

Abstract
The potassium channel accessory subunit KChIP2 associates with Kv4.2 channels in the cardiac myocyte and is involved in the regulation of the transient outward current (Ito) during the early phase of repolarization of the action potential. As a first step to biophysically probe the mechanism of KChIP2, we have chemically synthesized its minimal isoform, KChIP2d, using Boc chemistry solid phase peptide synthesis in conjunction with native chemical ligation. The synthetic KChIP2d protein is primarily alpha-helical as predicted and becomes more structured upon binding calcium as assessed by 1H-NMR and CD spectroscopy. Synthetic KChIP2d is in a monomer-dimer equilibrium in solution, and there is evidence for two monomer binding sites on an N-terminal peptide of Kv4.2. Planned future studies include the incorporation of fluorescent and spin labeled probes in KChIP2d to yield structural information in parallel with electrophysiologic studies to elucidate KChIP2d's mechanism of action.
Keywords: potassium channel, accessory subunit, total chemical synthesis, kinetically controlled ligation, Boc chemistry solid phase peptide synthesis
Potassium channels are critical in stabilizing the membrane potential of the cell. In electrically excitable cells, such as those in the heart, mutations in potassium channels or their auxiliary subunits are associated with arrhythmias and other conduction defects (Delisle et al. 2004). The expression and biophysical properties of potassium channels are controlled by different families of regulatory subunits, one class of which is the K channel interacting proteins (KChIPs) (An et al. 2000). KChIPs are soluble, cytoplasmic proteins that contain EF hand calcium binding motifs and that bind to the N termini of Kv4 channels to regulate them through an as-yet-unidentified mechanism (Burgoyne et al. 2004). KChIP2 is the predominant member of this family in the cardiac myocyte, where it associates with Kv4.2 and 4.3 channels and is responsible for regulation of the transient outward current (Ito) in the early phase of repolarization during the cardiac action potential (Pourrier et al. 2003). A mouse knockout of KChIP2 lacks Ito and has an increased action potential duration and an increased susceptibility to ventricular tachyarrythmias (Kuo et al. 2001), suggesting an important functional role for KChIP2 in the regulation of the myocyte membrane potential.
The KChIP2 gene has 10 exons, and eight known splice variants have been identified (Decher et al. 2004), the largest of which is KChIP2a, containing four EF hands, and the smallest of which is KChIP2d, a minimal isoform that corresponds to the C-terminal seventy residues of KChIP2a and contains only one EF hand (▶; Patel et al. 2002b). Early studies of KChIP2 showed that, in addition to increasing Kv4 current density by increasing its surface expression, KChIP2a modulates Kv4's biophysical parameters by shifting its inactivation voltage to more positive potentials, slowing Kv4 inactivation and accelerating its recovery from inactivation (An et al. 2000). KChIP2d, while less than a quarter of the size of KChIP2a, maintains most of the function of the larger isoform with some quantitative differences (Patel et al. 2002b). Using KChIP2d as a model system for structure-function studies, Patel et al. (2002b) have shown that the EF hand affects Kv4.3 inactivation but not recovery and have identified a stretch of residues responsible for Ca2+-independent effects on recovery. A more detailed electrophysiologic study (Patel et al. 2004) resulted in a gating model for KChIP2d's regulation of Kv4.3, but it is still unclear how the different open, closed, and inactivated states correspond to structural states in the KChIP2:Kv4 complex.
Figure 1.

KChIP2 protein isoforms. Variants of exons 1 and 10 are noted by filled shapes. There is considerable variability in the splicing of the first three exons, while exon 7 in KChIP2f is absent (removing EF-3). Modified from Decher et al. (2004).
We plan to probe the structural mechanism of Kv4.2 channel regulation by the KChIP2 accessory protein, using the tools of synthetic chemical biology applied to KChIP2d (Patel et al. 2002a,b). Many experiments have incorporated biophysical probes into proteins, through either semisynthesis (Hahn and Muir 2005) or incorporation of cysteine residues and subsequent reaction of the side-chain –SH moiety with a fluorescent or EPR-active probe (Hubbell et al. 2000). An alternate approach is the total chemical synthesis of the target protein, which allows simultaneous site-specific incorporation of a number of different probes in the same protein molecule in a versatile, controlled fashion (Dawson and Kent 2000). The first step in such a series of experiments is to demonstrate the feasibility of the total chemical synthesis of the target protein, KChIP2d. This requires us to be able to synthesize peptide segments comprising the amino acid sequence of the protein, ligate them to one another to obtain the full-length polypeptide in good yield and high purity, and to then fold this synthetic polypeptide to form the functional protein molecule. In this article, we demonstrate the feasibility of total chemical synthesis of KChIP2d, confirm that it folds into a functional protein with binding activity similar to that of the wild-type molecule, and probe its biophysical properties. This total synthetic access will allow future studies of the KChIP2d:Kv4.2 complex with site-specific labels designed to probe the structural basis of its biological function.
Results
Design of the synthesis
The primary structure of KChIP2d is shown in ▶: It contains only one cysteine, at position 53. In order to make the protein by native chemical ligation of two peptide segments at this cysteine residue, the synthesis of an impractically long 52-residue peptide would be required (Dawson et al. 1994). Rather than attempting to make such a large peptide segment, we decided to assemble the KChIP2d polypeptide chain from three segments, replacing Gln34 with a Cys, which was subsequently converted to a “pseudoglutamine” (ψGln) by alkylation with iodoacetamide (Kochendoerfer et al. 2003). This substitution is unlikely to result in any significant change in KChIP2d's structure or activity, as Gln34 is predicted to be surface-exposed and not in the KChIP2d:Kv4.2 binding interface (Zhou et al. 2004). The three peptide segments used to make KChIP2d are shown in ▶: They contain 33 amino acid residues, 19 residues, and 18 residues and are thus readily made by solid phase peptide synthesis. In order to join these peptide segments by native chemical ligation, the first two peptides must have C-terminal thioesters (1–33:COSR, 34–52:COSR), and the latter two N-terminal cysteines (Dawson et al. 1994). The strategy adopted for the synthesis of KChIP2d is shown in ▶. Selective alkylation of Cys34 required assembly of the protein from N terminus to C terminus, rather than the conventional C-terminal to N-terminal strategy used in most three segment syntheses (Bang et al. 2006b). This strategy necessitated a kinetically controlled ligation (Bang et al. 2006b) between the first two thioester-containing peptide segments to give a unique peptide-thioester product (Bang et al. 2006b). This reaction was accomplished by first exchanging the alkyl thioester of 1–33:COSR with mercaptophenylacetic acid to give a more reactive peptide:COSΦCH2COOH aryl thioester synthon (Johnson and Kent 2006a), thus favoring formation of the desired 1–52:COSR product (▶). Subsequent alkylation with iodoacetamide resulted in specific conversion of the sole Cys in the peptide, residue 34, to ψGln. After purification of the modified 1–52:COSR peptide, native chemical ligation reaction with the Cys53–70 peptide under standard conditions gave the desired KChIP2d synthetic product.
Figure 2.

KChIP2d synthetic strategy. (A) Primary structure of KChIP2d; ligation sites are bolded. Three peptides must be synthesized: 1–33:COSR, Cys34–52:COSR, and Cys53–70. The first two of these have C-terminal TAMPAG alkyl thioester groups; the latter two have N-terminal Cys residues. (B) Synthetic strategy. The peptide alkyl thioester 1–33:COSR is exchanged to 1–33:COSΦCH2COOH by treatment with mercaptophenylacetic acid, and purified. This is followed by the kinetically controlled ligation of 1–33:COSΦCH2COOH to Cys34–52:COSR, to give 1–52:COSR. Cys34 is then alkylated to from ψGln34, and the resulting peptide is ligated with the last peptide segments to form synthetic KChIP2d.
Synthetic data
The results obtained using this synthetic scheme are shown in ▶. Solid phase synthesis of the peptides was straightforward, with the major peaks of the HPLC chromatograms of the crude synthesis products corresponding to the desired peptides (1–33:COSR, 34–52:COSR, and 53–70) (Supplemental Fig. S1). After exchange of the 1–33:COSR peptide with mercaptophenylacetic acid to give 1–33:COSΦCH2COOH, a kinetically controlled reaction with purified Cys34–52:COSR was performed and was essentially complete in 30 min (▶, lower trace). Subsequent alkylation of Cys34 to ψGln by iodoacetamide was complete after 30 min; the observed gain in mass of 57 Da was consistent with the addition of the –CH2CONH2 moiety (▶, upper trace). The modified 1–52:COSR peptide was then purified and reacted with the Cys53–70 peptide under standard native chemical ligation conditions. The results of this ligation are shown in ▶: The reaction is complete in less than 24 h, and the major peak in the HPLC corresponded to the desired full-length product. KChIP2d was purified from the crude ligation mixture by preparative reverse-phase HPLC, resulting in a single peak whose mass corresponds to the expected mass of the KChIP2d(1–70) synthetic product (which we refer to as KChIP2d from hereon) (▶).
Figure 3.
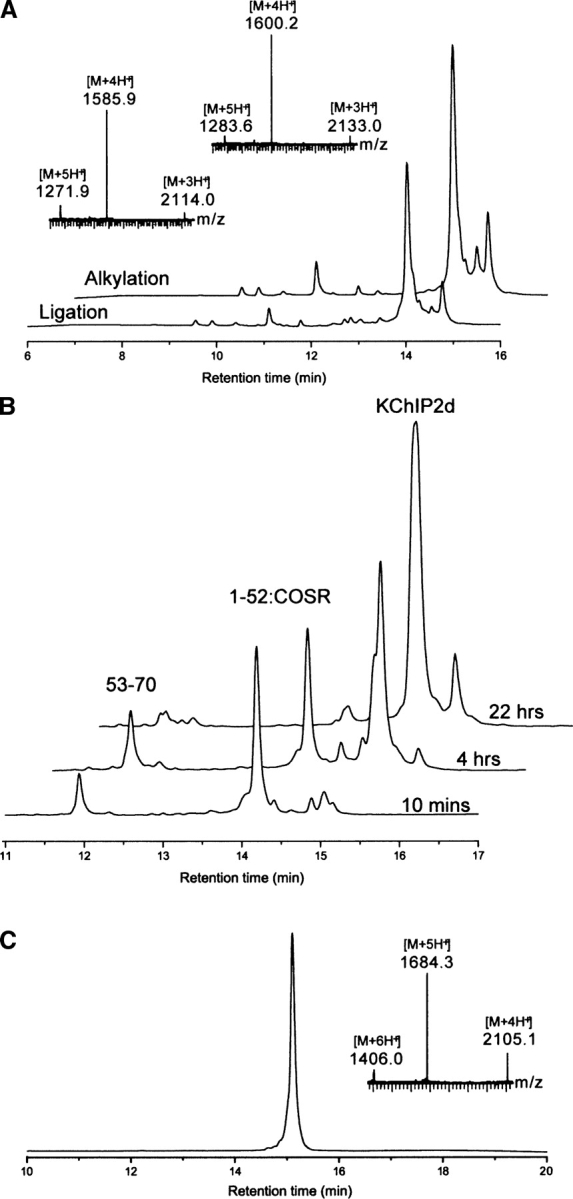
Synthetic data. (A) Kinetically controlled ligation between 1–33:COSΦCH2COOH and Cys34–52:COSR resulted in one major product; subsequent alkylation with ICH2CONH2 resulted in the expected mass shift of 72 Da (inset). (B) The final native chemical ligation reaction between 1–52:COSR and Cys53–70 was complete at 22 h, with the major product the full-length synthetic KChIP2d. (C) Purified synthetic KChIP2d after preparative reverse-phase HPLC. Observed mass: 8421 ± 2 Da; Calculated mass (average isotopes): 8418.6 Da.
Folding and characterization
Because KChIP2d contains no disulfide bonds, it was expected that the synthetic polypeptide would fold spontaneously under native conditions, and indeed, it adopts a predominantly helical conformation in solution as assessed by circular dichroism (▶, open boxes). Addition of 1 mM CaCl2 (▶, filled boxes) resulted in a 33% increase in helicity, likely associated with calcium binding to the EF hand motif. This observation is consistent with changes in CD absorption of recombinant KChIP2a, whose helicity also increases in the presence of calcium (Lin et al. 2004). In order to further probe these structural changes associated with calcium binding, we performed 1H-NMR spectroscopy. Unfortunately, two-dimensional NMR experiments were unsuccessful due to limited protein solubility and the inability to work with phosphate buffer solutions in the presence of calcium. However, one-dimensional 1H-NMR spectra of KChIP2d in the absence (▶) and presence (▶) of 1 mM CaCl2 demonstrated an increased dispersion of the spectrum in the presence of calcium, with peaks beyond 8 ppm visible in the sample with calcium, consistent with the adoption of a conformation with increased tertiary structure. From these experiments, we conclude that there is an increase in KChIP2d's secondary and tertiary structure associated with calcium binding to its EF hand motif.
Figure 4.
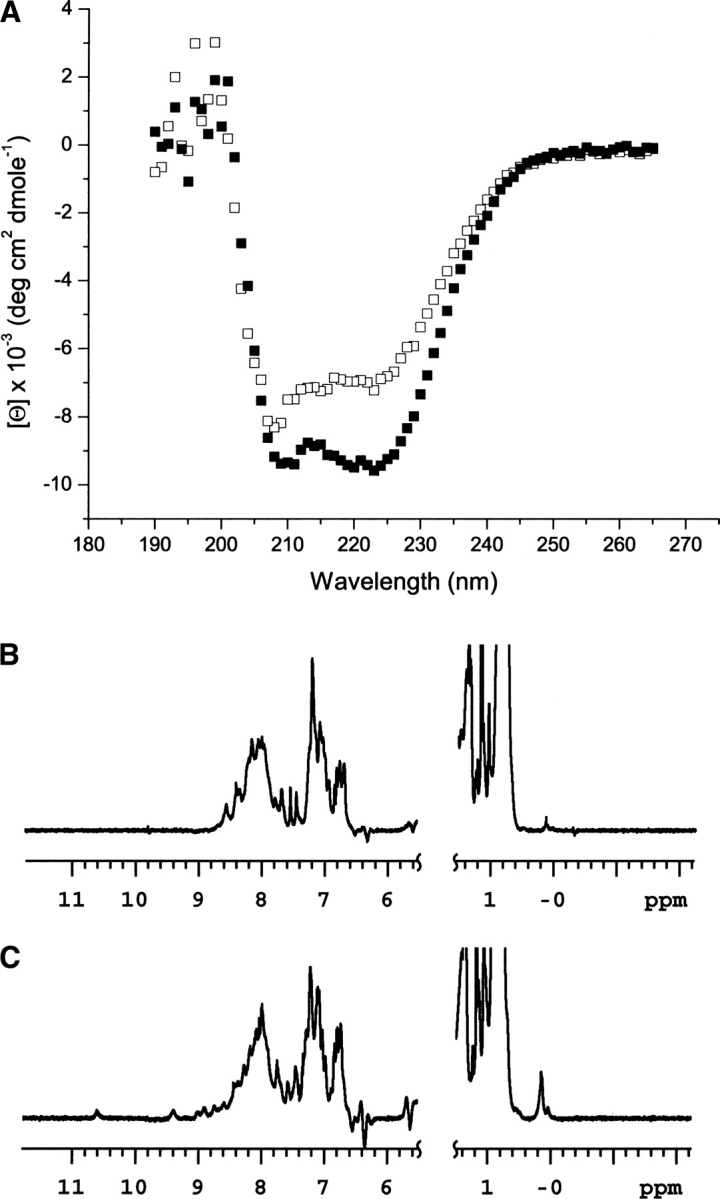
Increased secondary and tertiary structure in KChIP2d are observed upon calcium binding. (A) CD spectra of KChIP2d in the absence (open boxes) and presence (closed boxes) of 1 mM CaCl2. One-dimensional 1H-NMR spectra of KChIP2d in the absence (B) and presence (C) of 1 mM CaCl2. Note the peaks between 9 and 11 ppm that are clearly visible, as well as increased dispersion in the peaks between 6 and 9 ppm in C compared with B.
Effects of calcium binding on quaternary structure
Because calcium binding had a significant effect on the KChIP2d secondary and tertiary structure, we used sedimentation velocity analysis to probe what effect it had on quaternary structure. In both the absence and presence of calcium, KChIP2d's calculated sedimentation coefficient distribution is bimodal, consistent with a monomer-dimer equilibrium, with the dimer form clearly favored in the presence of calcium (▶). A calculated mass distribution by modeling the diffusion and sedimentation processes (Schuck 2003) has a similar shape (▶), with peaks at ∼8.6 and 17.7 kDa, consistent with a monomer-dimer equilibrium. A fit of the raw data with a monomer-dimer equilibrium model (▶) yields a good result using the parameters KD[No Ca2+] = 12 mM and KD[Ca2+] = 2 mM, with small, randomly distributed residuals (Supplemental Fig. S2), consistent with calcium favoring the dimerized form. It is likely that dimerization is also favored in the presence of Kv4.2, which would likely bind a KChIP2d dimer to interact in a manner similar to that observed in the KChIP1-Kv4 N terminus structure (Zhou et al. 2004). While the calculated sedimentation coefficients for the monomer and dimer forms are similar in the absence (1.05 S, 1.88 S) and presence (1.02 S, 1.94 S) of calcium, there is a more pronounced difference in their frictional ratios (f/f0[No Ca2+] = 1.41; f/f0[Ca2+] = 1.29). This is consistent with the adoption of a more compact structure of KChIP2d monomers and dimers in the presence of calcium, as suggested by the CD and NMR findings.
Figure 5.
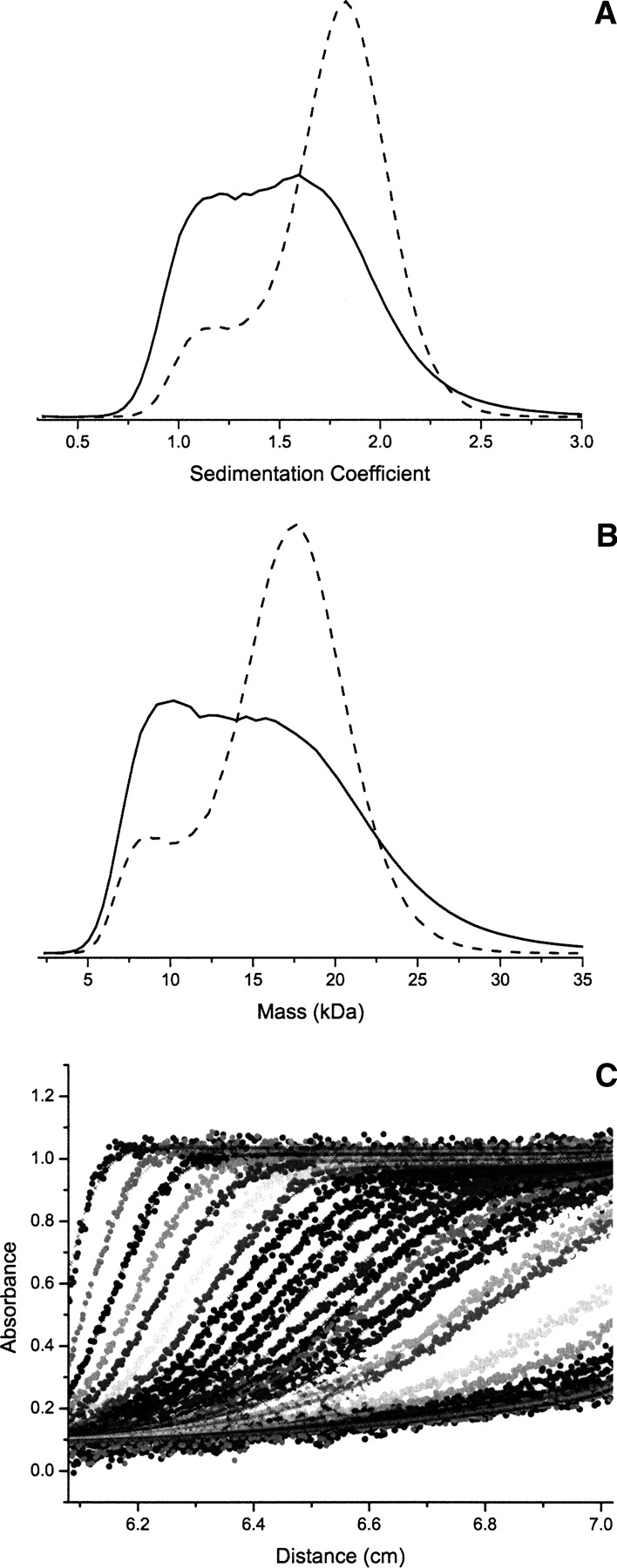
Oligomeric state of KChIP2d. (A) Bimodal sedimentation coefficient distribution consistent with a monomer (dashed line)-dimer (solid line) equilibrium in the absence (solid) and presence (dash) of 1 mM CaCl2. Calcium favors the dimer relative to the monomer. (B) The corresponding mass distribution is consistent with the predicted monomer (dashed line)-dimer (solid line) masses of KChIP2d of 8.6 and 17.7 kDa. (C) The experimental data are fit well with a monomer-dimer equilibrium model (see Materials and Methods).
Ligand binding studies
To assess the functional properties of synthetic KChIP2d, we measured its ability to bind to its peptide ligand, the N terminus of the Kv4.2 channel (Kv4.2NT). Electrophysiologic studies have identified residues 11–23 in the N terminus of Kv4.2 channel as being critical for this association, although other regions of the ion channel are also involved (Callsen et al. 2005). The peptide Kv4.2NT was synthesized and immobilized to a surface plasmon resonance (SPR) chip. KChIP2d associated in a reversible fashion with this surface bound peptide (▶), although the SPR response curve exhibits pronounced drift (see Materials and Methods). Binding curves generated from this data (▶) are clearly bimodal, with a pronounced second phase at high analyte concentration. The data were fit well using a two-site binding model with dissociation constants of 2.4 μM and 110 μM in the absence of calcium and 720 nM and 30 μM in the presence of calcium. Fits with one-site binding models were of poor quality (see Supplemental materials). These dissociation constants are only approximate because the stronger dissociation constants are near the lower limits of our data and the weaker dissociation constants are near or above the higher limits of our data.
Figure 6.
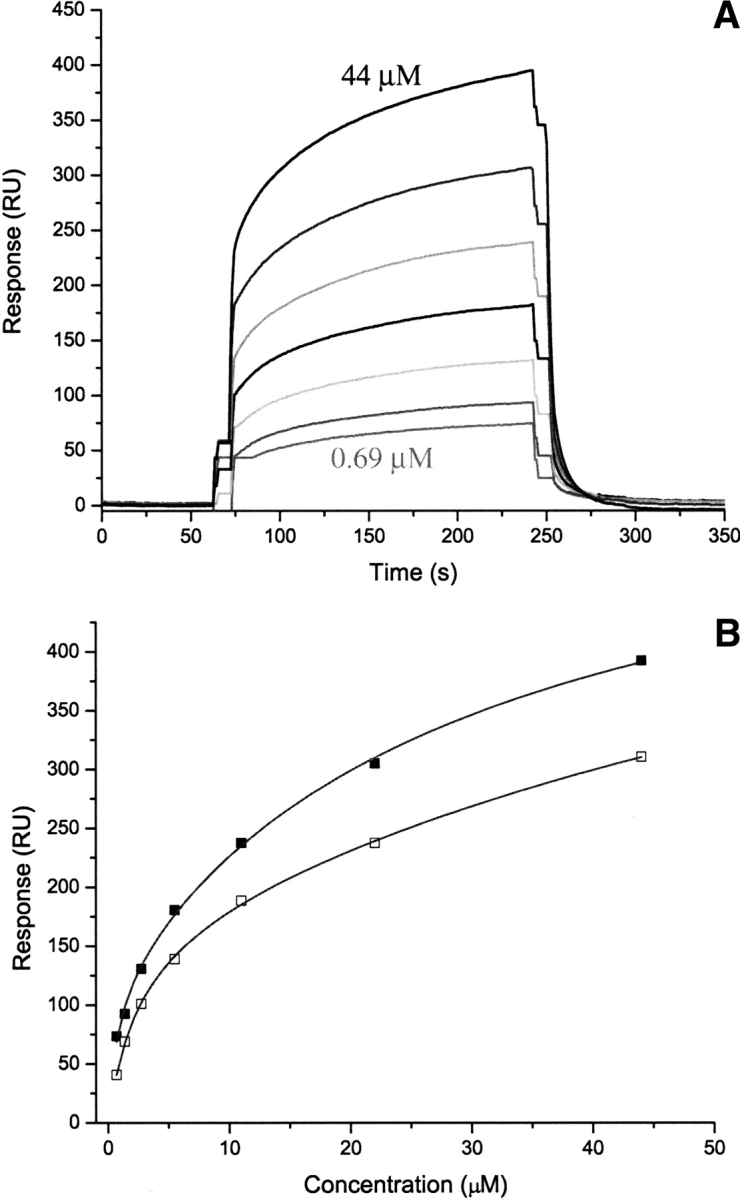
KChIP2d binding to Kv4.2 N-terminal peptide. (A) Surface plasmon resonance response curve of binding of KChIP2d (0.6875–44 μM) to immobilized Kv4.2NT in 1 mM CaCl2. (B) Binding curves in the absence (open boxes) and presence (closed boxes) of 1 mM CaCl2. Shown are fits to two-site models for interactions (see text).
These data suggest that calcium has only a minor effect on the nature of the KChIP2d:Kv4.2NT complex, as the addition of calcium has an approximately threefold effect on both binding constants, corresponding to ∼0.6 kcal/mol. As both binding constants are less than KChIP2d's dissociation constant, we are most likely observing binding of KChIP2d monomers to Kv4.2NT, and as the Kv4.2NT peptide can only accommodate binding of two KChIP2d monomers (Zhou et al. 2004), it is tempting to hypothesize that we are observing the binding of two KChIP2d monomers to two different sites on Kv4.2NT. However, such an interpretation does not explain the large changes in plasmon resonance units (RU) at longer times and at higher concentrations, which is possibly secondary to either drift in the instrument or oligomerization of KChIP2d. Thus we can safely say that KChIP2d is binding to Kv4.2NT, and this binding, by an unknown mechanism, is favored by calcium.
Discussion
Protein semi-synthesis by expressed protein ligation (Hahn and Muir 2005) or total synthesis by modern chemical ligation methods (Dawson and Kent 2000) provides versatile means for the precise labeling of proteins in ways that are not straightforward, or simply not possible, using recombinant DNA expression technologies. Chemical protein synthesis has previously been used to incorporate D-amino acids (Bang et al. 2006a), backbone-modified residues (Lu et al. 1999; Johnson and Kent 2006b), and spectroscopic probes (Cowburn et al. 2004) in a number of systems, including the MscL (Clayton et al. 2004) and KcsA (Valiyaveetil et al. 2004) ion channels. The major challenge in most of these studies is in the synthesis of the modified protein itself. KChIP2d is a small protein with no disulfides, which suggested that it would be relatively straightforward to synthesize and to fold. In order to facilitate the chemical synthesis of the necessary peptide segments, we introduced a cysteine at residue 34, which we then subsequently alkylated to from a “ψGln” residue (Kochendoerfer et al. 2003). The alkylation of only this Cys residue demanded that we use a strategy that involved ligating from the N terminus of the protein toward the C terminus (Bang et al. 2006b), rather than the typical C- to N-terminal ligation by native chemical ligation. The kinetically controlled ligation of segments 1–33:COSΦCH2COOH and Cys34–52:COSR was followed by alkylation with iodoacetamide in quantitative yield. Subsequent native chemical ligation of this product with the C-terminal peptide gave the desired synthetic KChIP2d product in good yield and high purity.
Synthetic KChIP2d folded into a conformation with defined secondary, tertiary, and quaternary structure, and the populations of all of these structures increased upon the binding of calcium to the protein's EF hand. Unlike other KChIP2 isoforms that contain as many as four EF hands, KChIP2d consists of only a single EF hand with two N-terminal helices, and it was thus of some interest as to how it is able to interact with its target ion channels (Patel et al. 2004). We found that KChIP2d is capable of associating with its biological ligand, the N terminus of the Kv4.2 ion channel, with two KChIP2d monomers likely binding to its target peptide. However, we have yet to explore how KChIP2d interacts with intact Kv4 channels.
It is still unclear from this initial study how KChIP2d regulates the function of Kv4.2 channels. In future studies, we plan to probe the dynamics of the KChIP2d:Kv4.2 complex using site-specific fluorescent or other biophysical probes, in conjunction with traditional ion channel electrophysiology, to develop structural and biophysical models for regulation of Kv4.2 channels by KChIPs. Much has been elucidated regarding the structural basis of activity of ion channels from crystallography and high-resolution spectroscopy (Chanda et al. 2005; Long et al. 2005a,b; Perozo 2006), but it is largely unclear how most accessory subunits interact with and regulate their cognate ion channels. Therefore it is of some interest to probe the dynamics of the KChIP2d:Kv4 channel interface. In the crystal structure of the KChIP1:Kv4.1 N-terminal peptide complex, the two N-terminal EF hands of KChIP1 that do not have calcium bound form a “clamshell” with a long hydrophobic groove in which the α-helical N terminus of Kv4.1 binds (Zhou et al. 2004). This association is mediated primarily by hydrophobic interactions between aromatic and hydrophobic residues in KChIP1 and aromatics on one face of the Kv4.1 N-terminal helix. As this crystal structure of the complex was not obtained in the context of full-length Kv4 channel, it is unclear how other portions of Kv4, which have been shown to play an important role in complex formation (Callsen et al. 2005), interact with KChIPs. A low-resolution EM structure of the intact 4:4 KChIP2-Kv4.2 complex (Kim et al. 2004a) yielded this general architecture: The complex is in a “hanging-gondola” conformation, with each of the four KChIP2 proteins binding to the cytoplasmic tetramerization domains of Kv4.2 (Kim et al. 2004b). This results in the formation of “fenestrations” connecting the pore to the cytoplasm in a general conformation similar to that seen in the Shaker crystal structure, in which these side portals between the tetramerization domain and the channel proper connect the cytoplasm to the pore (Long et al. 2005a), where it is likely that the ion flow is controlled by accessory subunits. These interactions could be intimately probed by the insertion of unnatural amino acids with chemical or biophysical reporter functionality into synthetic KChIP2d.
The success of the strategy used in the chemical synthesis of KChIP2d reported here is a further illustration of the utility of the kinetically controlled ligation approach to chemical protein synthesis. Incorporation of the artificial “pseudoglutamine” residue at position 34 constitutes proof of principle of the utility of the synthetic route for incorporation of spectroscopic probes: For example, use of 13C iodoacetamide would enable the site-specific incorporation of an NMR probe. Such specific modification of a single thiol moiety under mild aqueous conditions could, inter alia, be used to label the KChIP2 molecule with a commercially available thiol reactive fluorophore.
Materials and Methods
Materials
Nα-Boc amino acids with appropriate side-chain protection were obtained from Peptides International. Boc-Gly-OCH2-Pam- and Boc-Ile-OCH2-Pam-polystyrene resins were obtained from NeoMPS. Acetonitrile was from EMD Biosciences. N,N-diisopropylethylamine was from Applied Biosystems. HBTU was from Peptides International. p-Cresol, piperidine, iodoacetamide, and thiophenol were from Sigma. Dimethylformamide (DMF) was from Burdick and Jackson. Dichloromethane (DCM) was from Omnisolv. Trifluoroacetic acid (TFA) was from Halocarbon.
Total chemical synthesis of KChIP2d
Solid phase peptide synthesis
Peptides were manually synthesized by Boc chemistry on a 0.4-mmol scale using published “in situ neutralization” protocols (Schnolzer et al. 1992; Hackeng et al. 1999). Peptide 53–70 was made on Ile-OCH2-Pam-polystyrene resin. For the 1–33:COSR and 34–52:COSR peptides, a trityl-protected mercaptopropionic acid was added to the Gly-OCH2-PAM-polystyrene resin prior to chain elongation, which resulted in a C-terminal alkyl thioester peptide after HF cleavage as previously described (Hackeng et al. 1999). Side-chain protecting groups used were Arg(Tos), Asn(Xan), Cys(4-MeBzl), Glu(OcHex), His(Bom), Lys(ClZ), Ser(Bzl), Thr(Bzl), and Tyr(BrZ). After chain elongation was complete, the Nα-Boc group was removed by treatment with neat TFA; the peptide-resin was thoroughly washed with DCM and dried in a strong stream of air for 10 min. The peptide was cleaved from the resin with simultaneous removal of side-chain protecting groups by treatment with anhydrous HF with p-cresol (90/10 v/v) for 1 h at 0°C. After evaporation of HF under vacuum, the product was precipitated and washed with chilled diethyl ether and the crude peptide dissolved in 50/50 (v/v) CH3CN/H20, 0.1% TFA. Analytical HPLC of the crude products showed that each had one major peak corresponding to the desired product and could be readily purified by preparative reverse-phase HPLC (Supplemental Fig. S1). The electrospray ionization (ESI) mass spectrum of the purified peptide corresponded in each case to the calculated mass (average isotope composition) of the desired product (peptide 1–33:COSR—obs. 4144.8 ± 0.4 Da, calc. 4144.8 Da; peptide 34–52:COSR—obs. 2358.6 ± 0.3 Da, ∼2358.7 Da; peptide 53–70—obs. 2184.4 ± 0.2 Da, calc. 2184.5 Da).
Preparative reverse-phase HPLC purification
Peptides were purified using a Vydac C4 column (12 μm, 2.2 × 25 cm) at a flow rate of 10 mL/min using a gradient from 10% to 50% buffer B over 60–80 min (buffer A: H2O, 0.l% TFA; buffer B: CH3CN, 0.08% TFA) using a Waters 4000 Prep LC system. Fractions were analyzed by LC/MS (Agilent 1100 Series LC/MSD trap) or MALDI TOF mass spectrometry (Perseptive Biosystems Voyager-DE MALDI-TOF); fractions containing pure, desired peptide were combined and lyophilized (Supplemental Fig. S1).
Kinetically controlled ligation
A kinetically controlled ligation between segments 1–33:COSR and 34–52:COSR to yield 1–52:COSR was performed under similar conditions to those described by Bang et al. (2006b). The alkyl thioester group of crude 1–33:COSR at a concentration of 1.5 mM was exchanged with 100 mM of mercaptophenylacetic acid in 2 mL ligation buffer (6 M GuHCl, 100 mM phosphate buffer, pH adjusted to 6.8 with concentrated NaOH) to form 1–33:COSΦCH2COOH (Johnson and Kent 2006a; data not shown). This peptide thioarylester was purified by preparative reverse-phase HPLC and lyophilized, and 46 mg (∼11 μmol) was incubated with 26 mg (∼11 μmol) of pure 34–52:COSR in 4 mL of ligation buffer (final concentration of each peptide = 2.75 mM). The ligation reaction was essentially complete in 30 min.
Alkylation
To this reaction mixture, an equal volume of 100 mM of iodoacetamide in ligation buffer was added in order to alkylate Cys34 and to quench the ligation reaction (D. Bang, pers. comm.). Alkylation was complete in 30 min with an observed mass increase of 57 Da, consistent with a single alklylation by −CH2CONH2 (calc. 57.0 Da). The reaction mixture was then immediately run on preparative reverse-phase HPLC to give pure 1–52:COSR peptide containing a −CH2CONH2 modified Cys34 (data not shown). The overall yield of purified 1–52:COSR peptide for the kinetically controlled ligation reaction and alkylation from 1–33:COSR and 34–52:COSR was 17 mg (23%). This yield is consistent with previously reported values for kinetically controlled ligations of 40%–70% (Bang et al. 2006b) if we assume the alkylation reaction is ∼60% effective.
Native chemical ligation
Native chemical ligation (Dawson et al. 1994) between the modified 1–52:COSR peptide and the Cys53–70 peptide was performed under standard conditions (Dawson et al. 1997). The two peptides at concentrations of 0.7 mM (i.e., 11 mg 1–52:COSR; 3 mg Cys53-70) were stirred in 2 mL of degassed ligation buffer with 1.0% thiophenol added; the reaction was complete in 22 h (▶). The product synthetic KChIP2d(1–70) was purified by preparative reverse-phase HPLC from the ligation mixture, resulting in high purity product (▶). The isolated yield for purified synthetic KChIP2d from this reaction was 7 mg (58%).
Characterization of synthetic KChIP2d
Circular dichroism
Circular dichroism measurements from 190 to 265 nm were made at 25°C using an AVIV Circular Dichroism Spectrometer Model 202 at a protein concentration of 44 μM in 10 mM sodium cacodylate, 140 mM KCl, 2 mM MgCl2 in the absence an/or in the presence of 1 mM CaCl2.
NMR spectroscopy
KChIP2d at a concentration of 0.8 mg/mL (∼95 μM) in 10 mM HEPES, 300 mM KCl, 2 mM MgCl2, 5% D2O (v/v) ± 1 mM CaCl2 was used for NMR studies with a VARIAN Inova 600-MHz spectrometer equipped with a cryoprobe. We were unable to obtain samples of higher protein concentration due to limited protein solubility. Protein concentrations were calculated from A280. Two-dimensional 1H-NMR experiments were attempted but were not feasible due to insufficient protein concentration and our inability to work in phosphate buffer because of calcium phosphate precipitation. One-dimensional 1H-NMR data were analyzed in VNMR (Varian).
Analytical ultracentrifugation
Sedimentation velocity experiments were performed using a Beckman Optima XL-A analytical ultracentrifuge with an AN-60 rotor. Samples of OD230 ∼1.0 (∼20 μM KChIP2d in 10 mM HEPES, 300 mM KCl, 2 mM MgCl2) were injected into the 1.2-mm path length, two-channel aluminum centerpiece and centrifuged at 60K rpm for 24 h at 25°C while the absorbance was monitored at 230 nm. Data analysis was performed in SEDFIT and SEDPHAT (Schuck 2000, 2003). When fitting with the monomer-dimer equilibrium model, the molar extinction coefficient and monomer molecular weight were held constant. Residuals were small and randomly distributed (Supplemental Fig. S2).
Surface plasmon resonance
A peptide corresponding to the first 23 residues of Kv4.2 (Kv4.2NT: MAAGVAAWLPFARAAAIGWMPVA) was synthesized by Boc chemistry using the conditions described above. After HF cleavage, the formyl protecting group on Trp8 was removed by incubation with 10% piperidine (pH 11 in ligation buffer) for 10 min, followed by neutralization to slightly acidic pH with concentrated HCl and purification by preparative reverse-phase HPLC. The purified peptide was immobilized on a CM5 chip using standard amine coupling with EDC/NHS to ∼3000 RU followed by capping with ethanolamine. Binding experiments were performed with a Biacore 3000 system (Biacore) using HBS buffer (25 mM HEPES at pH 7.4, 150 mM NaCl, 0.005% Tween v/v) ± 1 mM CaCl2, with KChIP2d concentrations ranging from 0.6875 to 44 μM. Data analysis was performed in Scrubber 2 (Roden and Myszka 1996) and Origin 7.0 (OriginLabs). Although significant drift is present in the data, binding was clearly visible, and curves generated at both early and late times in the drift were consistent with one another.
Acknowledgments
We thank Duhee Bang for discussions regarding the kinetically controlled ligation and alkylation with iodoacetamide, Mohammed Yousuf for assistance with analytical ultracentrifugation and surface plasmon resonance, and Josh Kurutz for NMR data analysis and for assistance with NMR samples and instrumentation. We also thank Erik Johnson, Brad Pentelute, and Dave Boerema for valuable discussions. Research was funded by grants from the Department of Energy Genomes to Life Genomics Program (Grant DE-FG02-04ER63786) and the National Institutes of Health (Grant 1R01GM075993-01) to S.B.H.K.
Footnotes
Supplemental material: see www.proteinscience.org
Reprint requests to: Sudarshan Rajagopal, Department of Medicine, Duke University Medical Center, Durham, NC 27710, USA; e-mail: rajag007@mc.duke.edu; fax: (919) 681-6448.
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.072876107.
References
- An W.F., Bowlby, M.R., Betty, M., Cao, J., Ling, H.P., Mendoza, G., Hinson, J.W., Mattsson, K.I., Strassle, B.W., Trimmer, J.S., et al. 2000. Modulation of A-type potassium channels by a family of calcium sensors. Nature 403 553–556. [DOI] [PubMed] [Google Scholar]
- Bang D., Gribenko, A.V., Tereshko, V., Kossiakoff, A.A., Kent, S.B., and Makhatadze, G.I. 2006a. Dissecting the energetics of protein α-helix C-cap termination through chemical protein synthesis. Nat. Chem. Biol. 2 139–143. [DOI] [PubMed] [Google Scholar]
- Bang D., Pentelute, B., and Kent, S.B. 2006b. Kinetically controlled ligation for the convergent chemical synthesis of proteins. Angew. Chem. Int. Ed. Engl. 45 3985–3988. [DOI] [PubMed] [Google Scholar]
- Burgoyne R.D., O'Callaghan, D.W., Hasdemir, B., Haynes, L.P., and Tepikin, A.V. 2004. Neuronal Ca2+-sensor proteins: Multitalented regulators of neuronal function. Trends Neurosci. 27 203–209. [DOI] [PubMed] [Google Scholar]
- Callsen B., Isbrandt, D., Sauter, K., Hartmann, L.S., Pongs, O., and Bahring, R. 2005. Contribution of N- and C-terminal Kv4.2 channel domains to KChIP interaction. J. Physiol. 568 397–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chanda B., Asamoah, O.K., Blunck, R., Roux, B., and Bezanilla, F. 2005. Gating charge displacement in voltage-gated ion channels involves limited transmembrane movement. Nature 436 852–856. [DOI] [PubMed] [Google Scholar]
- Clayton D., Shapovalov, G., Maurer, J.A., Dougherty, D.A., Lester, H.A., and Kochendoerfer, G.G. 2004. Total chemical synthesis and electrophysiological characterization of mechanosensitive channels from Escherichia coli and Mycobacterium tuberculosis . Proc. Natl. Acad. Sci. 101 4764–4769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowburn D., Shekhtman, A., Xu, R., Ottesen, J.J., and Muir, T.W. 2004. Segmental isotopic labeling for structural biological applications of NMR. Methods Mol. Biol. 278 47–56. [DOI] [PubMed] [Google Scholar]
- Dawson P.E. and Kent, S.B. 2000. Synthesis of native proteins by chemical ligation. Annu. Rev. Biochem. 69 923–960. [DOI] [PubMed] [Google Scholar]
- Dawson P.E., Muir, T.W., Clark-Lewis, I., and Kent, S.B. 1994. Synthesis of proteins by native chemical ligation. Science 266 776–779. [DOI] [PubMed] [Google Scholar]
- Dawson P.E., Churchill, M.J., Ghadiri, M.R., and Kent, S.B.H. 1997. Modulation of reactivity in native chemical ligation through the use of thiol additives. J. Am. Chem. Soc. 119 4325–4329. [Google Scholar]
- Decher N., Barth, A.S., Gonzalez, T., Steinmeyer, K., and Sanguinetti, M.C. 2004. Novel KChIP2 isoforms increase functional diversity of transient outward potassium currents. J. Physiol. 557 761–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delisle B.P., Anson, B.D., Rajamani, S., and January, C.T. 2004. Biology of cardiac arrhythmias: Ion channel protein trafficking. Circ. Res. 94 1418–1428. [DOI] [PubMed] [Google Scholar]
- Hackeng T.M., Griffin, J.H., and Dawson, P.E. 1999. Protein synthesis by native chemical ligation: Expanded scope by using straightforward methodology. Proc. Natl. Acad. Sci. 96 10068–10073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn M.E. and Muir, T.W. 2005. Manipulating proteins with chemistry: A cross-section of chemical biology. Trends Biochem. Sci. 30 26–34. [DOI] [PubMed] [Google Scholar]
- Hubbell W.L., Cafiso, D.S., and Altenbach, C. 2000. Identifying conformational changes with site-directed spin labeling. Nat. Struct. Biol. 7 735–739. [DOI] [PubMed] [Google Scholar]
- Johnson E.C. and Kent, S.B. 2006a. Insights into the mechanism and catalysis of the native chemical ligation reaction. J. Am. Chem. Soc. 128 6640–6646. [DOI] [PubMed] [Google Scholar]
- Johnson E.C. and Kent, S.B. 2006b. Synthesis, stability and optimized photolytic cleavage of 4-methoxy-2-nitrobenzyl backbone-protected peptides. Chem. Commun. (Camb.) 1557–1559. [DOI] [PubMed]
- Kim L.A., Furst, J., Butler, M.H., Xu, S., Grigorieff, N., and Goldstein, S.A. 2004a. Ito channels are octomeric complexes with four subunits of each Kv4.2 and K+ channel-interacting protein 2. J. Biol. Chem. 279 5549–5554. [DOI] [PubMed] [Google Scholar]
- Kim L.A., Furst, J., Gutierrez, D., Butler, M.H., Xu, S., Goldstein, S.A., and Grigorieff, N. 2004b. Three-dimensional structure of I(to); Kv4.2-KChIP2 ion channels by electron microscopy at 21 angstrom resolution. Neuron 41 513–519. [DOI] [PubMed] [Google Scholar]
- Kochendoerfer G.G., Chen, S.Y., Mao, F., Cressman, S., Traviglia, S., Shao, H., Hunter, C.L., Low, D.W., Cagle, E.N., Carnevali, M., et al. 2003. Design and chemical synthesis of a homogeneous polymer-modified erythropoiesis protein. Science 299 884–887. [DOI] [PubMed] [Google Scholar]
- Kuo H.C., Cheng, C.F., Clark, R.B., Lin, J.J., Lin, J.L., Hoshijima, M., Nguyen-Tran, V.T., Gu, Y., Ikeda, Y., Chu, P.H., et al. 2001. A defect in the Kv channel-interacting protein 2 (KChIP2) gene leads to a complete loss of I(to) and confers susceptibility to ventricular tachycardia. Cell 107 801–813. [DOI] [PubMed] [Google Scholar]
- Lin Y.L., Chen, C.Y., Cheng, C.P., and Chang, L.S. 2004. Protein-protein interactions of KChIP proteins and Kv4.2. Biochem. Biophys. Res. Commun. 321 606–610. [DOI] [PubMed] [Google Scholar]
- Long S.B., Campbell, E.B., and Mackinnon, R. 2005a. Crystal structure of a mammalian voltage-dependent Shaker family K+ channel. Science 309 897–903. [DOI] [PubMed] [Google Scholar]
- Long S.B., Campbell, E.B., and Mackinnon, R. 2005b. Voltage sensor of Kv1.2: Structural basis of electromechanical coupling. Science 309 903–908. [DOI] [PubMed] [Google Scholar]
- Lu W., Randal, M., Kossiakoff, A., and Kent, S.B. 1999. Probing intermolecular backbone H-bonding in serine proteinase-protein inhibitor complexes. Chem. Biol. 6 419–427. [DOI] [PubMed] [Google Scholar]
- Patel S.P., Campbell, D.L., Morales, M.J., and Strauss, H.C. 2002a. Heterogeneous expression of KChIP2 isoforms in the ferret heart. J. Physiol. 539 649–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel S.P., Campbell, D.L., and Strauss, H.C. 2002b. Elucidating KChIP effects on Kv4.3 inactivation and recovery kinetics with a minimal KChIP2 isoform. J. Physiol. 545 5–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel S.P., Parai, R., and Campbell, D.L. 2004. Regulation of Kv4.3 voltage-dependent gating kinetics by KChIP2 isoforms. J. Physiol. 557 19–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perozo E. 2006. Gating prokaryotic mechanosensitive channels. Nat. Rev. Mol. Cell Biol. 7 109–119. [DOI] [PubMed] [Google Scholar]
- Pourrier M., Schram, G., and Nattel, S. 2003. Properties, expression and potential roles of cardiac K+ channel accessory subunits: MinK, MiRPs, KChIP, and KChAP. J. Membr. Biol. 194 141–152. [DOI] [PubMed] [Google Scholar]
- Roden L.D. and Myszka, D.G. 1996. Global analysis of a macromolecular interaction measured on BIAcore. Biochem. Biophys. Res. Commun. 225 1073–1077. [DOI] [PubMed] [Google Scholar]
- Schnolzer M., Alewood, P., Jones, A., Alewood, D., and Kent, S.B. 1992. In situ neutralization in Boc-chemistry solid phase peptide synthesis. Rapid, high yield assembly of difficult sequences. Int. J. Pept. Protein Res. 40 180–193. [DOI] [PubMed] [Google Scholar]
- Schuck P. 2000. Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling. Biophys. J. 78 1606–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuck P. 2003. On the analysis of protein self-association by sedimentation velocity analytical ultracentrifugation. Anal. Biochem. 320 104–124. [DOI] [PubMed] [Google Scholar]
- Valiyaveetil F.I., Sekedat, M., Muir, T.W., and MacKinnon, R. 2004. Semisynthesis of a functional K+ channel. Angew. Chem. Int. Ed. Engl. 43 2504–2507. [DOI] [PubMed] [Google Scholar]
- Zhou W., Qian, Y., Kunjilwar, K., Pfaffinger, P.J., and Choe, S. 2004. Structural insights into the functional interaction of KChIP1 with Shal-type K(+) channels. Neuron 41 573–586. [DOI] [PubMed] [Google Scholar]