

Abstract
As membrane transporter proteins, VGLUT1–3 mediate the uptake of glutamate into synaptic vesicles at presynaptic nerve terminals of excitatory neural cells. This function is crucial for exocytosis and the role of glutamate as the major excitatory neurotransmitter in the central nervous system. The three transporters, sharing 76% amino acid sequence identity in humans, are highly homologous but differ in regional expression in the brain. Although little is known regarding their three-dimensional structures, hydropathy analysis on these proteins predicts 12 transmembrane segments connected by loops, a topology similar to other members in the major facilitator superfamily, where VGLUT1–3 have been phylogenetically classified. In this work, we present a three-dimensional model for the human VGLUT1 protein based on its distant bacterial homolog in the same superfamily, the glycerol-3-phosphate transporter from Escherichia coli. This structural model, stable during molecular dynamics simulations in phospholipid bilayers solvated by water, reveals amino acid residues that face its pore and are likely to affect substrate translocation. Docking of VGLUT1 substrates to this pore localizes two different binding sites, to which inhibitors also bind with an overall trend in binding affinity that is in agreement with previously published experimental data.
Keywords: vesicular glutamate transporter, homology modeling, membrane protein structure, inhibitor, docking, molecular dynamics
The amino acid glutamate is used by the mammalian central nervous system as the major excitatory neurotransmitter. Following its exocytotic release into the synaptic cleft, glutamate binds post-synaptic receptors, ligand-gated ion channels that depolarize the post-synaptic neuron. However, for this to happen, a presynaptic transport system is needed to upload the neurotransmitter into small vesicles at the axon terminals. This uploading system uses vesicular glutamate transporters (VGLUTs) as its central component (Takamori 2006). Unlike plasma membrane glutamate transporters, which work with high affinity to terminate glutamate signaling by clearing the neurotransmitter molecules present in the synaptic cleft, the vesicular transporters transport glutamate with an affinity that is 100–1000-fold lower (Shigeri et al. 2004), but against much higher concentration gradients (Otis 2001). A total of three VGLUT subtypes have been identified in mammals: VGLUT1–3 (Aihara et al. 2000; Fremeau et al. 2002; Gras et al. 2002). These proteins are highly homologous in the amino acid sequence, but display markedly different and complementary regional expression patterns (Fremeau et al. 2004).
The human VGLUT1 protein comprises 560 amino acids. Due to its significant sequence homology (up to 30% identity) to mammalian Na+-dependent inorganic phosphate cotransporters, together with experiments demonstrating elevated levels of Na+-dependent Pi uptake after VGLUT1 mRNA injection into frog oocytes, VGLUT1 was originally identified as a brain-specific inorganic phosphate cotransporter (Ni et al. 1994). However, more recent observations have established vesicular glutamate transport as its predominant role in vivo (Bellocchio et al. 2000; Takamori et al. 2000). The functional difference between plasma membrane and vesicular glutamate transporters is also reflected in the phylogenetic separation between the two groups of proteins. The vesicular glutamate transporters have been classified as members of the anion-cation subfamily (ACS) of the major facilitator superfamily (MFS) (Pao et al. 1998), the largest family of secondary transporter proteins known today. With over 5000 members, the MFS proteins are represented in all kingdoms of life. They facilitate the transport across the membrane of a variety of hydrophilic solutes, such as ions, sugars, nucleosides, amino acids, small peptides, drugs, and neurotransmitters. Most MFS transporters are believed to feature 12 transmembrane helices (Abramson et al. 2003b; Lemieux et al. 2004), and their sequences display a weak internal homology between the N- and C-terminal halves of the protein, which are structurally related through a twofold psuedo-symmetry in the MFS structures (Abramson et al. 2003a; Huang et al. 2003; Yin et al. 2006a).
While structure determination through overexpression, purification, and crystallization of mammalian membrane proteins is notoriously challenging, inferring structural information from bacterial homologs of mammalian MFS transporters has been proven to be very successful (Almqvist et al. 2004; Vardy et al. 2004). Recently, a mutagenesis study on human VGLUT2 identified residues essential for full transport activity of L-glutamate (Juge et al. 2006), and the position of these residues in the backbone structure of a homology model was shown. This model was based on the X-ray crystal structure of a distant bacterial homolog—the glycerol-3-phosphate transporter GlpT from Escherichia coli. To gain further insight into the structure and mechanism of the VGLUTs, we present a three-dimensional model of VGLUT1 validated through molecular dynamics (MD) simulations, and describe its interaction with several known substrates and inhibitors.
Results
VGLUT domain parsing
The three human vesicular glutamate transporters are highly conserved in the amino acid sequence (▶). However, at the N- and C-terminal their sequences display significant lower homology. In the first 60 residues, although the degree of homology is low, there is some sequence conservation between all three proteins. The amino acid composition is indicative of water-soluble parts rather than membrane spanning, with roughly 50% of the residues being charged or polar. The overall charge, however, is close to neutral. In contrast, the last 60 C-terminal residues are the least conserved in the entire sequences, and heavily enriched in aspartate and glutamate, resulting in a net charge of −7 to –9 for this region. Given the high number of negative charges, this part of the sequence is also unlikely to span the vesicle membrane. Furthermore, for VGLUT1, this C-terminal region contains a very high number of proline residues (13 out of 43, or 30%). Outside a phospholipid membrane, the presence of tri- and tetra-proline peptides precludes secondary structure elements such as α-helices or β-sheets (Lise and Jones 2005), and suggests a flexible structure in this region. This is also confirmed in two recent studies (De Gois et al. 2006; Vinatier et al. 2006), which identifies protein–protein interactions between two isoforms of endophilins to a proline-rich motif (PPRPPPP) in the C-terminal region of VGLUT1. Finally, besides these two peripheral N- and C-terminal regions, another predominantly hydrophilic section is present in the center of the sequences. Situated in the middle of an overall hydrophobic milieu, a region of 40 amino acids (starting from S256 in VGLUT1) probably constitutes a loop connecting two transmembrane domains. Thus, three large regions of the sequences appear as extramembrane. With the alignment parsed in this way, the transmembrane segments and loops connecting them should be confined to the remaining two parts of the VGLUT sequences, which together consist of 468 residues with 79% sequence identity between the three VGLUT isoforms, in sharp contrast to the 17% for the N-terminal section, the 2% for the C-terminal section, and the 53% for the middle section. The alignment between VGLUT1 and GlpT (▶) was then used to build our three-dimensional model of the transmembrane region of VGLUT1. For clarity we have chosen to limit the modeling to VGLUT1. However, because of the high sequence identity between the three vesicular glutamate transporters in the modeled region, results should be representative for all the three human isoforms.
Figure 1.

Multiple sequence alignment between the three human vesicular glutamate transporters, VGLUT1–3, and the glycerol-3-phosphate transporter (GlpT) from E. coli. Solid lines mark the 12 predicted helical regions (H1–H12) and the predicted cytoplasmic loop in VGLUT 1–3. Gray tubes correspond to regions with transmembrane helices in the GlpT crystal structure, connected by cytoplasmic (convex) and periplasmic (concave) loops.
Proposed transmembrane topology
Transmembrane segment prediction software suggests 12 α-helical regions, with six helices in each of the two domains separated by the long, central loop in the middle section of the vesicular glutamate transporters sequences (▶). By combining these predicted transmembrane boundaries with those found in the GlpT crystal structure (both shown in ▶), we obtain a transmembrane topology for VGLUT1 (▶). In this topology model, both the N and C termini are located on the cytoplasmic side of the vesicular membrane. This sidedness is in agreement with a recent topology study on VGLUT2 expressed in COS7 cells (Jung et al. 2006), which suggests that both the N- and C-terminal regions reside in the cytoplasm and that the loop between transmembrane segments 1 and 2 faces the vesicle lumen. In addition, the proposed VGLUT1 helices follow the positive-inside rule (von Heijne 1989): 15 positively charged residues are situated near the start and end of the transmembrane helices on the cytoplasmic side, while only six such residues are found on the opposite side.
Figure 2.
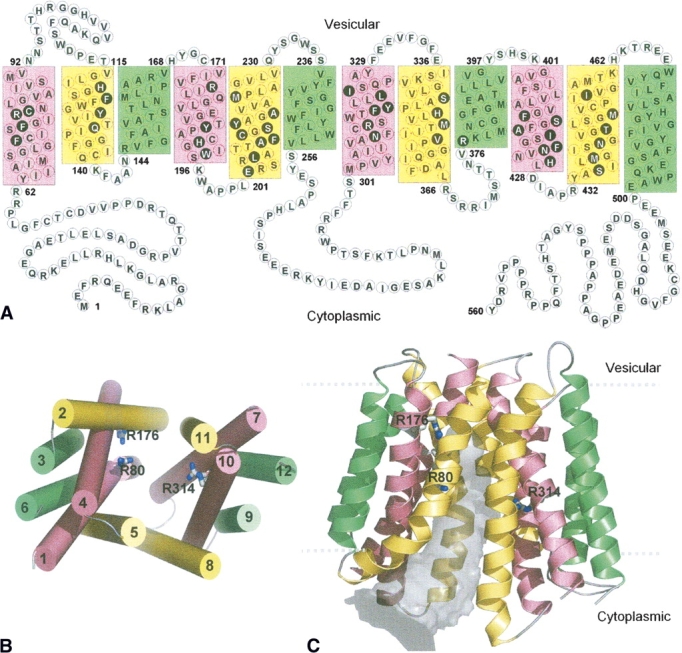
(A) Proposed topology of VGLUT1 based on transmembrane segment prediction and topology of bacterial MFS proteins. Residues in filled black circles face the center of the pore that separates the N- and C-terminal domains in 3D. The 12 transmembrane helices in this and the following figures are colored according to the grouping of helices in the oxalate transporter (Hirai et al. 2002), based on their symmetrical positions in the structure. Green: straight-spanning peripheral helices that are not involved in defining the pore. Yellow and pink: curved helices, at the interface of the two domains, lining the pore. (B) Packing of the helices viewed from the cytoplasmic side. The 12 helices are numbered sequentially. (C) Cartoon diagram of the human VGLUT1 model. The 12 transmembrane helices form pseudo-symmetrical domains, separated by a pore (gray volume) that is open to the cytoplasmic side. Three arginine residues (R80 from helix 1, R176 from helix 4, and R314 from helix 7) that are exposed to the pore are shown in sticks. The highly variable first and last 60 residues of the N- and C-terminal are not shown.
Overall three-dimensional structure
The spatial arrangement of the transmembrane helices is illustrated in ▶. The 12 helices (H1–H12) pack in four groups of three each (helices 1,5,6, 4,2,3, 7,11,12, and 10,8,9), with two curved helices (H1 and H5) and one straight spanning (H6) per group. Contacts within these groups are established by the packing of both hydrophobic and hydrophilic side chains. The first six helices at the N-terminal and the last six helices at the C-terminal constitute two separate pseudo-symmetrical halves, respectively. The overall three-dimensional VGLUT1 model features a conformation of the transporter closed to the vesicle lumen, with a water-accessible cavity between the two domain halves (▶). This funnel-shaped cavity is wide open at the cytoplasmic entrance (22 Å between H5 and H11 Cα) and narrows toward the center of the molecule (∼10 Å), which is also the presumed middle of the vesicular membrane. In this model, 21 residues from the N-terminal half and 24 residues from the C-terminal half face the inside of the pore (marked in ▶). Five of these residues are charged, 19 residues are polar, and 21 residues are hydrophobic. Remarkably, 44 of these 45 residues are fully conserved among VGLUT1–3 (▶).
Assessment of model stability
To validate the stability of the structural model, and further test if the modeling of this mammalian membrane protein based on a bacterial MFS structure is reasonable, we have carried out MD simulations for the model structure in DMPC bilayers surrounded by water. MD simulations on the GlpT template were carried out as a control. These simulations confirm that both the VGLUT1 model and the GlpT crystal structure template are stable in the simulated systems, with all transmembrane helices for both structures remaining intact after 10 nsec. The structural drift to the starting structure for transmembrane helices and loops (boundaries as in ▶) of the simulated VGLUT1 model is quantified through root-mean-square deviation (RMSD) values (▶). As expected, the transmembrane helices display the lowest RMSD fluctuations, with a flat curve after equilibration, indicating discrete structural change compared to the initial structure. When compared as subsets, helices from the N- and C-terminal domains of VGLUT1 display similar RMSD curves, suggesting a similar amount of structural variation in the two domains during the simulation. In contrast, the cytosolic loop connecting the N- and C-terminal domains display the largest coordinate displacement from the initial structure, followed by the vesicular loop between helices H1 and H2. When compared to the simulation of the template structure (data not shown), we notice a similar level of structural change from the starting structure, especially for the transmembrane helices. For both the VGLUT1 model and the GlpT template, all transmembrane helices remain stable after equilibration. After 10 nsec of MD simulation, secondary structure analysis by STRIDE reports 256 out of 305 residues as helical in the bilayer region for the VGLUT1 model, compared to 283 out of 311 for the GlpT template. The overall change in secondary structure during simulation is discrete for both systems.
Figure 3.

C-α RMSDs to the structure of the VGLUT1 model during molecular dynamics simulation (tm = transmembrane).
Surface properties of the model
Another important feature of the spatial distribution of residues in the VGLUT1 model is that its surface properties follow those observed for integral membrane proteins (Landolt-Marticorena et al. 1993; von Heijne 1994). In the region anticipated to face the fatty acid chains of the phospholipid membrane, the surface area is almost exclusively composed of hydrophobic side chains (92 hydrophobic and eight polar). Charged residues are found in the cytoplasm, periplasm, or in the cavity between the N- and C-terminal halves of the molecule, but not facing the anticipated membrane interface. In addition, the membrane–water interface regions of the model are rich in tryptophan and tyrosine residues. The VGLUT1 protein contains a total of 28 such residues in the region that we modeled (▶), and 16 of these are situated in the membrane–water interfaces, where they interact with phospholipid head groups during the MD simulation.
Dynamics of charged residues
Analysis of all the charged or possibly charged residues present in the membrane-embedded region of the VGLUT1 model reveals a total of eight residues: R80, H120, E183, and R176 in the N-terminal (▶, left), and R314, H348, E388, and H479 from the C-terminal (▶, right). Several of these residues protrude their side chains to the pore of the model. From the N-terminal domain, the side chains of R80 and E183 are found interacting with each other, while R176 and H120 project into the pore. In the C-terminal domain, the side chains of E388 and H479 also interact with each other, while R314 and H348 are found facing the center of the pore. Such highly conserved charged residues near the center of the transporter are also seen in two MFS crystal structures (Abramson et al. 2003a; Huang et al. 2003), and the presence of these residues in the VGLUT1 model suggests possible candidates for substrate binding and/or translocation. In ▶, the conformations of these charged residues before and after MD simulation are shown. While little change is seen for the residues interacting with each other during 10 nsec of MD simulation, R176 moves its guanidinium group from the pore of the model into the N-terminal domain (▶). This causes a slight opening of the N-terminal domain interface toward the vesicular side, so that R176 might become accessible to substrates and solvent in the vesicular lumen, although the central pore of the transporter remains closed. This structural flexibility also allows H120 to move toward the interior of the N-terminal domain (▶).
Figure 4.
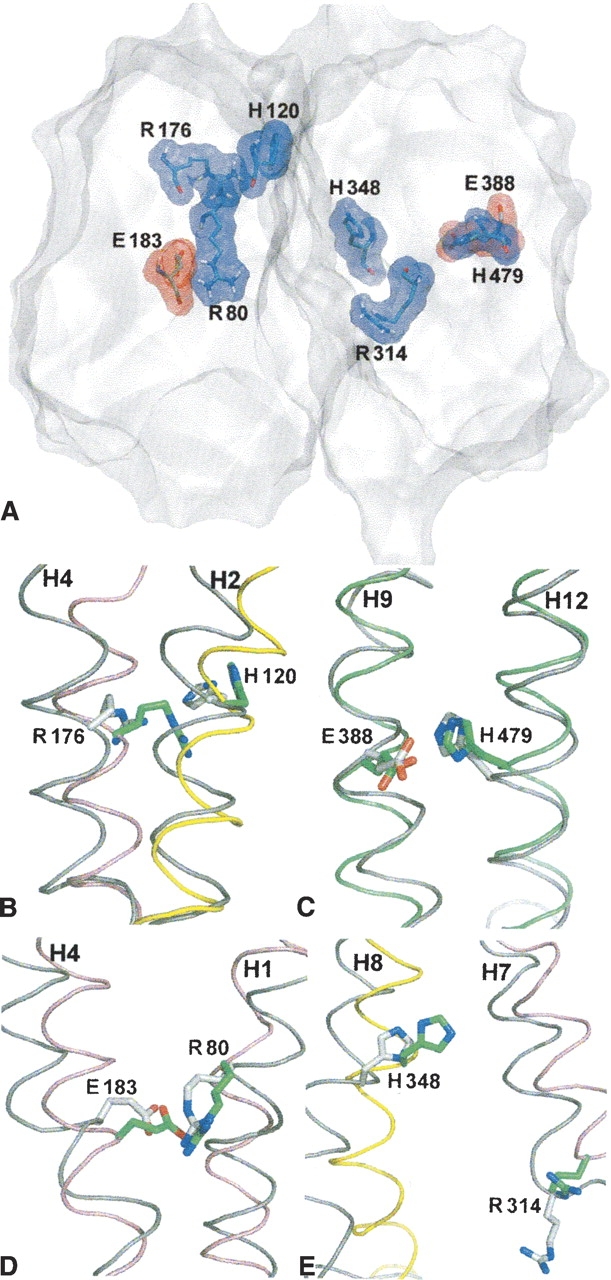
(A) Surface display of the VGLUT1 model cut in a plane parallel to the vesicle membrane, with all eight charged residues situated within the hydrophobic region of the membrane displayed. (B–E) The same residues viewed before (green) and after (gray) molecular dynamics simulation. Helices before MD simulation are colored as in ▶, helices after 10 nsec are colored in gray.
Two tentative substrate binding sites
To investigate substrate binding and translocation, we have carried out docking studies with the VGLUT substrates L-glutamate and inorganic phosphate (Pi). When L-glutamate is docked to the pore of our structural model, two structurally well-separated docking clusters are seen: one in level with R80 and R314, referred to as the central binding site, and the other near R176 and H120, the upper binding site (▶). For L-glutamate, the distribution of docked molecules between the two sites is ∼1–4, but the best dockings from the two different sites are similar in docked energy. On the other hand, when Pi is docked, a very different distribution of the docked conformations is observed. No molecules are found docked at the upper binding site; instead, practically all docked molecules are completely clustered around R314.
Docked inhibitor binding sites
A similar distribution of docked substrate molecules to that of L-glutamate is seen when the structurally related inhibitor 4-methyl-L-glutamate (Winter and Ueda 1993) and aspartate—which is not transported by the VGLUTs—are docked to the pore of our VGLUT1 model. We have also docked the azo dye molecules Trypan blue, Evans Blue, and Chicago Sky Blue (CSB) (Roseth et al. 1995, 1998), which, unlike L-glutamate, have very high affinity for the VGLUTs. When docked, the three molecules are also found at two different binding sites in the pore. The first site constitutes a volume from the cytoplasmic entrance of the pore to its center, where the two negatively charged sulphate groups in one end of the symmetrical dye molecules, Evans Blue (▶) and CSB (▶), are found docked at the central binding site, in level with R80 from the N-terminal domain of VGLUT1 and R314 from the C-terminal domain. Trypan blue also binds to the central binding site (▶), with an estimated Ki of 40 nM, but its lowest docked energy conformation is found at another site further into the pore. Around R176 the pore closes, and at this upper binding site Trypan blue is found docked (▶), with an estimated Ki of 0.35 nM. Two of its sulphate groups are found coordinated by R176 and R80 from the N-terminal domain of VGLUT1. Interestingly, neither Evans Blue nor CSB is found docked at this site. When all Trypan blue dockings to the upper binding site are visualized, a very tightly clustered mode of binding is seen for the upper half of the docked inhibitor. Since the volume in this part of the pore is quite small, there is little room for movement, and because of their shorter sulphate group spacing, Evans Blue and CSB cannot interact in a similar fashion without causing severe steric clashes to our model. While the upper half of the three symmetrical dye molecules interact firmly with the basic residues from the two sites, the half that is closer to the cytoplasmic side is found to be more flexible in all docking trials.
Figure 5.

Strong azo dye inhibitors of VGLUT1 docked to the pore of the model (oriented as in ▶). The lowest docked energy conformations are shown. Diameters of different intersections of the pore are labeled along the 27 Å deep cavity. Three arginine residues from VGLUT1 model are shown in green. (A) Evans Blue and (B) Chicago Sky Blue interacting with the central binding site. (C) Trypan blue in the central binding site and (D) Trypan blue in the upper binding site.
Finally, we have also docked an inhibitor structurally unrelated to L-glutamate, 4,4′-diisothiocyanatostilbene-2,2′-disulfonic acid (DIDS) (Hartinger and Jahn 1993). When docked, DIDS, like Evans Blue and CSB, only binds the central binding site. In contrast to the azo dyes, DIDS binds these residues in a horizontal fashion, so that the inhibitor molecule is extended parallel to the vesicular membrane. Its two sulphate groups interact very strongly with both R80 and R314, since the interatomic distances between the oxygens of the docked inhibitor (7.3 Å) allows firm hydrogen bonding to the amide protons of the two arginines (10.5 Å).
Discussion
The major difficulty in modeling the 3D structures of the VGLUT proteins from bacterial MFS structures is the very low sequence identity (10%–15%) between the distantly related proteins. To minimize the risk of matching structurally unrelated residues, we have used an alignment method that includes evolutionary information specific for the two proteins to be aligned (Wallner et al. 2004; Jaroszewski et al. 2005). Thus, our structural model for VGLUT1 is based on a profile–profile alignment to the GlpT template, which differs in several helices to the clustalX alignment used for the recent model of VGLUT2 (Juge et al. 2006).
When the topology and overall structure of our VGLUT1 model is compared to the template structure, the most noticeable differences are the longer hydrophilic loops between transmembrane helices 1–2 and 8–9 in the VGLUT1 model. Within the transmembrane domains, we observe local differences in the distribution of polar residues between the model and the template structure. When the region encompassing the pore between the N- and C-terminal domains is compared, interesting differences are found for residues that could be functionally conserved. In the template crystal structure, two arginines (R45 from helix 1 and R269 from helix 7) are believed to be important for the transport activity (Huang et al. 2003). In our VGLUT1 model, R80 is found at the same position as the first template structure arginine, and R314 is situated one helical turn below the second.
The MD simulations carried out in this work reinforce the assumption that the membrane-spanning region of human VGLUT1 proteins can be modeled from bacterial homologs. Although the membrane composition of synaptic vesicles differs considerably from the inner membrane of E. coli, both the VGLUT1 model and the bacterial template structure are stable in our DMPC simulation system. The overall structural stability of the VGLUT1 model is comparable to that of the template crystal structure, and the distribution of residues in the membrane- and membrane–water interface regions of the model are in agreement with that seen in integral membrane proteins (Landolt-Marticorena et al. 1993; von Heijne 1994). Because of its shorter, fully saturated carbon chains, a pure DMPC membrane bears more resemblance to the bacterial inner membrane, and the phospholipid contacts of the structural model could likely be further improved in a simulation system with a lipid composition more similar to the vesicular membrane (Benfenati et al. 1989).
Our VGLUT1 structural model represents a conformation of the transporter open to the cytoplasmic side of the neurotransmitter vesicle. The model allows docking and MD studies of L-glutamate binding prior to translocation into the neurotransmitter vesicle. For Pi uptake, the sidedness of transport is reversed; release occurs to the cytoplasmic side. The conformational cycle of MFS transporters is believed to feature at least two additional major structural states: a closed state with the substrate bound, and a state open to the periplasmic side. Since the transport cycle occurs on a millisecond timescale for secondary transporters, it is currently not practical to simulate transitions in the complete cycle using MD.
In addition to the overall structural flexibility, the most striking structural change after the MD simulation is perhaps the side-chain movement of R176 from the central pore to the interior of the N-terminal domain (▶). This causes an expansion of the N-terminal domain interface to the vesicle lumen, and could indicate that the upper binding site is accessible from that side. However, preliminary MD simulations indicate that when negatively charged VGLUT inhibitors are present in the central pore, R176 stays exposed to the central pore (data not shown).
The recent important findings of Juge et al. (2006) on VGLUT2 demonstrate that residues equivalent to R176, H120, and E183 in VGLUT1 (▶) are indeed essential for L-glutamate transport. On the other hand, mutations corresponding to R80A or R322A did not affect this uptake activity significantly. Viewed from our structural model, this strongly suggests that the L-glutamate transport process requires binding of L-glutamate to the upper binding site, where R176 and H120 are situated. Favorable interactions are found at the upper binding site in our docking studies (▶). Explaining the requirement for E183 is more intricate. In our model, E183 forms a salt bridge with R80 (▶) throughout the simulation, which could explain its intolerance to mutations. However, a mutation corresponding to R80A retained 74% uptake activity in VGLUT2. Clearly, more structural information is required to understand the role of these important residues.
Table 1.
Comparision of the predicted and measured affinities of VGLUT1 inhibitors

Interestingly, the Pi transport activity of VGLUT2 has been shown to be unaffected by mutations that affect the transport of L-glutamate (Juge et al. 2006), which implies that that the two processes utilize at least partly different sets of amino acid residues. Since the distribution of polar residues in the transmembrane helices of the VGLUT1 shows significant differences to the template structure, particularly for the peripheral helices 3, 6, 9, and 12, it is possible that the mechanism of Pi transport differs considerably to the template structure.
The L-glutamate transport activity of VGLUTs requires a proton electrochemical gradient generated by the vacuolar H+-ATPase (Forgac 2000; Juge et al. 2006). Because of the low pH in the synaptic vesicle, most histidine residues facing the vesicular side of our model are likely to be protonated in such an environment. If the central pore of the model indeed becomes open to the vesicle lumen during the transport cycle, several residues situated along the pore can be suggested as candidates for the protonation (▶). It is noteworthy that protonation of residues along the central pore have been extensively studied for LacY, where glutamate and histidine residues have been shown to bind protons and cause structural changes during substrate transport (Mirza et al. 2006; Yin et al. 2006b).
The molecular structures of the azo dyes contain charged groups that could mimic the structure of L-glutamate, and one hypothesis for their high affinities is that they partially span the transmembrane “tunnel” while binding both outer and inner domains (Thompson et al. 2005). The results from the docking trials of these inhibitors to our model of VGLUT1 suggest that the molecules operate by effectively inhibiting access to positively charged residues situated inside the pore of the vesicular glutamate transporters, and that the volume of the pore is large enough to host the full volume of the azo dye molecule. Interestingly, the docked conformation of CSB binds significantly weaker than Evans Blue (estimated Ki = 74 nM vs. 2.4 nM). From experimental data, Evans Blue inhibits VGLUT1 30 times more efficiently than CSB, although the only chemical difference between the two molecules is the change of a methyl group to a metoxy group on the biphenyl linker. However, structural modeling of the free dye molecules has previously shown that this change causes CSB to adopt a less planar conformation (Roseth et al. 1998), which is also seen when the docked structures of the different azo dye inhibitors are compared (▶). Besides having a less planar structure, the docked conformations of CSB molecules are markedly less clustered (data not shown), especially in the region of the dye molecule that is found docked below the central binding site arginines. It is possible that the weaker inhibition by CSB, compared to the other azo dyes, stems from its incapability of adopting a planar structure in the pore of the transporter.
Since inhibition by DIDS is prevented by high Cl− concentration, it has been proposed that the VGLUTs possess a DIDS-sensitive chloride-binding site on the cytoplasmic side, distinct from the substrate-binding site, which regulates transport activity (Hartinger and Jahn 1993). When docked to our structural model, DIDS binds to both arginines of the central binding site in a horizontal mode different from the other inhibitors. Although structurally distinct from the upper binding site, further experimental data is needed to verify the relevance of this mode of binding.
While we see a reasonable correlation to experimental Ki values, it is important to keep in mind that the estimated inhibition constants obtained from the docking studies are associated with large errors even for dockings to high-resolution crystal structures (Morris et al. 1998). Thus, the estimated Ki values obtained from docking simulations carried out in this study cannot alone be used to legitimize the modeling method. Nevertheless, the docking method itself is useful in the search for possible binding sites, given a reasonable homology model. An interesting example is the recent study on the glucose transporter GLUT1 (Cunningham et al. 2005), where docking to a structure model based on GlpT suggests several substrate binding sites situated near residues involved in disease-causing missense mutations.
Taken together, what can MD and docking simulations of a model built from a distant bacterial homolog tell us about glutamate transport by VGLUT1? Most importantly, the three-dimensional modeling reveals charged residues facing a pore in the transporter, and subsequent docking trials locate two sites from the residues constituting this pore. The relevance of these predicted sites for glutamate transport, as well as for its inhibition, can be tested experimentally. Furthermore, since 44 out of the 45 amino acid residues that face this pore are conserved among the three human VGLUT isoforms, the predictions presented here should also hold for VGLUT2 and VGLUT3. In the absence of a crystal structure, we hope that the three-dimensional model described here will be useful as a structural framework for further mechanistic studies of these important neurotransmitter transporters.
Coordinates of the model docked with inhibitors can be obtained from the corresponding author.
Material and Methods
Justification of the modeling methodology
So far no crystal structures have been obtained for any vesicular neurotransmitter transporter, but a three-dimensional model based on homology with bacterial MFS proteins has been proposed for the vesicular monoamine transporter, VMAT2, from the rat (Vardy et al. 2004). For the vesicular glutamate transporters, their bacterial homologs in the major facilitator superfamily include the glucarate porter, GudP (21%–30% sequence identity, depending on bacterial source), the hexuronate porter, ExuT (13%–18%), and noteworthy, the glycerol-3-phosphate/inorganic phosphate antiporter, GlpT, from E. coli (15%). The GlpT structure has been solved to 3.3 Å by X-ray crystallography (Huang et al. 2003). The other two high-resolution structures of MFS transporters available are for the lactose:H+ symporter lactose permease (Abramson et al. 2003a), LacY, and the multidrug resistance protein D (Yin et al. 2006a), EmrD, both from E. coli. Despite sharing only 10% sequence identity, these three high-resolution structures are very similar, and feature the same fold: 12 transmembrane α-helices separated into two pseudo-symmetrical domains of six helices each. Since this topology is repeatedly found for MFS members which share very low sequence identity, it has been proposed that all MFS members share this fold (Vardy et al. 2004). In addition, weak sequential homology exists between the two six-helix bundles in MFS proteins, which probably reflects a common evolutionary origin and the structural similarity observed in the crystal structures.
Choice of template for structural modeling
Among the three solved bacterial MFS structures, GlpT, LacY, and EmrD, the human VGLUTs are sequentially more similar to GlpT; the VGLUT1–GlpT alignment features up to 21 more identical residues and 32 less gaps. Although the average sequence identity between a predicted VGLUT helix and its corresponding region in GlpT is only 17%, this varies from 4% up to 27%. The lowest homology for the helical region to the crystal structure sequence is found in helices 3 and 9, and the highest in helices 4 and 7. In the MFS crystal structures, helices 3 and 9 are situated at the rim of the protein, and residues from these helices predominantly make contact to lipids. In contrast, helices 4 and 7 in GlpT harbor residues that face the substrate translocation pore.
Sequence alignment and topology prediction
The VGLUT1–GlpT sequence alignment was created using the fold and function assignment system (FFAS) protocol for profile–profile alignments (Jaroszewski et al. 2005). Transmembrane topology prediction was performed with TMHMM 2.0 (Melen et al. 2003), HMMTOP (Tusnady and Simon 2001), and DAS (Cserzo et al. 1997). All three programs suggested 12 transmembrane α-helices. The boundaries of the transmembrane segments predicted by TMHMM were used in our study (see ▶). When aligned to GlpT, the 12 hydrophobic segments in the amino acid sequences of the VGLUTs overlap with the 12 α-helices found in the X-ray structure of GlpT (▶). Loop regions that connect the helices are also of similar length, with exception for the longer loops between the predicted transmembrane segments 1–2 and 8–9. In the ACS subfamily, loop 1–2 is five to 30 residues longer in proteins from animals, and loop 8–9 contains eight inserted residues in about half of its currently identified members, including VGLUT1–3.
Homology modeling
Three-dimensional structural models for the predicted membrane-spanning region of VGLUT1 (residues 60–443) were constructed using MODELLER8v2 (Sali and Blundell 1993) based on a VGLUT1-GlpT alignment obtained from the FFAS03 Web server (Jaroszewski et al. 2005). From this initial alignment, 10 preliminary alignments were constructed, each corresponding to a different orientation of the predicted helix 9. The final alignment was chosen with aid of the helical boundaries obtained from transmembrane segment prediction, as well as from visual inspection of the corresponding preliminary models. From the final alignment, 100 structural models were generated using different random seeds, and the five models with the lowest MODELLER objective function were kept and further evaluated with ProQ (Wallner and Elofsson 2003). Both MaxSub (0.234) and LG (2.786) scores calculated by ProQ for the final model were similar to those observed for correct models of other proteins (Wallner and Elofsson 2003), although it should be kept in mind that ProQ is optimized for water-soluble proteins.
Docking of substrates and inhibitors
Coordinate files for substrate and inhibitors were built using the PRODRG server (Schuttelkopf and van Aalten 2004), and docked to the VGLUT1 structural model with AUTODOCK 3.0 (Morris et al. 1998) using standard grid spacing. For each ligand, 1000 searches with the Lamarckian Genetic Algorithm were carried out in the volume secluded by the pore of the model (54 × 60 × 126 grid points in x, y, and z, respectively). The resulting conformations with lowest docked energy were chosen as the final. Inhibition constant (Ki) values for each conformation were estimated by AUTODOCK.
Simulation system for MD
An all-atom simulated system was constructed which consisted of the human VGLUT1 model (totally 4348 atoms including hydrogen) dissolved in a dimyristoyl phosphatidylcholine (DMPC) bilayer. The DMPC bilayer structure was constructed by adding four replicas of 128 DMPC molecules from a recent work by Karttunen et al. (Gurtovenko et al. 2004) (http://www.apmaths.uwo.ca/∼mkarttu/). The VGLUT1 model was placed in the bilayer by manually removing lipid molecules. The number of lipids that remained was 460 (totaling 21,160 atoms). The protein and its hosting bilayer were placed in a simulation cell of 130 Å × 130 Å × 90 Å with the bilayer situated in the x–y plane. The remaining volume of the simulation cell was filled with water molecules, upon which water molecules found between the protein and the lipid tails were removed. The total number of water molecules included was 28,999. To electroneutralize the system, 10 randomly chosen water molecules were replaced with Cl− ions. The grand total number of atoms in the cell prior to the simulation was 112,485. The final box size fluctuated around 128.2 Å × 128.2 Å × 88.7 Å during the simulation. The same method was used to generate a simulation system for the template structure GlpT.
MD simulations
All MD simulations were carried out with the GROMACS package (version 3.3.1) using the gmxff force field for the protein (Berendsen et al. 1995; Lindahl et al. 2001). For the DMPC system the force field was complemented with the topology files dmpc.itp and lipid.itp from Tieleman's Web site (http://moose.bio.ucalgary.ca/). The GROMACS SPC potential model was used for water molecules. Before simulations were started, an initial energy minimization was carried out, followed by a short position-restrained MD simulation of 20 psec to relax the system. During equilibration, the volume was kept constant for 100 psec followed by NPT simulation. Ewald summation was employed using a Particle Mesh Ewald scheme (Essman et al. 1995). An integration time step of 2 fsec was used. The fast internal water degrees of freedom were constrained by using the SETTLE algorithm (Miyamoto and Kollman 1992) while the LINCS scheme was used to freeze the hydrogen atoms (Hess et al. 1997). An isotropic external pressure of 1 atmosphere was maintained during the entire simulation. The simulations were performed at 300 K.
Visualization of the model
Cavity calculation for display purposes was performed with program Caver (Petrek et al. 2006) and PyMOL (DeLano 2002). Structure analysis and molecular surface rendering were carried out with VMD (Humphrey et al. 1996).
Acknowledgments
A.L. and S.H. express their gratitude to the Swedish Science Council (VR) for financial support and the Swedish National Infrastructure for Computing (SNIC) for allocation of computing time. D.N.W. thanks NIDDK for financial support (DK-53973).
Footnotes
Reprint requests to: Jonas Almqvist, Structural Chemistry, Arrhenius Laboratory, Stockholm University, S-10691 Stockholm, Sweden; e-mail: jonasa@struc.su.se; fax: 46-8-163118.
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.072944707.
References
- Abramson J., Smirnova, I., Kasho, V., Verner, G., Kaback, H.R., and Iwata, S. 2003a. Structure and mechanism of the lactose permease of Escherichia coli . Science 301 610–615. [DOI] [PubMed] [Google Scholar]
- Abramson J., Smirnova, I., Kasho, V., Verner, G., and Iwata, S. 2003b. The lactose permease of Escherichia coli: Overall structure, the sugar-binding site, and the alternating access model for transport. FEBS Lett. 555 96–101. [DOI] [PubMed] [Google Scholar]
- Aihara Y., Mashima, H., Onda, H., Hisano, S., Kasuya, H., Hori, T., Yamada, S., Tomura, H., Yamada, Y., Inoue, I., et al. 2000. Molecular cloning of a novel brain-type Na-dependent inorganic phosphate cotransporter. J. Neurochem. 74 2622–2625. [DOI] [PubMed] [Google Scholar]
- Almqvist J., Huang, Y., Hovmoller, S., and Wang, D.N. 2004. Homology modeling of the human microsomal glucose 6-phosphate transporter explains the mutations that cause the glycogen storage disease type Ib. Biochemistry 43 9289–9297. [DOI] [PubMed] [Google Scholar]
- Bellocchio E.E., Reimer, R.J., Fremeau Jr, R.T., and Edwards, R.H. 2000. Uptake of glutamate into synaptic vesicles by an inorganic phosphate transporter. Science 289 957–960. [DOI] [PubMed] [Google Scholar]
- Benfenati F., Greengard, P., Brunner, J., and Bahler, M. 1989. Electrostatic and hydrophobic interactions of synapsin I and synapsin I fragments with phospholipid bilayers. J. Cell Biol. 108 1851–1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berendsen H.J.C., van der Spoel, D., and van Drunen, R. 1995. GROMACS: A message-passing parallel molecular dynamics implementation. Comp. Phys. Commun. 91 43–56. [Google Scholar]
- Cserzo M., Wallin, E., Simon, I., von Heijne, G., and Elofsson, A. 1997. Prediction of transmembrane alpha-helices in prokaryotic membrane proteins: The dense alignment surface method. Protein Eng. 10 673–676. [DOI] [PubMed] [Google Scholar]
- Cunningham P., Afzal-Ahmed, I., and Naftalin, R.J. 2005. Docking studies show that D-glucose and quercetin slide through the transporter GLUT1. J. Biol. Chem. 281 5797–5803. [DOI] [PubMed] [Google Scholar]
- De Gois S., Jeanclos, E., Morris, M., Grewal, S., Varoqui, H., and Erickson, J.D. 2006. Identification of endophilins 1 and 3 as selective binding partners for VGLUT1 and their co-localization in neocortical glutamatergic synapses: Implications for vesicular glutamate transporter trafficking and excitatory vesicle formation. Cell. Mol. Neurobiol. 26 679–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLano W.L. 2002. The PyMOL molecular graphics system, DeLano Scientific, Palo Alto, CA, http://www.pymol.org.
- Essman U., Perela, L., Berkowitz, M.L., Darden, T., Lee, H., and Pedersen, L.G. 1995. A smooth particle mesh Ewald method. J. Chem. Phys. 103 8577–8592. [Google Scholar]
- Forgac M. 2000. Structure, mechanism and regulation of the clathrin-coated vesicle and yeast vacuolar H(+)-ATPases. J. Exp. Biol. 203 71–80. [DOI] [PubMed] [Google Scholar]
- Fremeau R.T., Burman, J., Qureshi, T., Tran, C.H., Proctor, J., Johnson, J., Zhang, H., Sulzer, D., Copenhagen, D.R., Storm-Mathisen, J., et al. 2002. The identification of vesicular glutamate transporter 3 suggests novel modes of signaling by glutamate. Proc. Natl. Acad. Sci. 99 14488–14493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fremeau R.T., Voglmaier, S., Seal, R.P., and Edwards, R.H. 2004. VGLUTs define subsets of excitatory neurons and suggest novel roles for glutamate. Trends Neurosci. 27 98–103. [DOI] [PubMed] [Google Scholar]
- Gras C., Herzog, E., Bellenchi, G.C., Bernard, V., Ravassard, P., Pohl, M., Gasnier, B., Giros, B., and El Mestikawy, S. 2002. A third vesicular glutamate transporter expressed by cholinergic and serotoninergic neurons. J. Neurosci. 22 5442–5451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurtovenko A.A., Patra, M., Karttunen, M., and Vattulainen, I. 2004. Cationic DMPC/DMTAP lipid bilayers: Molecular dynamics study. Biophys. J. 86 3461–3472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartinger J. and Jahn, R. 1993. An anion binding site that regulates the glutamate transporter of synaptic vesicles. J. Biol. Chem. 268 23122–23127. [PubMed] [Google Scholar]
- Hess B., Bekker, H., Berendsen, H.J.C., and Fraaije, J.G.E.M. 1997. LINCS: A linear contraints solver for molecular simulations. J. Comput. Chem. 18 1463–1472. [Google Scholar]
- Hirai T., Heymann, J.A., Shi, D., Sarker, R., Maloney, P.C., and Subramaniam, S. 2002. Three-dimensional structure of a bacterial oxalate transporter. Nat. Struct. Biol. 9 597–600. [DOI] [PubMed] [Google Scholar]
- Huang Y., Lemieux, M.J., Song, J., Auer, M., and Wang, D.N. 2003. Structure and mechanism of the glycerol-3-phosphate transporter from Escherichia coli . Science 301 616–620. [DOI] [PubMed] [Google Scholar]
- Humphrey W., Dalke, A., and Schulten, K. 1996. VMD—Visual molecular dynamics. J. Mol. Graph. 14 33–38. [DOI] [PubMed] [Google Scholar]
- Jaroszewski L., Rychlewski, L., Li, Z., Li, W., and Godzik, A. 2005. FFAS03: A server for profile–profile sequence alignments. Nucleic Acids Res. 33 W284–W288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juge N., Yoshida, Y., Yatsushiro, S., Omote, H., and Moriyama, Y. 2006. Vesicular glutamate transporter contains two independent transport machineries. J. Biol. Chem. 281 39499–39506. [DOI] [PubMed] [Google Scholar]
- Jung S.K., Morimoto, R., Otsuka, M., and Omote, H. 2006. Transmembrane topology of vesicular glutamate transporter 2. Biol. Pharm. Bull. 29 547–549. [DOI] [PubMed] [Google Scholar]
- Landolt-Marticorena C., Williams, K.A., Deber, C.M., and Reitmeier, R.A.F. 1993. Non-random distribution of amino acids in the transmembrane segments of human type I single-span membrane proteins. J. Mol. Biol. 229 602–608. [DOI] [PubMed] [Google Scholar]
- Lemieux M.J., Huang, Y., and Wang, D.N. 2004. The structural basis of substrate translocation by the Escherichia coli glycerol-3-phosphate transporter: A member of the major facilitator superfamily. Curr. Opin. Struct. Biol. 14 405–412. [DOI] [PubMed] [Google Scholar]
- Lindahl E., Hess, B., and van der Spoel, D. 2001. GROMACS 3.0: A package for molecular simulation and trajectory analysis. J. Mol. Model. 7 306–317. [Google Scholar]
- Lise S. and Jones, D.T. 2005. Sequence patterns associated with disordered regions in proteins. Proteins 58 144–150. [DOI] [PubMed] [Google Scholar]
- Melen K., Krogh, A., and von Heijne, G. 2003. Reliability measures for membrane protein topology prediction algorithms. J. Mol. Biol. 327 735–744. [DOI] [PubMed] [Google Scholar]
- Mirza O., Guan, L., Verner, G., Iwata, S., and Kaback, H.R. 2006. Structural evidence for induced fit and a mechanism for sugar/H+ symport in LacY. EMBO J. 25 1177–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto S. and Kollman, P.A. 1992. SETTLE: An analytical version of the SHAKE and RATTLE algorithms for rigid water models. J. Comput. Chem. 13 952–962. [Google Scholar]
- Morris G.M., Goodsell, D.S., Halliday, R.S., Huey, R., Hart, W.E., Belew, R.K., and Olson, A.J. 1998. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 19 1639–1662. [Google Scholar]
- Ni B., Rosteck Jr, P.R., Nadi, N.S., and Paul, S.M. 1994. Cloning and expression of a cDNA encoding a brain-specific Na-dependent inorganic phosphate cotransporter. Proc. Natl. Acad. Sci. 91 5607–5611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otis T.S. 2001. Vesicular glutamate transporters in cognito. Neuron 29 11–14. [DOI] [PubMed] [Google Scholar]
- Pao S.S., Paulsen, I.T., and Saier Jr, M.H. 1998. Major facilitator superfamily. Microbiol. Mol. Biol. Rev. 62 1–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrek M., Otyepka, M., Banás, P., Kosinova, P., Koca, J., and Damborsky, J. 2006. CAVER: A new tool to explore routes from protein clefts, pockets and cavities. BMC Bioinformatics 7 316. doi: 10.1186/1471-2105-7-316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roseth S., Fykse, E.M., and Fonnum, F. 1995. Uptake of L-glutamate into rat brain synaptic vesicles: Effect of inhibitors that bind specifically to the glutamate transporter. J. Neurochem. 65 96–103. [DOI] [PubMed] [Google Scholar]
- Roseth S., Fykse, E.M., and Fonnum, F. 1998. Uptake of L-glutamate into synaptic vesicles: Competitive inhibition by dyes with biphenyl and amino- and sulphonic acid-substituted naphthyl groups. Biochem. Pharmacol. 56 1243–1249. [DOI] [PubMed] [Google Scholar]
- Sali A. and Blundell, T.L. 1993. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234 779–815. [DOI] [PubMed] [Google Scholar]
- Schuttelkopf A.W. and van Aalten, D.M. 2004. PRODRG: A tool for high-throughput crystallography of protein–ligand complexes. Acta Crystallogr. D Biol. Crystallogr. 60 1355–1363. [DOI] [PubMed] [Google Scholar]
- Shigeri Y., Seal, R.P., and Shimamoto, K. 2004. Molecular pharmacology of glutamate transporters, EAATs and VGLUTs. Brain Res. Brain Res. Rev. 45 250–265. [DOI] [PubMed] [Google Scholar]
- Takamori S. 2006. VGLUTs: “Exciting” times for glutamatergic research? Neurosci. Res. 55 343–351. [DOI] [PubMed] [Google Scholar]
- Takamori S., Rhee, J.S., Rosenmund, C., and Jahn, R. 2000. Identification of a vesicular glutamate transporter that defines a glutamatergic phenotype in neurons. Nature 407 189–194. [DOI] [PubMed] [Google Scholar]
- Thompson C.M., Davis, E., Carrigan, C.N., Cox, H.D., Bridges, R.J., and Gerdes, J.M. 2005. Inhibitor of the glutamate vesicular transporter (VGLUT). Curr. Med. Chem. 12 2041–2056. [DOI] [PubMed] [Google Scholar]
- Tusnady G.E. and Simon, I. 2001. The HMMTOP transmembrane topology prediction server. Bioinformatics 17 849–850. [DOI] [PubMed] [Google Scholar]
- Vardy E., Arkin, I.T., Gottschalk, K.E., Kaback, H.R., and Schuldiner, S. 2004. Structural conservation in the major facilitator superfamily as revealed by comparative modeling. Protein Sci. 13 1832–1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinatier J., Herzog, E., Plamont, M.A., Wojcik, S.M., Schmidt, A., Brose, N., Daviet, L., El Mestikawy, S., and Giros, B. 2006. Interaction between the vesicular glutamate transporter type 1 and endophilin A1, a protein essential for endocytosis. J. Neurochem. 97 1111–1125. [DOI] [PubMed] [Google Scholar]
- von Heijne G. 1989. Control of topology and mode of assembly of a polytopic membrane protein by positively charged residues. Nature 341 456–458. [DOI] [PubMed] [Google Scholar]
- von Heijne G. 1994. Membrane proteins: From sequence to structure. Annu. Rev. Biophys. Biomol. Struct. 23 167–192. [DOI] [PubMed] [Google Scholar]
- Wallner B. and Elofsson, A. 2003. Can correct protein models be identified? Protein Sci. 12 1073–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallner B., Fang, H., Ohlson, T., Frey-Skott, J., and Elofsson, A. 2004. Using evolutionary information for the query and target improves fold recognition. Proteins 54 342–350. [DOI] [PubMed] [Google Scholar]
- Winter H.C. and Ueda, T. 1993. Glutamate uptake system in the presynaptic vesicle: Glutamic acid analogs as inhibitors and alternate substrates. Neurochem. Res. 18 79–85. [DOI] [PubMed] [Google Scholar]
- Yin Y., He, X., Szewczyk, P., Nguyen, T., and Chang, G. 2006a. Structure of the multidrug transporter EmrD from Escherichia coli . Science 312 741–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Y., Jensen, M.O., Tajkhorshid, E., and Schulten, K. 2006b. Sugar binding and protein conformational changes in lactose permease. Biophys. J. 91 3972–3985. [DOI] [PMC free article] [PubMed] [Google Scholar]