of high-risk breast cancer patients and those at risk of late relapse. J Clin Oncol. 2006;24:5373–5380. doi: 10.1200/JCO.2006.05.9584. [DOI] [PubMed] [Google Scholar]
- Azuma T, Takahashi T, Kunisato A, Kitamura T, Hirai H. Human CD4+ CD25+ regulatory T cells suppress NKT cell functions. Cancer Res. 2003;63:4516–4520. [PubMed] [Google Scholar]
- Lim HW, Hillsamer P, Banham AH, Kim CH. Cutting edge: direct suppression of B cells by CD4+ CD25+ regulatory T cells. J Immunol. 2005;175:4180–4183. doi: 10.4049/jimmunol.175.7.4180. [DOI] [PubMed] [Google Scholar]
- Trzonkowski P, Szmit E, Mysliwska J, Dobyszuk A, Mysliwski A. CD4+CD25+ T regulatory cells inhibit cytotoxic activity of T CD8+ and NK lymphocytes in the direct cell-to-cell interaction. Clin Immunol. 2004;112:258–267. doi: 10.1016/j.clim.2004.04.003. [DOI] [PubMed] [Google Scholar]
- Romagnani C, Della Chiesa M, Kohler S, Moewes B, Radbruch A, Moretta L, Moretta A, Thiel A. Activation of human NK cells by plasmacytoid dendritic cells and its modulation by CD4+ T helper cells and CD4+ CD25hi T regulatory cells. Eur J Immunol. 2005;35:2452–2458. doi: 10.1002/eji.200526069. [DOI] [PubMed] [Google Scholar]
- Chen W. Dendritic cells and (CD4+)CD25+ T regulatory cells: crosstalk between two professionals in immunity versus tolerance. Front Biosci. 2006;11:1360–1370. doi: 10.2741/1889. [DOI] [PubMed] [Google Scholar]
- Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, Evdemon-Hogan M, Conejo-Garcia JR, Zhang L, Burow M, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 2004;10:942–949. doi: 10.1038/nm1093. [DOI] [PubMed] [Google Scholar]
- Ellner JJ. Suppressor cells of man. Clin Immunol Rev. 1981;1:119–214. [PubMed] [Google Scholar]
- Columba-Cabezas S, Serafini B, Ambrosini E, Sanchez M, Penna G, Adorini L, Aloisi F. Induction of macrophage-derived chemokine/CCL22 expression in experimental autoimmune encephalomyelitis and cultured microglia: implications for disease regulation. J Neuroimmunol. 2002;130:10–21. doi: 10.1016/S0165-5728(02)00170-4. [DOI] [PubMed] [Google Scholar]
- Goodwin JS, Ceuppens J. Regulation of the immune response by prostaglandins. J Clin Immunol. 1983;3:295–315. doi: 10.1007/BF00915791. [DOI] [PubMed] [Google Scholar]
- Ribatti D, Ennas MG, Vacca A, Ferreli F, Nico B, Orru S, Sirigu P. Tumor vascularity and tryptase-positive mast cells correlate with a poor prognosis in melanoma. Eur J Clin Invest. 2003;33:420–425. doi: 10.1046/j.1365-2362.2003.01152.x. [DOI] [PubMed] [Google Scholar]
- Esposito I, Menicagli M, Funel N, Bergmann F, Boggi U, Mosca F, Bevilacqua G, Campani D. Inflammatory cells contribute to the generation of an angiogenic phenotype in pancreatic ductal adenocarcinoma. J Clin Pathol. 2004;57:630–636. doi: 10.1136/jcp.2003.014498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yano H, Kinuta M, Tateishi H, Nakano Y, Matsui S, Monden T, Okamura J, Sakai M, Okamoto S. Mast cell infiltration around gastric cancer cells correlates with tumor angiogenesis and metastasis. Gastric Cancer. 1999;2:26–32. doi: 10.1007/s101200050017. [DOI] [PubMed] [Google Scholar]
- Sparmann A, Bar-Sagi D. Ras-induced interleukin-8 expression plays a critical role in tumor growth and angiogenesis. Cancer Cell. 2004;6:447–458. doi: 10.1016/j.ccr.2004.09.028. [DOI] [PubMed] [Google Scholar]
- Coussens LM, Raymond WW, Bergers G, Laig-Webster M, Behrendtsen O, Werb Z, Caughey GH, Hanahan D. Inflammatory mast cells up-regulate angiogenesis during squamous epithelial carcinogenesis. Genes Dev. 1999;13:1382–1397. doi: 10.1101/gad.13.11.1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Visser KE, Korets LV, Coussens LM. De novo carcinogenesis promoted by chronic inflammation is B lymphocyte dependent. Cancer Cell. 2005;7:411–423. doi: 10.1016/j.ccr.2005.04.014. [DOI] [PubMed] [Google Scholar]
- Lin EY, Nguyen AV, Russell RG, Pollard JW. Colony-stimulating factor 1 promotes progression of mammary tumors to malignancy. J Exp Med. 2001;193:727–740. doi: 10.1084/jem.193.6.727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin EY, Pollard JW. Macrophages: modulators of breast cancer progression. Novartis Found Symp. 2004;256:158–168. discussion 168–172, 259–269. [PubMed] [Google Scholar]
- Imada A, Shijubo N, Kojima H, Abe S. Mast cells correlate with angiogenesis and poor outcome in stage I lung adenocarcinoma. Eur Respir J. 2000;15:1087–1093. doi: 10.1034/j.1399-3003.2000.01517.x. [DOI] [PubMed] [Google Scholar]
- Bingle L, Brown NJ, Lewis CE. The role of tumour-associated macrophages in tumour progression: implications for new anticancer therapies. J Pathol. 2002;196:254–265. doi: 10.1002/path.1027. [DOI] [PubMed] [Google Scholar]
- Benitez-Bribiesca L, Wong A, Utrera D, Castellanos E. The role of mast cell tryptase in neoangiogenesis of premalignant and malignant lesions of the uterine cervix. J Histochem Cytochem. 2001;49:1061–1062. doi: 10.1177/002215540104900816. [DOI] [PubMed] [Google Scholar]
- Snell GD. Incompatibility reactions to tumor homotransplants with particular reference to the role of the tumor; a review. Cancer Res. 1957;17:2–10. [PubMed] [Google Scholar]
- Kaliss N. Immunological enhancement of tumor homografts in mice: a review. Cancer Res. 1958;18:992–1003. [PubMed] [Google Scholar]
- Agassy-Cahalon L, Yaakubowicz M, Witz IP, Smorodinsky NI. The immune system during the precancer period: naturally-occurring tumor reactive monoclonal antibodies and urethane carcinogenesis. Immunol Lett. 1988;18:181–189. doi: 10.1016/0165-2478(88)90017-X. [DOI] [PubMed] [Google Scholar]
- Siegel CT, Schreiber K, Meredith SC, Beck-Engeser GB, Lancki DW, Lazarski CA, Fu YX, Rowley DA, Schreiber H. Enhanced growth of primary tumors in cancer-prone mice after immunization against the mutant region of an inherited oncoprotein. J Exp Med. 2000;191:1945–1956. doi: 10.1084/jem.191.11.1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber H, Wu TH, Nachman J, Rowley DA. Immunological enhancement of primary tumor development and its prevention. Semin Cancer Biol. 2000;10:351–357. doi: 10.1006/scbi.2000.0331. [DOI] [PubMed] [Google Scholar]
- Barbera-Guillem E, Nyhus JK, Wolford CC, Friece CR, Sampsel JW. Vascular endothelial growth factor secretion by tumor-infiltrating macrophages essentially supports tumor angiogenesis, and IgG immune complexes potentiate the process. Cancer Res. 2002;62:7042–7049. [PubMed] [Google Scholar]
- Barbera-Guillem E, May KF, Jr, Nyhus JK, Nelson MB. Promotion of tumor invasion by cooperation of granulocytes and macrophages activated by anti-tumor antibodies. Neoplasia. 1999;1:453–460. doi: 10.1038/sj.neo.7900054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogarth PM. Fc receptors are major mediators of antibody based inflammation in autoimmunity. Curr Opin Immunol. 2002;14:798–802. doi: 10.1016/S0952-7915(02)00409-0. [DOI] [PubMed] [Google Scholar]
- Ravetch JV, Bolland S. IgG Fc receptors. Annu Rev Immunol. 2001;19:275–290. doi: 10.1146/annurev.immunol.19.1.275. [DOI] [PubMed] [Google Scholar]
- Maglione JE, Moghanaki D, Young LJ, Manner CK, Ellies LG, Joseph SO, Nicholson B, Cardiff RD, MacLeod CL. Transgenic Polyoma middle-T mice model premalignant mammary disease. Cancer Res. 2001;61:8298–8305. [PubMed] [Google Scholar]
- Lin EY, Li JF, Gnatovskiy L, Deng Y, Zhu L, Grzesik DA, Qian H, Xue XN, Pollard JW. Macrophages regulate the angiogenic switch in a mouse model of breast cancer. Cancer Res. 2006;66:11238–11246. doi: 10.1158/0008-5472.CAN-06-1278. [DOI] [PubMed] [Google Scholar]
- Society AC. Tumour-educated macrophages promote tumour progression and metastasis. Nat Rev Cancer. 2004;4:71–78. doi: 10.1038/nrc1256. [DOI] [PubMed] [Google Scholar]
- Hayakawa Y, Takeda K, Yagita H, Smyth MJ, Van Kaer L, Okumura K, Saiki I. IFN-gamma-mediated inhibition of tumor angiogenesis by natural killer T-cell ligand, alpha-galactosylceramide. Blood. 2002;100:1728–1733. [PubMed] [Google Scholar]
- Smyth MJ, Crowe NY, Godfrey DI. NK cells and NKT cells collaborate in host protection from methylcholanthrene-induced fibrosarcoma. Int Immunol. 2001;13:459–463. doi: 10.1093/intimm/13.4.459. [DOI] [PubMed] [Google Scholar]
- Zou W. Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nat Rev Cancer. 2005;5:263–274. doi: 10.1038/nrc1586. [DOI] [PubMed] [Google Scholar]
- Finke J, Ferrone S, Frey A, Mufson A, Ochoa A. Where have all the T cells gone? Mechanisms of immune evasion by tumors. Immunol Today. 1999;20:158–160. doi: 10.1016/S0167-5699(98)01435-2. [DOI] [PubMed] [Google Scholar]
- Almand B, Clark JI, Nikitina E, van Beynen J, English NR, Knight SC, Carbone DP, Gabrilovich DI. Increased production of immature myeloid cells in cancer patients: a mechanism of immunosuppression in cancer. J Immunol. 2001;166:678–689. doi: 10.4049/jimmunol.166.1.678. [DOI] [PubMed] [Google Scholar]
- Serafini P, De Santo C, Marigo I, Cingarlini S, Dolcetti L, Gallina G, Zanovello P, Bronte V. Derangement of immune responses by myeloid suppressor cells. Cancer Immunol Immunother. 2004;53:64–72. doi: 10.1007/s00262-003-0443-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabrilovich DI, Velders MP, Sotomayor EM, Kast WM. Mechanism of immune dysfunction in cancer mediated by immature Gr-1+ myeloid cells. J Immunol. 2001;166:5398–5406. doi: 10.4049/jimmunol.166.9.5398. [DOI] [PubMed] [Google Scholar]
- Yang L, Debusk LM, Fukuda K, Fingleton B, Green-Jarvis B, Shyr Y, Matrisian LM, Carbone DP, Lin PC. Expansion of myeloid immune suppressor Gr+CD11b+ cells in tumor-bearing host directly promotes tumor angiogenesis. Cancer Cell. 2004;6:409–421. doi: 10.1016/j.ccr.2004.08.031. [DOI] [PubMed] [Google Scholar]
- Pistoia V, Corcione A. Relationships between B cell cytokine production in secondary lymphoid follicles and apoptosis of germinal center B lymphocytes. Stem Cells. 1995;13:487–500. doi: 10.1002/stem.5530130506. [DOI] [PubMed] [Google Scholar]
- Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002;23:549–555. doi: 10.1016/S1471-4906(02)02302-5. [DOI] [PubMed] [Google Scholar]
- Mantovani A, Allavena P, Sica A. Tumour-associated macrophages as a prototypic type II polarised phagocyte population: role in tumour progression. Eur J Cancer. 2004;40:1660–1667. doi: 10.1016/j.ejca.2004.03.016. [DOI] [PubMed] [Google Scholar]