

Abstract
The link between the NF-κB signal transduction pathway and cancer is now well established. Inhibiting this pathway is therefore a promising approach in the treatment of certain cancers through a pro-apoptotic effect in malignant cells. Owing to its central role in the pathway, the IκB kinase (IKK) complex is a privileged target for designing inhibitors. Previously, we showed that oligomerization of NEMO is necessary for IKK activation and defined a minimal oligomerization domain (CC2-LZ) for NEMO, and we developed NEMO peptides inhibiting NF-κB activation at the level of the IKK complex. To improve the low-affinity inhibitors, we used ribosome display to select small and stable proteins with high affinity against the individual CC2-LZ because the entire NEMO protein is poorly soluble. Several binders with affinities in the low nanomolar range were obtained. When expressed in human cells, some of the selected molecules, despite their partial degradation, inhibited TNF-α-mediated NF-κB activation while having no effect on the basal activity. Controls with a naive library member or null plasmid had no effect. Furthermore, we could show that this NF-κB inhibition occurs through a specific interaction between the binders and the endogenous NEMO, resulting in decreased IKK activation. These results indicate that in vitro selections with the NEMO subdomain alone as a target may be sufficient to lead to interesting compounds that are able to inhibit NF-κB activation.
Keywords: NEMO, IKK complex, NF-κB pathway, ribosome display, protein evolution
The NF-κB signaling pathway is involved in the regulation of a wide variety of physiological and pathological processes (Hayden and Ghosh 2004). While its prominent role in the regulation of inflammatory, innate, and adaptative immune responses is extensively studied, it has been shown more recently that this pathway also provides a mechanistic link between inflammation and oncogenic progression (Karin 2006). Many genes that control six essential alterations characterizing a tumor cell (Hanahan and Weinberg 2000) are closely regulated by the NF-κB transcription factor family. This underscores the importance of this pathway to provide attractive targets for cancer prevention and therapy. However, as the NF-κB pathway also serves an important immune function, its inhibition could result in severe immunodeficiency. Thus, the clinical success of potential NF-κB inhibitors will arise from lead compounds that specifically block the hyperactivation of the pathway in malignant cells without totally suppressing the innate and adaptative responses in inflammatory cells. In this context, regulatory proteins or protein domains may become more attractive targets than their catalytic counterparts. The NF-κB essential modulator protein termed NEMO (also called IKKγ) belongs to this class of regulatory proteins and plays a central role in signal-induced NF-κB activation (Rothwarf et al. 1998; Yamaoka et al. 1998). The variety of signals that lead to NF-κB activation proceed through at least three main pathways, which are referred to as the “classical” (Israel 2000), the “alternative” (Claudio et al. 2002), and the “DNA damage induced” pathways (Huang et al. 2003). Each of these pathways was shown to be associated with some cancer cell types or with therapeutic resistance (Kim et al. 2006). Signals transduce through two central IκB kinases, IKKα and IKKβ, which interact with the essential regulatory protein NEMO. The alternative pathway, which is required for the generation of secondary lymphoid organ and for B-cell maturation, is strictly dependent on IKKα (Bonizzi and Karin 2004), whereas the classical pathway, which is rapidly activated in response to pro-inflammatory stimuli and antigen–receptor engagement, is strictly dependent on NEMO and to a large extent on the activity of IKKβ (Hacker and Karin 2006). While the NEMO-dependent signal leading to kinase activation proceeds from outside the cell to the cytosol in the classical pathway, it can also occur from the nucleus to the cytosol in an again NEMO-dependent manner upon genotoxic stress. NEMO, owing to its central role, thus provides an attractive target for the development of new NF-κB inhibitors since it integrates and coordinates most of the NF-κB stimuli.
NEMO oligomerization has been shown to be an important step in signal-induced activation of the IKK kinases (Poyet et al. 2000; Agou et al. 2002). Moreover, it has recently been shown that NEMO also serves as a sensor of K63-linked-polyubiquitin chains, allowing the specific targeting of the IKK kinases to activated receptors (Ea et al. 2006; Wu et al. 2006). However, the precise mechanistic role of NEMO in this process is still unclear and may differ depending on the pathway.
The NEMO protein is made of an α-helical scaffold that contains several coiled-coil motifs. The N-terminal half, containing a large coiled-coil motif, is necessary for interaction with the IKKs. It also contains a short, α-helical subdomain that participates in NEMO dimerization (Marienfeld et al. 2006). The C-terminal domain is composed of two shorter coiled-coil motifs, termed CC2 and LZ, as well as a zinc finger motif at the extreme C terminus. It is involved in the integration of the upstream signals and mainly contributes to NEMO oligomerization. Within the C-terminal part, the CC2-LZ domain, which is essential for full NF-κB activation, exhibits a dual activity: It is responsible for NEMO oligomerization (Tegethoff et al. 2003; Agou et al. 2004b) and also serves as a binding domain to specifically recruit NEMO to K63-linked-polyubiquitin chains (Ea et al. 2006; Wu et al. 2006). Based on biochemical and biophysical data, we proposed a structural model that consists of a trimer of heterodimers, the whole forming a six-α-helix bundle (Filipe-Santos et al. 2006). Cell-permeable peptides were derived from this model and were shown to inhibit the NF-κB pathway by blocking NEMO oligomerization (Agou et al. 2004a). In addition, the most potent NEMO peptide inhibitor, called LZ, was shown to induce apoptosis in high-risk myelodysplastic syndrome and acute myeloid leukemia (Carvalho et al. 2006). However, although the inhibition effect in cellulo is significant, the in vitro binding constant of peptides for the CC2-LZ domain is relatively weak, with IC50 values in the micromolar range (Agou et al. 2004a). Our goal was, therefore, to develop a new strategy to find stronger inhibitors of IKK activation targeting the CC2-LZ domain of NEMO. For this, we used ribosome display (Zahnd et al. 2007) to select binders with high affinity from a protein library. In ribosome display, a synthetic DNA library coding for the protein library is transcribed in vitro into mRNA and further translated in vitro to give ternary complexes of polypeptide, ribosome, and mRNA. Because the mRNA bears no stop codon, the ternary complex does not fall apart under appropriate experimental conditions, allowing coupling of the phenotype to the genotype. These complexes are then used for binding selection on the immobilized target. After washing, the mRNA from the bound complexes is rescued by dissociating the ribosome with EDTA and is used for the next selection round after reverse transcription followed by PCR amplification and in vitro transcription.
We performed the selection of CC2-LZ binders using a library of designed ankyrin-repeat proteins (DARPins) bearing small proteins (14 kDa), which exhibit very high thermodynamic stability (Binz et al. 2006). The library was constructed by consensus design from naturally occurring ankyrin repeats (Sedgwick and Smerdon 1999). Within one ankyrin module, all amino acids are allowed at the six variable positions, except Pro, Cys, and Gly, providing a theoretical diversity of 7.2 × 107 per repeat (Binz et al. 2003). We used the DARPin library N2C, which consists of two variable repeats and of two capping modules, generating therefore a maximal diversity of ∼1010 to 1011. The binding interface provided by the N2C DARPin, which was evaluated from the crystal structure of N3C DARPin (PDB:1MJO), was considered large enough to eventually form a tight complex with the CC2-LZ monomer.
Results
Synthesis of a biotinylated polypeptide that mimics the K63-polyubiquitin- binding/oligomerization domain of NEMO
Our target for the selection of binders was the K63-polyubiquitin-binding/oligomerization domain of NEMO, namely, the so-called CC2-LZ, a domain corresponding to the murine sequence 253–336 of the NEMO protein. We chemically synthesized the entire CC2-LZ domain and conjugated it at the N terminus with the NHS-PEO4 activated biotine. The specific labeling was achieved when the peptide was still attached to the resin, resulting in a quantitative and selective N-terminal labeling. The biotin was separated from the α-amine of the target by a long hydrophilic spacer of 29 Å to minimize any steric hindrance. The stoichiometry and the homogeneity of the biotinylated CC2-LZ were verified by mass spectrometry as described in Materials and Methods. We also used gel filtration and CD spectroscopy to ensure that the trimerization properties and the helicity of the synthetic target were similar to those of the previously reported His-tagged recombinant CC2-LZ (Agou et al. 2002, 2004b; data not shown).
Selection of CC2-LZ binders by in vitro evolution using ribosome display
In an attempt to inhibit NEMO function, and therefore, the activation of the IKK kinases, we decided to search for high-affinity binders that preferentially target the monomeric state of the ubiquitin-binding/oligomerization domain of NEMO. Such binders could inhibit the NF-κB signaling pathway by disrupting NEMO oligomerization or K63-polyubiquitin-binding properties through steric means. New generations of protein scaffolds, specifically selected for binding any target, were recently developed (see Hosse et al. 2006). Among the novel binding molecules, the designed ankyrin-repeat proteins (DARPins) exhibit an α-helical framework that has been successfully used to generate high-affinity binders against a variety of targets (Binz et al. 2004). We used ribosome display for evolving in vitro small and stable proteins that bind to CC2-LZ with high affinity.
We started from the DARPin library called N2C, which includes a diversity of ∼1010 to 1011 members. Proteins expressed from the N2C library bear two variable ankyrin repeats with seven randomized positions located between two capping modules (▶). As we wanted to select DARPins against the CC2-LZ monomer, we used a low concentration of the target CC2-LZ protein (50 nM) for the selection to minimize the trimer formation on the microtiter plate. ▶ shows the enrichment of specific DARPin cDNAs, which was monitored by the number of cycles used for the RT-PCR step. The specificity is shown by the difference between the unspecific band resulting from panning on Neutravidin and BSA only, and the specific band resulting from panning to Neutravidin, BSA, and biotinylated CC2-LZ. In all selection rounds, the total washing time was increased from 25 min in the first round to 190 min in the fourth round. After the first two rounds of selection, with 40 and 35 PCR cycles, respectively, there was hardly any difference between the two amplified DNA fragments. Round 3 with 30 PCR cycles brought up a very clear enrichment. After four rounds, no unspecific DNA band could be visualized in the absence of the CC2-LZ target (▶, round 4). Single clones were expanded to 1 mL of culture, and expression was induced with IPTG. Binding of the DARPins was first evaluated by a direct ELISA assay using clonal crude extract, in which 80% of all clones showed specific binding. ▶ shows the sequence analysis of 42 positive DARPin clones. It reveals one large family, 2A1, to which 38% of all binders belonged. In addition, there were several small families as well as sequences that were found only once. We also found a small amount of clones (12%) bearing deletions of up to the first variable module or frameshifts (data not shown). To estimate the affinity constants in solution, we also carried out competition ELISA assays. DARPins with the ability to compete against 40 nM free CC2-LZ were considered strong binders. Analysis of individual ankyrin sequences indicated that they predominantly contained acidic (mostly aspartate) and aromatic (mostly tryptophan) amino acids at the variable positions (▶).
Table 1.
Alignment of DARPin sequences with improved affinity by in vitro evolution
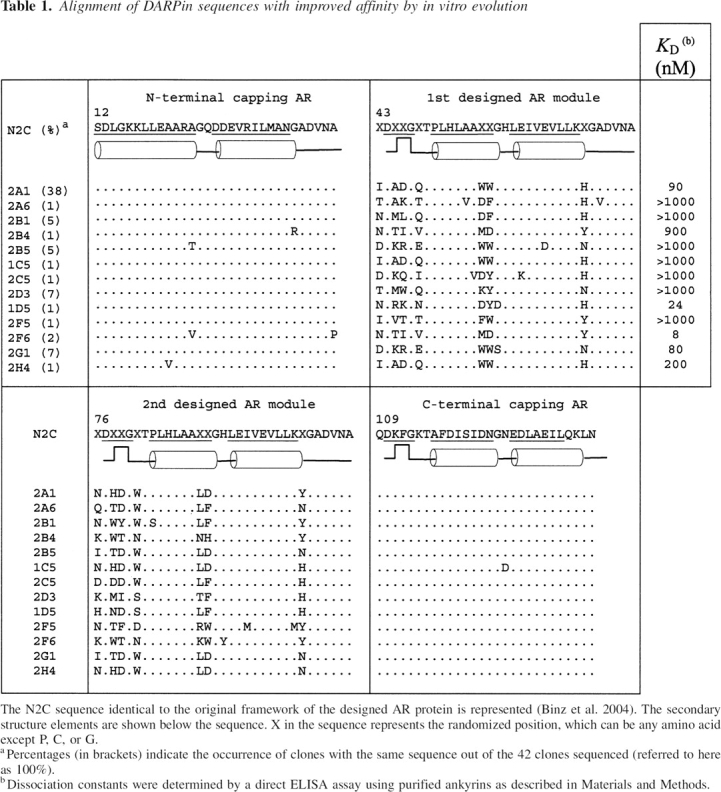
Figure 1.

Enrichment of CC2-LZ binders from a RNA library using ribosome display. The in vitro translation mixture was applied either to a specific surface coated with BSA and the target CC2-LZ protein, or to a surface coated with BSA only. Enrichment of specific binders can be evaluated by comparing the quantity of cDNA obtained after RT-PCR between the two samples. The unspecific binders can be detected until round three.
In vitro affinities of selected binders to CC2-LZ
Next, we expressed and purified 16 ankyrin binders from Escherichia coli to measure the binding constants of ankyrins with a precise accuracy. These ankyrins were chosen based on sequences as well as IC50 values obtained from the competition ELISA assay described above. It is, of course, not clear whether high in vitro affinities correlate with the inhibition effect of ankyrins in vivo. However, we presumed that strong affinities were required to interfere with CC2-LZ oligomerization at low inhibitor concentrations. The affinities were measured by direct ELISA on microtiter plates. Four of the 16 binders (2A1, 1D5, 2F6, 2G1) had affinities (K D values) below 100 nM, including a member of the predominant 2A1 sequence family. Two other binders (2H4, 2B4) displayed a K D between 100 nM and 1 μM, while the others showed weak binding (K D > 1 μM) (▶). ▶ shows the purity levels and the binding curves for three ankyrin binders—2F6, 2A1, and 1D5. With the exception of 2B4, which bears three mutations in the framework, all ankyrin sequences with nanomolar to micromolar affinities share similar sequence features. They all contain at least 50% of hydrophobic and aromatic amino acids at the randomized positions within the two α-helices of the variable ankyrin modules. Among these amino acids, 60% are aromatic residues. There does not appear to be any correlation between the total hydrophobic pattern of the ankyrin interface and the affinity for the CC2-LZ target. For example, ankyrin 1D5, which has the most hydrophilic interface (only 23% of hydrophobic amino acids), displays a similar free energy of binding as ankyrin 2F6, which represents the most hydrophobic protein interface (46%). This might reflect the recognition of different regions with respect to the binding of CC2-LZ (see Discussion).
Figure 2.

Ankyrins with improved affinity for the CC2-LZ domain of NEMO. (A) Purification of the three selected ankyrins (2F6, 1D5, and 2A1), which exhibit high affinity for the CC2-LZ domain. The purified ankyrins were examined by SDS-PAGE and Coomassie staining. (B) The in vitro binding affinity was determined by a direct ELISA assay. The binding curves were fitted using the Langmuir binding equation as described in Materials and Methods. 2F6 ankyrin (circles) exhibits the best affinity, with a K D value of 8 nM. K D values for 1D5 (diamonds) and 2A1 (squares) were 24 and 90 nM, respectively.
Out of 16 purified ankyrins, six ankyrins (2A1, 2F6, 1D5, 2H4, 2G1, and 2B4) displayed an affinity for CC2-LZ below the micromolar range (▶). While no correlation appears between the total hydrophobic pattern of the ankyrin interface and the binding strength, a clear correlation between the degree of hydrophobicity in the two α-helices of the variable modules and the affinity can be drawn. Indeed, all binders with affinities less than micromolar contain at least 60% of aromatic and hydrophobic residues, whereas those with K D greater than micromolar have only 50% of these residues. For example, the 2B4 ankyrin differs from 2F6 only by the presence of polar residues in the second α-helix. It has an identical sequence with 2F6 in the β-turn and loop regions, as well as in the first α-helix. Despite this sequence similarity, the affinity of 2B4 is ∼100 times lower than that of 2F6.
Among the selected ankyrins, all sequences except for 2A1 contain one or several mutations that are not present in the conserved original framework, indicating that these mutations probably accumulated during extensive PCR. We observed that the presence of a single mutation in the framework can affect binding. Indeed, the ankyrin 2H4, which has the same sequence as 2A1 in the randomized positions, contains one valine mutation in the N-terminal cap. This A21V mutation leads to an affinity of 200 nM, which is lower than that of 2A1 (K D = 90 nM). For this reason, the 2H4 ankyrin was not selected for further studies. Similarly, the 2G1 (K D = 80 nM) as well as 2B4 (K D = 900 nM) ankyrins, which have similar or lower affinities compared to 2A1, were not further analyzed.
To better visualize how the various ankyrin interfaces evolved with improved affinity against the CC2-LZ domain, structural models of three ankyrins (2A1, 2F6, 1D5) were built using the X-ray structure of N3C ankyrin (PDB entry 2BKG) as a template (▶). Taking advantage of the models, the ankyrins could be divided into at least two classes. One class, including 2A1 and 2F6, displays similar hydrophobic interfaces with a hydrophobic content of 42% and 46%, respectively. The main difference between these two ankyrins is the charged residues, which are mainly basic in 2F6 and rather acidic in 2A1. The second class, illustrated by 1D5, exhibits a more hydrophilic ankyrin interface, suggesting that this ankyrin recognizes a different CC2-LZ conformation or a different CC2-LZ-binding site.
Figure 3.

Protein interface evolution of DARPin binding to the ubiquitin-binding/oligomerization domain of NEMO. The N2C scaffold AR as well as the three DARPins 1D5, 2A1, and 2F6 with an affinity in the nanomolar range are shown in surface representation. (Gray) The conserved surface of N2C; (red) the variable protein interface. Residues in each ankyrin that contribute to an improved affinity of the CC2-LZ domain are highlighted in different colors: (black) hydrophobic; (red) acidic; (blue) basic; (green) polar. All figures were prepared using the program PyMOL (DeLano 2002).
Inhibition of NF-κB activation in vivo by in vitro selected DARPins
We next addressed the question whether the DARPins could inhibit stimulus-dependent NF-κB activation in cell culture. For this we used an inhibition assay in HEK293T cells following TNF-α stimulation. An expression plasmid containing the sequence of 2F6, 2A1, or 1D5 DARPin under the control of the constitutive EF-1α promoter was cotransfected with two reporter plasmids: The Igκ-luc NF-κB reporter, which was included to monitor NF-κB activation, while the β-galactosidase reporter, under the constitutive EF-1α promoter, served to normalize transfections. Thus, we could monitor differences in the number of living cells, as well as normalize deviations in the preparation of the crude extract used to determine luciferase and β-galactosidase activity. As controls, we also transiently transfected HEK293T cells with a null plasmid or with a plasmid encoding a naive ankyrin that was not selected in vitro by successive rounds of ribosome display. As shown in ▶, the null plasmid control showed a very low NF-κB basal activity in the absence of TNF-α, whereas an 80-fold increase in luciferase activity was observed upon addition of TNF-α for 4 h, indicating that our NF-κB activation assay was very sensitive. While having no effect on the basal activity, the expressions of the two selected ankyrins, 2A1 and 2F6, significantly inhibited the NF-κB-dependent luciferase expression, since the NF-κB activation level was reduced by about two- and sevenfold, respectively, in response to TNF-α. This inhibitory effect was very specific because no NF-κB inhibition was observed when cells were transfected with the naive ankyrin. In contrast, another selected DARPin 1D5, which displays an in vitro affinity for the CC2-LZ domain in the nanomolar range, showed no inhibition on NF-κB activation (▶). This was not due to the absence of the ankyrin expression because Western blotting indicated that 1D5 as well as the naive DARPin were expressed at similar levels (▶). On the contrary, expression levels of the two NF-κB inhibitors, 2A1 and 2F6, were significantly lower. These low levels of DARPin expression were highly reproducible and were not TNF-α-dependent (data not shown).
Figure 4.

Effect of selected ankyrins on TNF-α-induced NF-κB activation. (A) HEK293T cells were transiently cotransfected with the null plasmid (pEF6) or pEF6 plasmids (9 μg) expressing the naive ankyrin (random), 2A1, 2F6, or 1D5 ankyrins, together with 0.5 μg of β-galactosidase(βGal)-encoding pEF1 plasmid and 0.5 μg of the luciferase reporter plasmid pIgκ-luc. After 24 h, cells were stimulated with TNF-α (10 ng/mL), and cell lysates were assayed for luciferase and galactosidase activities. (Error bars represent the mean SD of two samples, which is <10% in all experiments.) Samples were normalized according to the expression level of ankyrins as judged by Western blot analysis against Flag. (B) Expression of ankyrins in 293T cells stimulated with 10 ng/mL of TNF-α. Western blot analysis was performed with anti-Flag antibodies. An equal amount of total protein was applied to each lane.
DARPins selected for binding to the CC2-LZ domain target the native, full-length NEMO in vivo
Our next step was to determine whether the best NF-κB inhibitor 2F6 binds to the native, full-length NEMO in cell culture and whether its inhibition of the NF-κB pathway depends on NEMO interaction. To address this question, we developed a fluorescence-based assay in which we expressed a Flag-2F6 ankyrin construct and a EGFP-NEMO fusion protein in 293T cells. We previously showed, using a genetic complementation assay (Fontan et al. 2007), that fluorescent EGFP protein fused to the N terminus of NEMO does not interfere with NEMO function. As a control, we transiently cotransfected the EGFP vector alone with the Flag-2F6 ankyrin (▶). At 24 h post-transfection, the cotransfected cells were lysed, and Flag-2F6 was immunoprecipitated from crude extract with Flag antibodies bound to protein G immobilized on Sepharose beads. The Flag ankyrins were eluted with an excess of Flag-peptide, and the presence of ankyrin was evaluated by measuring fluorescence (▶) or by performing a Western blot using NEMO antibodies (▶). As shown in ▶, when cells were cotransfected with EGFP-NEMO and Flag-ankyrin 2F6, the intensity of fluorescence was fivefold higher than the control coexpressing GFP and Flag-ankyrin 2F6. This shows that the 2F6 ankyrin specifically interacts with the EGFP-NEMO protein in vivo. This interaction was also confirmed by analyzing the Flag peptide eluates by Western blotting using anti-NEMO antibodies (▶).
Figure 5.

NF-κB inhibition by DARPin occurs through the specific interaction with NEMO. (A,B) Specific binding of 2F6 to GFP-NEMO. 293 HEK cells were transfected with the indicated expression vectors. After 24 h, cell lysates were prepared and an anti-Flag immunopurification experiment was performed using anti-Flag antibody prior to the elution by Flag peptide. The peptide eluate was then analyzed by (A) fluorescence at 535 nm or by (B) immunoblot with anti-NEMO antibodies. For the immunoblot, equivalent amounts of input (one-fourth of the eluates) were loaded on each lane of the SDS-PAGE gel. (C) 2F6 and 1D5 bind to YFP-NEMO through the specific recognition of the CC2-LZ domain. An experiment similar to that described for B was performed after transient transfection of 293 HEK cells with the indicated Flag-DARPins in combination with YFP-NEMO or YFP-NEMO deleted of the CC2-LZ domain (YFP-NEMO Mu). (IB) Immunoblot; (IP) immunoprecipitate. (D) 2F6 and 1D5 specifically bind to the endogenous NEMO. The indicated Flag-DARPins or the Flag naive DARPin (Co.) were individually immunopurified after transient expression in 293 HEK cells as described in A. The eluted DARPins were then incubated with crude extracts prepared from MEFs or NEMO-deficient MEFs, and a second anti-Flag immunoprecipitation experiment was performed. The resulting protein complexes were subjected to an immunoblot (IB) analysis using the indicated antibodies. (HC) Heavy chain antibody; (E) whole-cell extracts.
To further verify the specificity of the interaction with the full-length NEMO, and to evaluate whether the lack of NF-κB inhibition by 1D5 was due to the lack of interaction with NEMO, similar experiments were performed with the inhibitory DARPin 2F6 and the noninhibitory DARPin 1D5 coexpressed together with NEMO (EYFP-NEMO) or a variant of NEMO deleted of the CC2-LZ domain (EYFP-NEMO Mu). As shown in ▶, specific binding to EYFP-NEMO was observed with both DARPins, whereas no interaction was detected with EYFP-NEMO Mu, indicating that both DARPins specifically bind to NEMO through the specific recognition to the CC2-LZ domain. Furthermore, since the DARPin–NEMO interaction was detected under overexpression conditions in HEK293T cells, we next investigated if similar interaction can occur with the endognous NEMO expressed at the physiological level. To this end, the Flag-DARPin 2F6, 1D5, or the naive DARPin (Co.) were individually immunopurified from the HEK293T cells and incubated with crude extracts from mouse embryonic fibroblasts (MEFs) or from NEMO-deficient MEFs (control). The interaction with the endogenous NEMO was then monitored by Western blot using anti-NEMO (▶). Again, we observed a specific binding to NEMO with both Flag-DARPins 2F6 and 1D5 in MEFs, whereas no interaction was detected either with the naive DARPin in MEFs or with 2F6 and 1D5 in NEMO-deficient MEFs. This interaction was specific because the NRP protein, which displays a significant homology with the NEMO CC2-LZ, was not able to bind to any DARPins (▶). Taken together, these results demonstrate that, although 2F6 and 1D5 specifically interact with the endogenous NEMO, only 2F6 DARPin was able to inhibit the NF-κB pathway by interfering with NEMO function.
Discussion
NEMO is a crucial regulatory protein of ∼50 kDa that modulates signal-induced NF-κB activation. Like many signaling proteins, it exhibits a flexible and modular architecture that can adopt several conformations. In this study, we selected designed ankyrin-repeat proteins (DARPins) with high affinity against the individual CC2-LZ, which serves as a minimal oligomerization domain (Tegethoff et al. 2003; Agou et al. 2004b), and, more recently, as a specific K63-linked-polyubiquitin chain-binding domain (Ea et al. 2006; Wu et al. 2006). The specific interaction of DARPins with isolated CC2-LZ was shown in vitro by direct as well as by competition ELISA assays using a synthetic domain and purified proteins. In addition, the specific binding of the DARPins to the whole NEMO protein was demonstrated in vivo by immunoprecipitation, indicating that the isolated CC2-LZ domain adopts a conformation very similar to that of the CC2-LZ domain within the entire NEMO protein. We also showed that some, but not all DARPins, specifically inhibit the TNF-α-induced NF-κB activation in an NF-κB-dependent reporter-gene assay.
Several DARPins showed high affinity constants of 100 nM or better. Among these DARPins, 1D5 binds to CC2-LZ with a high affinity of 22 nM. However, this DARPin does not affect NF-κB activation when expressed in human cells. In contrast, 2A1 and 2F6 DARPins reduced the level of TNF-α-induced NF-κB activation by 2.5- and fivefold, respectively. In the case of these two DARPins, the inhibition correlates with the in vitro affinity for CC2-LZ, since both the affinity and inhibition strength of 2F6 are stronger than those of 2A1. The absence of inhibition for 1D5, as compared to 2F6 and 2A1, may result from a different mode of binding to CC2-LZ. Interestingly, modeling showed that the inhibitory DARPins 2F6 and 2A1 belong to the same structural class as judged by the hydrophobic content of the ankyrin interface, while the ankyrin interface of 1D5 is more hydrophilic. We thus hypothesize that these two ankyrin classes recognize different epitopes or different conformations of the CC2-LZ domain. In the light of the structural models shown in ▶, it is tempting to speculate that the inhibition strength is linked to the hydrophobic nature of the DARPin interface. In this context, DARPins with a predominantly hydrophobic interface may prevent hydrophobic protein–protein interactions that govern either NEMO oligomerization or K63-polyubiquitin-binding. However, although the hydrophobic content of the interface is of similar size, 2A1 and 2F6 DARPins may have a very different mode of action. Indeed, one DARPin might inhibit NF-κB activation by blocking NEMO oligomerization, while the other might alter NEMO function by disrupting its binding to K63-polyubiquitin chains. Further studies are needed to clarify the mechanism of action of these ankyrins that also shed light on the NEMO-dependent mechanism of IKK complex activation.
The effect of DARPin expression in human cells and a possibly inhibitory action has, so far, never been reported. Upon expression in human 293T cells or HeLa cells, we systematically observed partial degradation of the inhibitory DARPins 2A1 and 2F6. This DARPin degradation was not observed with a naive ankyrin or with the 1D5 DARPin, which has no ability to inhibit TNF-α-induced NF-κB activation although it also binds to the CC2-LZ in vitro nanomolar affinity. This specific partial DARPin degradation of 2A1 and 2F6 could reflect an important control mechanism to prevent inhibition of the NEMO-dependent NF-κB pathway. We hypothesize that the degradation could be a consequence of the ubiquitin-dependent proteosomal and/or lysosomal degradation pathways. However, we were not able to impair this partial DARPin proteolysis by cell treatment with lactacystin-β-lactone, a specific proteasome inhibitor, or with bafilomycin A, a lysosome inhibitor. Similarly, an inhibitor of autophagy (3-methyladenine) (Kroemer and Jaattela 2005) did not affect DARPin degradation in vivo, indicating that the susceptibility of DARPin inhibitors to degradation is linked to other not-yet-identified intracellular proteolytic enzymes. A partial degradation of NEMO-derived peptides was also observed when we transduced cell-permeable NEMO inhibitors into the cells (Agou et al. 2004a). However, the peptide degradation following its internalization was less pronounced than that of a similar peptide produced by DNA transfection (data not shown). Similarly, we hypothesize that the susceptibility of DARPins to proteolysis differs when the DARPin is directly transduced into the cells or when it is produced by DNA transfection. To make the 2F6 DARPin permeable to the plasma membrane, we purified from E. coli a variant of 2F6 with an N-terminal tag containing the nona-arginine cell-permeable sequence (R9). Unfortunately, the presence of this polyarginine tag results in a strong loss of solubility so that we were unable to evaluate the inhibition strength of 2F6 by transduction.
Finally, the results presented here confirm the previous approach showing that peptides binding to the CC2-LZ serve as effective inhibitors of the NF-κB pathway. Beyond the pharmacological interest for searching specific inhibitors of the pathway, DARPins with affinities from nanomolar to micromolar may also provide benefits to help the crystallization of the CC2-LZ domain. To date, crystallization of this individual domain has been unsuccessful, presumably because of its flexibility. It is generally considered that rigid protein structures are easier to crystallize and form better diffraction-quality crystals. Binding of DARPins to the CC2-LZ domain may lead to a more rigid structure of the CC2-LZ, thereby facilitating its crystallization. In this context, the inhibitory 2F6 and 2A1 DARPins are more attractive than 1D5, as these DARPins induce an inactive conformation of the CC2-LZ. Structure determination of DARPin inhibitor complexed with CC2-LZ would provide an important structural basis to search for small molecules antagonizing NEMO oligomerization and/or K63-linked polyubiquitin-chain binding.
Materials and Methods
Ribosome display
The selection was initiated using a naive DARPin N2C library (Binz et al. 2004). Ribosome display selection rounds were essentially performed as previously described (Binz et al. 2004; Zahnd et al. 2007).
Analysis of selected binders
After selection with the ribosome display method, individual binders were isolated from the pool by cloning cDNA into pQE-30 (QIAGEN). After transformation in E. coli XL1-blue, single clones were isolated and expression was induced with IPTG for 4 h. After cell lysis with B-PER II (Pierce), the crude extract was used for ELISA to test specific and unspecific binding on CC2-LZ/Neutravidin/BSA and Neutravidin/BSA, respectively. Antibodies against RGS-His6 (QIAGEN) and anti-mouse-Ig conjugated to alkaline phosphatase (Pierce) were used for detection. For competition ELISA, crude extracts were preincubated with non-biotinylated CC2-LZ for 1 h at 4°C prior to competition. Quantitative ELISAs were performed to determine binding constants as described above, except that various concentrations of purified DARPins were used.
Purification of DARPins for affinity determination
DARPins were expressed in E. coli XL1-blue in 500 mL of LB/1% glucose with 50 μg/mL ampicillin. Cells were induced with 1 mM IPTG at OD600 = 0.6–0.8 for 4 h at 37°C. The cells were harvested by centrifugation and lysed using a French press. After purification with Ni-NTA-Agarose (QIAGEN) on an Äkta system, 10% glycerol was added, and the proteins were stored at −80°C for further use.
Protein modeling and bioinformatics
The structural models of the 1D5, 2F6, and 2A1 DARPins were built by homology modeling from the X-ray structure of E3_5 N3C ankyrin (PDB:1MJ0) using the InsightII98 software package (Accelrys Inc.). Energy minimization was performed with the GROMOS96 parameter set (van Gunsteren et al. 1996). The models were evaluated using the program PROCHEK (Laskowski et al. 1993). PyMOL was used for structural alignment and molecular display (DeLano 2002).
Electronic supplemental material
Descriptions of synthesis and purification of biotinylated CC2-LZ, preparation of microtiter plates for ELISA, ribosome display, NF-κB reporter assay in human cells, immunopurification, Western blotting, and fluorescence quantification are available in the Supplemental material.
Acknowledgments
We thank F. Traincard for providing the YFP-NEMO plasmids and J.M. Betton, F. Pecorari, P. Forrer, and A. Plückthun for their very valuable technical support and fruitful discussions; R. Weil and A. Israël for constant support; and O. Grubisha for critical review of this manuscript. This work was supported in part by grants from the Association pour la Recherche sur le Cancer, the Cancéropôle Ile de France, the BNP-Paribas Foundation, and the Direction des Applications de la Recherche et Relations Industrielles at the Pasteur Institute. M.K. is supported by a Bourse Roux fellowship from the Pasteur Institute.
Footnotes
Supplemental material: see www.proteinscience.org
Reprint requests to: Fabrice Agou, Unité Régulation Enzymatique des Activités Cellulaires, Institut Pasteur, CNRS URA 2185, 25-28 rue du Dr. Roux, 75724 Paris Cedex 15, France; e-mail: fagou@pasteur.fr; fax: 33-1-33145688399.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.072924907.
References
- Agou F., Ye, F., Goffinont, S., Courtois, G., Yamaoka, S., Israel, A., and Veron, M. 2002. NEMO trimerizes through its coiled-coil C-terminal domain. J. Biol. Chem. 277 17464–17475. [DOI] [PubMed] [Google Scholar]
- Agou F., Courtois, G., Chiaravalli, J., Baleux, F., Coic, Y.M., Traincard, F., Israel, A., and Veron, M. 2004a. Inhibition of NF-κB activation by peptides targeting NEMO oligomerization. J. Biol. Chem. 279 54248–54257. [DOI] [PubMed] [Google Scholar]
- Agou F., Traincard, F., Vinolo, E., Courtois, G., Yamaoka, S., Israel, A., and Veron, M. 2004b. The trimerization domain of NEMO is composed of the interacting C-terminal CC2 and LZ coiled-coil subdomains. J. Biol. Chem. 279 27861–27869. [DOI] [PubMed] [Google Scholar]
- Binz H.K., Stumpp, M.T., Forrer, P., Amstutz, P., and Pluckthun, A. 2003. Designing repeat proteins: Well-expressed, soluble, and stable proteins from combinatorial libraries of consensus ankyrin-repeat proteins. J. Mol. Biol. 332 489–503. [DOI] [PubMed] [Google Scholar]
- Binz H.K., Amstutz, P., Kohl, A., Stumpp, M.T., Briand, C., Forrer, P., Grutter, M.G., and Pluckthun, A. 2004. High-affinity binders selected from designed ankyrin-repeat protein libraries. Nat. Biotechnol. 22 575–582. [DOI] [PubMed] [Google Scholar]
- Binz H.K., Kohl, A., Pluckthun, A., and Grutter, M.G. 2006. Crystal structure of a consensus-designed ankyrin-repeat protein: Implications for stability. Proteins 65 280–284. [DOI] [PubMed] [Google Scholar]
- Bonizzi G. and Karin, M. 2004. The two NF-κB activation pathways and their role in innate and adaptive immunity. Trends Immunol. 25 280–288. [DOI] [PubMed] [Google Scholar]
- Carvalho G., Fabre, C., Braun, T., Grosjean, J., Ades, L., Agou, F., Tasdemir, E., Boehrer, S., Israel, A., Veron, M., et al. 2006. Inhibition of NEMO, the regulatory subunit of the IKK complex, induces apoptosis in high-risk myelodysplastic syndrome and acute myeloid leukemia. Oncogene 26 2299–2307. [DOI] [PubMed] [Google Scholar]
- Claudio E., Brown, K., Park, S., Wang, H., and Siebenlist, U. 2002. BAFF-induced NEMO-independent processing of NF-κB2 in maturing B cells. Nat. Immunol. 3 958–965. [DOI] [PubMed] [Google Scholar]
- DeLano W.L. 2002. The PyMOL user's guide. DeLano Scientific LLC, San Carlos, CA, http://www.pymol.org.
- Ea C.K., Deng, L., Xia, Z.P., Pineda, G., and Chen, Z.J. 2006. Activation of IKK by TNFα requires site-specific ubiquitination of RIP1 and polyubiquitin binding by NEMO. Mol. Cell 22 245–257. [DOI] [PubMed] [Google Scholar]
- Filipe-Santos O., Bustamante, J., Haverkamp, M.H., Vinolo, E., Ku, C.L., Puel, A., Frucht, D.M., Christel, K., von Bernuth, H., Jouanguy, E., et al. 2006. X-linked susceptibility to mycobacteria is caused by mutations in NEMO impairing CD40-dependent IL-12 production. J. Exp. Med. 203 1745–1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontan E., Traincard, F., Levy, S.G., Yamaoka, S., Véron, M., and Agou, F. 2007. NEMO oligomerization in the dynamic assembly of the IκB kinase core complex. FEBS J. 274 2540–2551. [DOI] [PubMed] [Google Scholar]
- Hacker H. and Karin, M. 2006. Regulation and function of IKK and IKK-related kinases. Sci. STKE 2006 re13. doi: 10.1126/stke.3572006re13. [DOI] [PubMed] [Google Scholar]
- Hanahan D. and Weinberg, R.A. 2000. The hallmarks of cancer. Cell 100 57–70. [DOI] [PubMed] [Google Scholar]
- Hayden M.S. and Ghosh, S. 2004. Signaling to NF-κB. Genes & Dev. 18 2195–2224. [DOI] [PubMed] [Google Scholar]
- Hosse R.J., Rothe, A., and Power, B.E. 2006. A new generation of protein display scaffolds for molecular recognition. Protein Sci. 15 14–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang T.T., Wuerzberger-Davis, S.M., Wu, Z.H., and Miyamoto, S. 2003. Sequential modification of NEMO/IKKγ by SUMO-1 and ubiquitin mediates NF-κB activation by genotoxic stress. Cell 115 565–576. [DOI] [PubMed] [Google Scholar]
- Israel A. 2000. The IKK complex: An integrator of all signals that activate NF-κB? Trends Cell Biol. 10 129–133. [DOI] [PubMed] [Google Scholar]
- Karin M. 2006. Nuclear factor-κB in cancer development and progression. Nature 441 431–436. [DOI] [PubMed] [Google Scholar]
- Kim H.J., Hawke, N., and Baldwin, A.S. 2006. NF-κB and IKK as therapeutic targets in cancer. Cell Death Differ. 13 738–747. [DOI] [PubMed] [Google Scholar]
- Kroemer G. and Jaattela, M. 2005. Lysosomes and autophagy in cell death control. Nat. Rev. Cancer 5 886–897. [DOI] [PubMed] [Google Scholar]
- Laskowski R.A., MacArthur, M.W., Moss, D.S., and Thornton, J.M. 1993. PROCHECK—A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26 283–291. [Google Scholar]
- Marienfeld R.B., Palkowitsch, L., and Ghosh, S. 2006. Dimerization of the IκB kinase-binding domain of NEMO is required for tumor necrosis factor α-induced NF-κB activity. Mol. Cell. Biol. 26 9209–9219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poyet J.-L., Srinivasula, S.M., Lin, J.-H., Fernandes-Alnemri, T., Yamaoka, S., Tsichlis, P.N., and Alnemri, E.S. 2000. Activation of the IκB kinases by RIP via IKKγ/NEMO-mediated oligomerization. J. Biol. Chem. 275 37966–37977. [DOI] [PubMed] [Google Scholar]
- Rothwarf D.M., Zandi, E., Natoli, G., and Karin, M. 1998. IKK-γ is an essential regulatory subunit of the IκB kinase complex. Nature 395 297–300. [DOI] [PubMed] [Google Scholar]
- Sedgwick S.G. and Smerdon, S.J. 1999. The ankyrin repeat: A diversity of interactions on a common structural framework. Trends Biochem. Sci. 24 311–316. [DOI] [PubMed] [Google Scholar]
- Tegethoff S., Behlke, J., and Scheidereit, C. 2003. Tetrameric oligomerization of IκB kinase γ (IKKγ) is obligatory for IKK complex activity and NF-κB activation. Mol. Cell. Biol. 23 2029–2041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Gunsteren W.F., Billeter, S.R., Eising, A.A., Hünenberger, P.H., Krüger, P., Mark, A.E., Scott, W.R.P., and Tironi, I.G. 1996. Biomolecular simulation: The GROMOS manual and user guide. ETH Zürich, vdf Hochschulverlag, Switzerland.
- Wu C.J., Conze, D.B., Li, T., Srinivasula, S.M., and Ashwell, J.D. 2006. Sensing of Lys 63-linked polyubiquitination by NEMO is a key event in NF-κB activation. Nat. Cell Biol. 8 398–406. [DOI] [PubMed] [Google Scholar]
- Yamaoka S., Courtois, G., Bessia, C., Whiteside, S.T., Weil, R., Agou, F., Kirk, H.E., Kay, R.J., and Israel, A. 1998. Complementation cloning of NEMO, a component of the IκB kinase complex essential for NF-κB activation. Cell 93 1231–1240. [DOI] [PubMed] [Google Scholar]
- Zahnd C., Amstutz, P., and Pluckthun, A. 2007. Ribosome display: Selecting and evolving proteins in vitro that specifically bind to a target. Nat. Methods 4 269–279. [DOI] [PubMed] [Google Scholar]