

Abstract
Mycobacterium leprae protein ML2640c belongs to a large family of conserved hypothetical proteins predominantly found in mycobacteria, some of them predicted as putative S-adenosylmethionine (AdoMet)-dependent methyltransferases (MTase). As part of a Structural Genomics initiative on conserved hypothetical proteins in pathogenic mycobacteria, we have determined the structure of ML2640c in two distinct crystal forms. As expected, ML2640c has a typical MTase core domain and binds the methyl donor substrate AdoMet in a manner consistent with other known members of this structural family. The putative acceptor substrate-binding site of ML2640c is a large internal cavity, mostly lined by aromatic and aliphatic side-chain residues, suggesting that a lipid-like molecule might be targeted for catalysis. A flap segment (residues 222–256), which isolates the binding site from the bulk solvent and is highly mobile in the crystal structures, could serve as a gateway to allow substrate entry and product release. The multiple sequence alignment of ML2640c-like proteins revealed that the central α/β core and the AdoMet-binding site are very well conserved within the family. However, the amino acid positions defining the binding site for the acceptor substrate display a higher variability, suggestive of distinct acceptor substrate specificities. The ML2640c crystal structures offer the first structural glimpses at this important family of mycobacterial proteins and lend strong support to their functional assignment as AdoMet-dependent methyltransferases.
Keywords: S-adenosylmethionine-dependent methyltransferase, Mycobacterium leprae, X-ray crystallography, function discovery
Mycobacterial genomics has revealed that a significant fraction of the predicted proteome (∼40%) lacks functional annotation. A comparative analysis of different mycobacterial genomes, including those of the important human pathogens Mycobacterium tuberculosis (Cole et al. 1998) and M. leprae (Cole et al. 2001), led to the identification of several conserved families of hypothetical proteins that are largely restricted to mycobacteria or actinomycetes. The M. leprae protein ML2640c belongs to one of these families, which is highly represented in some mycobacterial genomes. Thus, there are as many as 22 ML2640c-like genes in M. avium, 17 in M. tuberculosis, and 13 in M. ulcerans. Some of these proteins (classified into the Cluster of Orthologous Groups [COG] named COG3315; Tatusov et al. 2001) are annotated as S-adenosylmethionine (AdoMet)-dependent methyltransferases (MTases) involved in polyketide biosynthesis, because they share a conserved motif found at the N terminus of polyketide synthesis O-MTases (PFAM entry Omt_N); (Bateman et al. 2004). However, direct biochemical or structural data validating this functional assignment is missing for the entire ML2640c-like mycobacterial family.
As part of our Structural Genomics initiative that was focused on conserved hypothetical proteins of mycobacteria (Alzari et al. 2006; Fogg et al. 2006; Shepard et al. 2007), we have determined the crystal structure of ML2640c to obtain novel insights into the functional role of this protein family. The structure reveals a typical MTase core domain, which is highly conserved in the entire family and binds AdoMet in a similar way as observed in other known class I MTase structures. The acceptor substrate-binding site is occluded from the solvent and displays a largely apolar surface, suggesting that ML2640c-like enzymes might methylate lipid-like molecules. The higher variability of the amino acid residues defining this binding site in the ML2640c-like family also indicates that different members of the family could target distinct acceptor substrates. The crystal structures of ML2640c further validate the functional assignment of this protein family as AdoMet-dependent methyltransferases and provide useful hints on the structural basis of substrate binding and specificity.
Results and Discussion
The overall structure
The structure of seleno-L-methionine-substituted ML2640c has been determined using single-wavelength anomalous diffraction (SAD) methods in a tetragonal crystal form and refined to a final Rfactor of 19.9% (Rfree = 25.5%) at 2.8 Å resolution. This crystal form has two molecules in the asymmetric unit, and the final model comprises residues 8–310 from one monomer and 13–310 from the other. A second crystal form (hexagonal space group with one molecule in the asymmetric unit) was subsequently obtained, which diffracted to 1.7 Å resolution. The structure was determined by molecular replacement methods using the first model as a search probe, and refined to a final Rfactor of 20.8% (Rfree = 23.4%) (▶). In addition to the N terminus, the final hexagonal model lacks two protein loops (residues 58–69 and 241–250), which are presumably disordered in this form but are visible in the tetragonal crystal form.
Table 1.
Data collection, phasing, and refinement statistics

The overall structure shows an N-terminal helical domain followed by an α/β C-terminal domain made up of a central β-sheet flanked by α-helices (▶). The N-terminal helical domain is formed by the antiparallel association of three helices (α1–α3) packed at a right angle against a helical hairpin formed by α4 and the first part of the long α5. This domain presents a concave surface that is covered by an insertion from the C-terminal domain including helix α10 (see below). The α/β C-terminal domain contains a central seven-stranded β-sheet, with strand β7 antiparallel to the other six strands, and three helices on each side (▶). This topology corresponds to that observed for the highly conserved structural fold of AdoMet-dependent methyltransferases (Martin and McMillan 2002; Schubert et al. 2003). The major deviation from this fold in the C-terminal domain is a 30-residue insertion between β5 and α11 (222–256, ▶), which includes helix α10 and folds over the N-terminal helical domain. This region displays large temperature factors in the tetragonal structure (▶) and is disordered in the hexagonal crystal form, suggesting that it could serve as a flap for ligand binding to the acceptor substrate-binding site.
Figure 1.
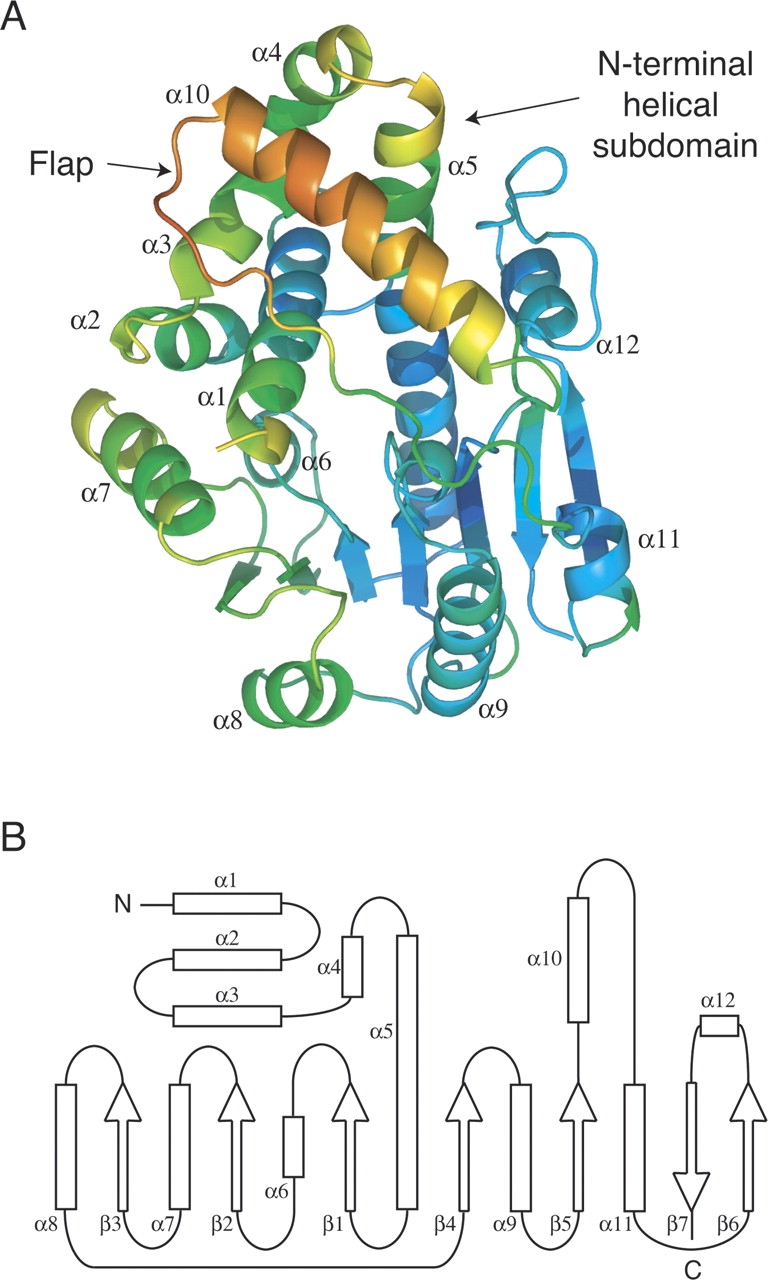
(A) ML2640c structure, in cartoon representation, colored by crystallographic temperature factors. Note the high mobility of the flap region. (B) Secondary structure topology of ML2640c.
Structural similarity searches
Similarity searches in structural databases confirmed the resemblance of the C-terminal domain with that of AdoMet-dependent methyltransferases (MTases). Searches using DALI (Holm and Sander 1998) revealed a large number of significant hits, all of them MTases acting upon a wide range of substrates that include small molecules, nucleic acids, and proteins (▶). Other search programs, like VAST at NCBI (Gibrat et al. 1996) and SSM at EBI (Krissinel and Henrick 2004), produced similar lists of structural neighbors. The central core of the AdoMet-dependent MTase fold, composed of the central β-sheet and flanking helices, matches the equivalent region from several eukaryotic and bacterial methyltransferases (▶). All methods, however, retrieved the same closest structural neighbor of ML2640c, namely the protein phosphatase methyltransferase 1 (PPM1) from yeast (PDB code 1RJD) (Leulliot et al. 2004). On the other hand, no significant hits were obtained with any of the above programs when using only the N-terminal domain of ML2640c (helices α1–α5) as a search probe, suggesting that the helical arrangement of this region in ML2640c has not been previously observed in other protein structures.
Table 2.
The 10 closest structural neighbors of ML2640c, as determined by DALI (Holm and Sander 1998)

The structural superposition of PPM1 and ML2640c reveals a well-conserved AdoMet-dependent MTase fold (including the binding pocket for the methyl donor substrate) as well as a partially conserved N-terminal helical domain (▶). However, there are significant structural changes in the putative binding site for the methyl acceptor substrate. In PPM1 the acceptor-binding site is a long tunnel open to the bulk solvent (▶), in agreement with its biochemical function of carboxyl-terminal protein methyltransferase. In contrast, a distinct disposition of the flap segment covers the equivalent site in ML2640c (▶), converting the entry tunnel of PPM1 into an internal, solvent-inaccessible cavity in ML2640c.
Figure 2.

(A) Structural superposition (RMSD 2.1 Å for 221 aligned residues) of the protein backbones of ML2640c (light brown) and yeast PPM1 (cyan), looking down along the methyl acceptor substrate-binding site. The proposed flap (including helix α10) is shown in darker color and AdoMet as yellow spheres. (B) Molecular surface representation of PPM1. (C) Molecular surface of ML2640c. Note that a different position of the flap (shown in dark color) covers the putative acceptor substrate-binding site in ML2640c, whereas the equivalent region in PPM1 has a different orientation and the binding site is open to the bulk solvent.
Multiple sequence alignment of ML2640c-like proteins
ML2640c belongs to a large family of conserved hypothetical proteins, mostly found in mycobacteria where their genes often occur in tandem. Using a relatively stringent E-value cutoff in a Blast search (Altschul et al. 1997), 70 nonidentical protein sequences could be retrieved and all of them are from mycobacterial species. The multiple sequence alignment (Supplemental Fig. 1) revealed 36 invariant amino acid positions. These positions are not equally distributed along the sequence, since the N-terminal helix α1 and the first part of the AdoMet-dependent MTase fold (from strands β1 to β5, see ▶) are highly conserved, whereas a more important variability is observed for the rest of the N-terminal helical domain and the second half of the AdoMet-dependent MTase fold. When the invariant residues were mapped into the 3D structure, all amino acid residues defining the AdoMet-binding pocket as seen in the PPM1 structure were observed to be strictly or largely conserved (▶). This conservation pattern, which probably reflects functional rather than structural constraints, is a strong indicator that most ML2640c-like proteins share the capability of binding the methyl donor substrate AdoMet.
Figure 3.

(A) Residue conservation from the multiple alignment of ML2640c-like sequences (Supplemental Fig. 1) mapped onto the ML2640c molecular surface, looking down the AdoMet-binding pocket. The methyl donor substrate is shown in yellow sticks as seen in the structurally equivalent PPM1 structure (PDB code 1RJD). Color code goes from blue (fully conserved) to red (poorly conserved). (B) Enlarged view of the AdoMet-binding site (color-coded as in A), showing that all protein residues close to the substrate (distance <4 Å) are invariant or largely conserved in the ML2640c-like family.
ML2640c binds S-adenosylmethionine
To further validate the above hypothesis, we carried out calorimetric and structural studies of ML2640c–AdoMet complexes. Binding measurements using isothermal titration calorimetry (ITC) demonstrated that AdoMet did bind to ML2640c with a 1:1 stoichiometry and an affinity constant in the micromolar range (▶). The binding reaction is both enthalpically and entropically favorable and conducted by the enthalpy term (ΔH°/ΔG° = 77.5%). The large value of the enthalpy term, the low K d value, and the 1:1 stoichiometry support a specific binding mechanism of the methyl donor substrate to the putative ML2640c active site.
Figure 4.

(A) Isothermal titration calorimetry measurement of the binding of AdoMet to ML2640c protein at 25°C. Upper panel, row calorimetric data of the titration of AdoMet into ML2640c corrected for the heat of dilution of the ligand. Lower panel, integrated heats of injections with the solid line corresponding to the best fit to the data using MicroCal software (N = 1.0 ± 0.1, K d = 2.1 ± 0.3 μM, ΔH° = −6.1 ± 0.7 kcal/mol−1, TΔS° = +1.7 ± 0.6 kcal/mol−1). (B) Electron density (2Fo–Fc) map (contoured at 1 σ) of the bound substrate. Protein-substrate hydrogen bonding interactions are indicated, and other important residues (see text). (C) The methyl donor substrate occupies the same binding pocket in ML2640c (yellow) and PPM1 methyltransferase (cyan) (PDB code 1RJG).
Tetragonal or hexagonal ML2640c crystals soaked with AdoMet cracked and eventually dissolved, suggesting that substrate binding promotes conformational changes in the protein. However, we were able to freeze one of the hexagonal crystal fragments and determined the crystal structure of the complex at 1.8 Å resolution (▶). The adenosyl moiety of AdoMet was clearly defined in the electron density map (▶), and its position and orientation with respect to the α/β C-terminal domain were closely similar to those observed in other MTases such as PPM1 (▶). The amino acid moiety of AdoMet was disordered in the ML2640c complex, probably because the protein complex failed to reach a final stable state in the crystalline state. Although our attempts to co-crystallize the protein–ligand complex were unsuccessful, the superposition of the ML2640c and PPM1 structures shows that the conformation of the amino acid moiety, as seen in the PPM1 structure, can be accommodated with no steric clashes within the ML2640c binding pocket (▶).
No significant structural differences are observed between the apo form of ML2640c and the AdoMet bound complex (root mean square deviation is 0.4 Å for all Cα positions). The ligand is bound within a deep pocket at the center of the protein, with contributions from the C-terminal ends of strands β1 to β4 and the N-terminal helix α1. In ML2640c, AdoMet binding follows the same mode as class I methyltransferases (Martin and McMillan 2002). The adenine base of AdoMet is stabilized by van der Waals interactions with Leu162, Leu188 and the aliphatic portion of Gln133, and by hydrogen-bonding interactions of the N6 and N1 positions with the Asp161 side chain and the Leu162 main-chain NH, respectively. The ribose moiety is close to protein residues Ala110 and Gly112, which are strictly conserved in ML2640c-like proteins (Supplemental Fig. 1) and correspond to the conserved motif GxG of class I MTases (Martin and McMillan 2002). The carboxylate oxygens of Asp132 are hydrogen bonded to the ribose O2 and O3 hydroxyls (▶), and the side-chain residues Arg25, Arg87, and Glu186 could make electrostatic interactions with the charged tail of the AdoMet residue.
The structure of the ML2640c–AdoMet complex confirmed the prediction from sequence analysis (▶), since all residues in contact with AdoMet are highly conserved in the family (Supplemental Fig. 1). The motif VGxTAxxVAxxRA in helix α1 (positions 14–26) includes the contact residues Gly15, Ala18, Val21, and Arg25 and is conserved in the family (except for Val21, replaced by Ile in some sequences), even though helix α1 seems to make mostly nonspecific contacts with the ligand. Other invariant residues interacting with the methyl-donor substrate in the ML2640c complex involve Ala110 and Gly112 between β1 and α6, Glu130 and Asp132 at the C terminus of β2, Asp161–Leu162–Arg163 between β3 and α8, and Glu186–Gly187–Leu188 between β4 and α9. This high conservation pattern, together with the binding and structural studies of the ML2640c–AdoMet complex, strongly validates the functional assignment of the ML2640c-like family of mycobacterial homologs as AdoMet-dependent MTases.
The architecture of the AdoMet-binding site is remarkably similar in ML2640c and PPM1. In the two proteins, the entrance to the substrate-binding site is partially occluded by the N terminus of helix α1 and by loops β2–α7 and β3–α8. However, the solvent-exposed surface of bound substrate is higher in the ML2640c complex (67 Å2) than in the PPM1 complex (8 Å2), as calculated with the program AreaIMol from the CCP4 package (Collaborative Computational Project, Number 4 1994). The difference is mostly due to the phenol group of PPM1 Tyr129 (replaced by Gln133 in ML2640c), which makes stacking interactions with the adenine base and occludes it from the solvent in PPM1. Although we cannot exclude the existence of a more open form of the apoprotein in solution, the crystal structure suggests that a relatively large conformational change would be necessary for AdoMet to enter (or for the reaction product to exit) the binding cavity. Indeed, such structural changes could explain the observed cracking of ML2640c crystals upon soaking with substrate. They could also involve the N-terminal segment of the protein (before α1), which is disordered in the two crystal forms of apo-ML2640c and might undergo a structural rearrangement upon AdoMet binding.
The binding site for the methyl acceptor substrate
The ML2640c structure clearly revealed the presence of two large pockets or cavities within the 3D structure (▶). One of these pockets is occupied by the methyl donor substrate AdoMet, as seen in the crystal structure of the complex. The second cavity is connected to the AdoMet-binding pocket at the position of the transferable methyl group and should therefore represent the acceptor substrate-binding site. It is primarily defined by residues from helices α1 and α5, the C-terminal ends of strands β4 and β5, and the protein loop immediately preceding β7. Furthermore, flap residues 230–234 (helix α10) and 248–252 cover the site and isolate it from the solvent (▶). Mobility of the flap (as seen in the hexagonal crystal form) connects the binding site to the bulk solvent, suggesting that the flap could serve as a gateway for substrate entry and product release.
Figure 5.

Electrostatic surfaces showing substrate-binding sites in different MTases. (A) ML2640c. (B) Chemotaxis receptor MTase CheR from Salmonella typhimurium (PDB code 1BC5) (Djordjevic and Stock 1998). (C) Yeast carboxy MTase for protein phosphatase 2A (1RJG) (Leulliot et al. 2004). (D) Catechol-O-MTase (1H1D) (Bonifacio et al. 2002). (E) Glycine N-MTase (1KIA) (Takata et al. 2003). (F) Mycolic acid cyclopropane synthase CmaA1 from M. tuberculosis (1KPG) (Huang et al. 2002).
Several amino acid residues lining the putative binding site for the acceptor substrate have aliphatic or aromatic side chains and, in contrast with residues defining the AdoMet-binding site, display a higher variability in ML2640c homologs (Supplemental Fig. 1), indicating that different members of the family may have different acceptor substrate specificities. The only exceptions are Arg87 and Glu217, which are both close to the region connecting the two substrate-binding cavities. Indeed, Arg87 is part of a group of deeply buried charged residues surrounding the AdoMet carboxylate group (together with Asp114, Arg116, and Glu186), which is strictly conserved in ML2640c homologs (Supplemental Fig. 1). Interestingly, these buried charged residues are also conserved in PPM1 (Arg81, Asp109, Arg111, and Glu201), where it has been proposed that Arg81 might play a role in catalysis by stabilizing a catalytically competent conformation of the AdoMet substrate (Leulliot et al. 2004).
The wide diversity in the biological roles of methylation is paralleled by the baffling number of methyltransferase enzymes that catalyze the methylation reaction. While a great majority of these enzymes use AdoMet as the methyl donor substrate, the binding site for the acceptor substrate is highly variable in AdoMet-dependent MTases (Martin and McMillan 2002). More than 120 members of this family (EC 2.1.1.X) have been identified, based on acceptor substrate specificity (small molecules, lipids, proteins, and nucleic acids) and the atom targeted for methylation (nitrogen, oxygen, carbon, and sulfur). Although we can only speculate on the putative nature of the ML2640c acceptor substrate, some insights can be gathered from a structural comparison of the acceptor substrate-binding site in other MTases. Depending on the nature of the substrate, the shape, size, and electrostatic properties of the binding site may vary widely. Thus, in MTases that transfer a methyl group to macromolecules such as DNA or proteins (▶), the binding site is a rather shallow region of the molecular surface. In PPM1 (▶), which methylates the C-terminal leucine in protein phosphatase 2A, the binding site is a relatively deep conical cavity (14 Å) that connects the catalytic center with the bulk solvent. On the other hand, MTases that use smaller soluble molecules as substrates may bind them in solvent-exposed grooves (one example is catechol-O-MTase; ▶) or in protein buried cavities (as in glycine MTase; ▶). Finally, lipids or similar hydrophobic acceptor substrates are expected to bind internal cavities of mostly apolar character, as observed for instance in mycolic acid cyclopropane synthase (▶).
In ML2640c, the solvent inaccessibility of the putative acceptor substrate-binding site, its rather apolar character, and large internal volume (∼400 Å3, similar to that of cyclopropane synthases [Huang et al. 2002], see ▶) suggest that the protein could methylate small or medium-sized lipid-like molecules. The large number of ML2640c-like paralogs in some mycobacterial genomes could thus reflect the participation of these enzymes at several stages in the biosynthesis of lipid-like metabolites. The structural data are also in agreement with the predicted functional assignment of the COG3315 family as MTases involved in polyketide (i.e., lipid-like) biosynthesis (Tatusov et al. 2001). Indeed, the ML2640c family of proteins appears to be largely restricted to bacteria in the order Actinomycetales, which are among the organisms known to produce a diversity of complex polyketides as secondary metabolites (Hopwood and Sherman 1990; Katz and Donadio 1993). However, it is puzzling that in no case is an ML2640c ortholog linked to a known gene cluster involved in polyketide or lipid biosynthesis in the sequenced mycobacterial genomes and this may suggest that other metabolites are the substrate. This is currently the subject of investigation.
Materials and Methods
Gene cloning and protein production
The ML2640c coding sequence was amplified by two-step PCR from the genomic DNA of M. leprae NT (Cole et al. 2001) and cloned into the expression vector pDEST17 using the Gateway recombination system (Invitrogen). Transformation of DH5α cells was done in 50 μL and two transformants were screened for recombinant insert analysis. After sequencing, a streak of freshly transformed BL21(DE3)pLysS cells by pDEST-ML2640c was used to inoculate 500 mL of LB medium containing 100 μg/mL ampicillin and 25 μg/mL chloramphenicol. The culture was then grown for 3.5 h at 30°C and induced with 1 mM isopropopyl β-D-thiogalactoside (IPTG) at OD600 of 0.8–0.9. After 1.5 h of induced growth, cells were harvested by centrifugation, resuspended, and frozen in 50 mM Tris-HCl buffer containing 150 mM NaCl and 10 mM imidazole, pH 8.0. Cellular suspension was thawed and cells lyzed in the French press at 14,000 psi. After centrifugation, the supernatant was loaded on Ni-NTA resin (Qiagen) and the protein eluted by a linear gradient of imidazole. The purified protein, diluted in 50 mM Tris-HCl buffer containing 0.1 M NaCl and 1 mM β-mercaptoethanol, pH 8.0, at 0.5 mg/mL, was incubated with His6-tagged TEV protease at a protein-TEV ratio (w/w) of 1:7 for 6 h at 30°C. After centrifugation, the supernatant was loaded on Ni-NTA resin, as above. The flow-through containing the tag-free protein was collected, analyzed on SDS-PAGE, and concentrated for crystallization. After cleavage with TEV, ML2640c contains a single Met → Gly substitution at the N terminus. The selenomethionine-labeled protein was produced in BL21(DE3) Escherichia coli cells (Novagen) as described (Wingfield 2000) and purified as described above for the nonlabeled protein.
Crystallization
Crystallization screens at 18°C were carried out using a Cartesian Technology workstation. Sitting-drops were composed of 200 nL of native ML2640c protein (12 mg/mL) and 200 nL of mother liquor equilibrated against 150 μL of the well solution on Greiner plates. Two different crystal forms were obtained from drops containing either sodium citrate or ammonium sulfate as precipitant. After manual optimization, the best crystals were obtained in 1.6 M ammonium sulfate, 50 mM MgCl2, and 100 mM Na-Hepes, pH 7.5 (tetragonal space group P43212), and 0.8 M sodium citrate, 100 mM Bicine, pH 9.0 (hexagonal space group P65). Crystals of seleno-L-methionine-labeled protein were obtained in the tetragonal space group and used for structure determination by SAD methods. Hexagonal ML2640c crystals were also soaked with 5 mM AdoMet. For data collection, crystals were transferred to a cryoprotectant solution containing the mother liquor +25% (v/v) of glycerol. All diffraction data sets were collected at the ESRF (Grenoble) using beamlines ID14.4 (SeMet-labeled protein) and ID29 (all other data sets).
Structure determination and refinement
The structure was determined using single-wavelength anomalous diffraction (SAD) data from a tetragonal crystal of SeMet-labeled ML2640c, measured at the K edge of selenium (▶). The substructure of the Se atoms was solved by direct methods (Weeks and Miller 1999) locating 16 sites, which were thereafter refined using SHARP (de La Fortelle and Bricogne 1997), allowing the identification of two additional minor sites. The electron density maps calculated with the solvent-flipped (Abrahams and Leslie 1996) SAD phases to 3.2 Å resolution allowed tracing the entire polypeptide chain for the two molecules in the asymmetric unit, except for the first seven (first molecule) or 12 (second molecule) N-terminal residues.
Model optimization was carried out by alternating crystallographic refinement cycles with the program REFMAC (Murshudov et al. 1999) and manual rebuilding with the programs O (Jones et al. 1991) and COOT (Emsley and Cowtan 2004) against a 2.8 Å resolution data set collected for the unlabeled protein. The refined model was subsequently used to solve the structure of the hexagonal crystal form, which contains only one molecule in the asymmetric unit, using molecular replacement methods (Navaza 1994). The crystallographic refinements of the protein alone to 1.7 Å resolution and its complex with AdoMet to 1.8 Å resolution were carried out as above, except that water molecules were now introduced in the model. In the hexagonal crystal form, two protein loops (residues 58–69 and 241–250) are disordered and were excluded from the final models. The parameters for the final refinement cycles are shown in ▶.
Isothermal titration calorimetry
Isothermal titration calorimetry (ITC) was performed using a VP-ITC (MicroCal). ML2640c was dialyzed into Hepes 0.1 M pH 7.0 buffer and AdoMet (Sigma) was resuspended into the same batch of the same buffer. Titration was performed by injecting 23 consecutive aliquots (10 μL) of AdoMet (100 μM) into the ITC cell containing ML2640c (3 μM) at 25°C. The heat of dilution of AdoMet was determined by performing the same experiment with the ITC cell containing buffer alone. The raw calorimetric data corrected for the heat of dilution was analyzed using the ORIGIN™ software provided by the manufacturer. The molar binding stoichiometry (N), association constant (K a; K d = 1/K a), and molar binding enthalpy (ΔH°) were determined by fitting the binding isotherm to a model with one set of sites. The binding entropy change (TΔS) was calculated (ΔG = ΔH – TΔS).
Bioinformatics tools and procedures
Sequence database searches were performed with BLAST at NCBI (Altschul et al. 1997). Fold searches and comparisons were done with DALI (Holm and Sander 1998), SSM (Krissinel and Henrick 2004), and VAST at NCBI (Gibrat et al. 1996). The APBS program, which solves the Poisson-Boltzmann equation in vacuum medium (Baker et al. 2001), was used for electrostatic calculations. Figures were prepared with PyMOL (http://pymol.sourceforge.net).
Protein Data Bank deposition
The atomic coordinates and structure factors have been deposited in the Protein Data Bank with accession codes 2CKD (tetragonal crystal form), 2UYO (hexagonal crystal form), and 2UYQ (ML2640c–AdoMet complex, hexagonal crystal form).
Acknowledgments
We thank Roman Laskowski, James D. Watson, Héctor Romero, and Gustavo Salinas for helpful discussions, and the staff of the ESRF (Grenoble, France) for technical assistance. This work has been partially supported by grants from the Institut Pasteur (GPH-Tuberculose), the Réseau National des Génopoles (RNG, France), and the European Commission contracts nos. QLG2-CT-2002-00988 (SPINE) and LSHP-CT-2005-018923 (NM4TB).
Footnotes
Supplemental material: see www.proteinscience.org
Reprint requests to: Pedro M. Alzari, Unité de Biochimie Structurale, Institut Pasteur, 25 rue du Dr. Roux, F-75724 Paris Cedex 15, France; e-mail: alzari@pasteur.fr; fax: 33-1-45688604.
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.072982707.
References
- Abrahams J.P. and Leslie, A.G.W. 1996. Methods used in the structure determination of bovine mitochondrial F1 ATPase. Acta Crystallogr. D Biol. Crystallogr. 52 30–42. [DOI] [PubMed] [Google Scholar]
- Altschul S.F., Madden, T.L., Schaffer, A.A., Zhang, J., Zhang, Z., Miller, W., and Lipman, D.J. 1997. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 25 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alzari P.M., Berglund, H., Berrow, N.S., Blagova, E., Busso, D., Cambillau, C., Campanacci, V., Christodoulou, E., Eiler, S., Fogg, M.J., et al. 2006. Implementation of semi-automated cloning and prokaryotic expression screening: The impact of SPINE. Acta Crystallogr. D Biol. Crystallogr. 62 1103–1113. [DOI] [PubMed] [Google Scholar]
- Baker N.A., Sept, D., Joseph, S., Holst, M.J., and McCammon, J.A. 2001. Electrostatics of nanosystems: Application to microtubules and the ribosome. Proc. Natl. Acad. Sci. 98 10037–10041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman A., Coin, L., Durbin, R., Finn, R.D., Hollich, V., Griffiths-Jones, S., Khanna, A., Marshall, M., Moxon, S., Sonnhammer, E.L., et al. 2004. The Pfam protein families database. Nucleic Acids Res. 32 D138–D141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonifacio M.J., Archer, M., Rodrigues, M.L., Matias, P.M., Learmonth, D.A., Carrondo, M.A., and Soares-da-Silva, P. 2002. Kinetics and crystal structure of catechol-O-methyltransferase complex with co-substrate and a novel inhibitor with potential therapeutic application. Mol. Pharmacol. 62 795–805. [DOI] [PubMed] [Google Scholar]
- Cole S.T., Brosch, R., Parkhill, J., Garnier, T., Churcher, C., Harris, D., Gordon, S.V., Eiglmeier, K., Gas, S., Barry III, C.E., et al. 1998. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393 537–544. [DOI] [PubMed] [Google Scholar]
- Cole S.T., Eiglmeier, K., Parkhill, J., James, K.D., Thomson, N.R., Wheeler, P.R., Honore, N., Garnier, T., Churcher, C., Harris, D., et al. 2001. Massive gene decay in the leprosy bacillus. Nature 409 1007–1011. [DOI] [PubMed] [Google Scholar]
- Collaborative Computational Project, Number 4 1994. The CCP4 suite: Programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50 760–763. [DOI] [PubMed] [Google Scholar]
- de La Fortelle E. and Bricogne, G. 1997. Maximum-likelihood heavy-atom parameter refinement for multiple isomorphous replacement and multiwavelength anomalous diffraction methods. Methods Enzymol. 276 472–494. [DOI] [PubMed] [Google Scholar]
- Djordjevic S. and Stock, A.M. 1998. Chemotaxis receptor recognition by protein methyltransferase CheR. Nat. Struct. Biol. 5 446–450. [DOI] [PubMed] [Google Scholar]
- Emsley P. and Cowtan, K. 2004. Coot: Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60 2126–2132. [DOI] [PubMed] [Google Scholar]
- Fogg M.J., Alzari, P.M., Bahar, M., Bertini, I., Betton, J.M., Burmeister, W.P., Cambillau, C., Canard, B., Carrondo, M., Coll, M., et al. 2006. Application of the use of high-throughput technologies to the determination of protein structures of bacterial and viral pathogens. Acta Crystallogr. D Biol. Crystallogr. 62 1196–1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibrat J.F., Madej, T., and Bryant, S.H. 1996. Surprising similarities in structure comparison. Curr. Opin. Struct. Biol. 6 377–385. [DOI] [PubMed] [Google Scholar]
- Holm L. and Sander, C. 1998. Touring protein fold space with DALI/FSSP. Nucleic Acids Res. 26 316–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopwood D.A. and Sherman, D.H. 1990. Molecular genetics of polyketides and its comparison to fatty acid biosynthesis. Annu. Rev. Genet. 24 37–66. [DOI] [PubMed] [Google Scholar]
- Huang C.C., Smith, C.V., Glickman, M.S., Jacobs, W.R., and Sacchettini, J.C. 2002. Crystal structures of mycolic acid cyclopropane synthases from Mycobacterium tuberculosis . J. Biol. Chem. 277 11559–11569. [DOI] [PubMed] [Google Scholar]
- Jones T.A., Zou, J.Y., Cowan, S.W., and Kjeldgaard, M. 1991. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A 47 110–119. [DOI] [PubMed] [Google Scholar]
- Katz L. and Donadio, S. 1993. Polyketide synthesis: Prospects for hybrid antibiotics. Annu. Rev. Microbiol. 47 875–912. [DOI] [PubMed] [Google Scholar]
- Krissinel E. and Henrick, K. 2004. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr. D Biol. Crystallogr. 60 2256–2268. [DOI] [PubMed] [Google Scholar]
- Leulliot N., Quevillon-Cheruel, S., Sorel, I., de La Sierra-Gallay, I.L., Collinet, B., Graille, M., Blondeau, K., Bettache, N., Poupon, A., Janin, J., et al. 2004. Structure of protein phosphatase methyltransferase 1 (PPM1), a leucine carboxyl methyltransferase involved in the regulation of protein phosphatase 2A activity. J. Biol. Chem. 279 8351–8358. [DOI] [PubMed] [Google Scholar]
- Martin J.L. and McMillan, F.M. 2002. SAM (dependent) I AM: The S-adenosylmethionine-dependent methyltransferase fold. Curr. Opin. Struct. Biol. 12 783–793. [DOI] [PubMed] [Google Scholar]
- Murshudov G.N., Vagin, A.A., Lebedev, A., Wilson, K.S., and Dodson, E.J. 1999. Efficient anisotropic refinement of macromolecular structures using FFT. Acta Crystallogr. D Biol. Crystallogr. 55 247–255. [DOI] [PubMed] [Google Scholar]
- Navaza J. 1994. AMoRe: An automated package for molecular replacement. Acta Crystallogr. A 50 157–163. [Google Scholar]
- Schubert H.L., Blumenthal, R.M., and Cheng, X. 2003. Many paths to methyltransfer: A chronicle of convergence. Trends Biochem. Sci. 28 329–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepard W., Haouz, A., Graña, M., Buschiazzo, A., Betton, J.M., Cole, S.T., and Alzari, P.M. 2007. The crystal structure of Rv0813c from Mycobacterium tuberculosis reveals a new family of fatty acid binding protein-like proteins in bacteria. J. Bacteriol. 189 1899–1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takata Y., Huang, Y., Komoto, J., Yamada, T., Konishi, K., Ogawa, H., Gomi, T., Fujioka, M., and Takusagawa, F. 2003. Catalytic mechanism of glycine N-methyltransferase. Biochemistry 42 8394–8402. [DOI] [PubMed] [Google Scholar]
- Tatusov R.L., Natale, D.A., Garkavtsev, I.V., Tatusova, T.A., Shankavaram, U.T., Rao, B.S., Kiryutin, B., Galperin, M.Y., Fedorova, N.D., and Koonin, E.V. 2001. The COG database: New developments in phylogenetic classification of proteins from complete genomes. Nucleic Acids Res. 29 22–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weeks C.M. and Miller, R. 1999. The design and implementation of SnB v2.0. J. Appl. Crystallogr. 32 120–124. [Google Scholar]
- Wingfield P. 2000. Production of recombinant proteins. In Current protocols in protein science (eds. J. Coligan et al.), pp. 5.3.9–14. Wiley, New York.