

Abstract
The tissue inhibitors of metalloproteinases (TIMPs) are endogenous inhibitors of the matrix metalloproteinases (MMPs). Since unregulated MMP activities are linked to arthritis, cancer, and atherosclerosis, TIMP variants that are selective inhibitors of disease-related MMPs have potential therapeutic value. The structures of TIMP/MMP complexes reveal that most interactions with the MMP involve the N-terminal pentapeptide of TIMP and the C–D β-strand connector which occupy the primed and unprimed regions of the active site. The loop between β-strands A and B forms a secondary interaction site for some MMPs, ranging from multiple contacts in the TIMP-2/membrane type-1 (MT1)-MMP complex to none in the TIMP-1/MMP-1 complex. TIMP-1 and its inhibitory domain, N-TIMP-1, are weak inhibitors of MT1-MMP; inhibition is not improved by grafting the longer AB loop from TIMP-2 into N-TIMP-1, but this change impairs binding to MMP-3 and MMP-7. Mutational studies with N-TIMP-1 suggest that its weak inhibition of MT1-MMP, as compared to other N-TIMPs, arises from multiple (>3) sequence differences in the interaction site. Substitutions for Thr2 of N-TIMP-1 strongly influence MMP selectivity; Arg and Gly, that generally reduce MMP affinity, have less effect on binding to MMP-9. When the Arg mutation is added to the N-TIMP-1(AB2) mutant, it produces a gelatinase-specific inhibitor with Ki values of 2.8 and 0.4 nM for MMP-2 and -9, respectively. Interestingly, the Gly mutant has a Ki of 2.1 nM for MMP-9 and >40 μM for MMP-2, indicating that engineered TIMPs can discriminate between MMPs in the same subfamily.
Keywords: matrix metalloproteinase, metalloproteinase inhibition, protein engineering, inhibitory specificity, membrane-type matrix metalloproteinase, interaction interface
The matrix metalloproteinases (MMPs) are metalloendopeptidases that catalyze the proteolysis of extracellular matrix (ECM) and cell surface proteins. Their activities affect cell growth, migration, apoptosis, and cell–cell communication (McCawley and Matrisian 2001). The 23 MMPs of humans can be subdivided into collagenases, gelatinases, stromelysins, matrilysins, and membrane type MMPs, MT-MMPs (Page-McCaw et al. 2007). MMPs share a catalytic domain with a zinc-binding motif, but the subgroups differ in content of hemopexin, fibronectin, and transmembrane domains (Page-McCaw et al. 2007); classification into subgroups is based on substrate specificity and domain structure.
Gelatinases A (MMP-2) and B (MMP-9) are of special interest in cancer biology because of their roles in tumor invasion and angiogenesis (Brooks et al. 1996, 1998; Coussens et al. 2000). They are unique in having three fibronectin type II domains (FnII) inserted within the catalytic domain that facilitate binding of collagen-like substrates (Steffensen et al. 1995). They have closely similar active site clefts and typical MMP folds, but their S1′ specificity pockets are larger than those of other MMPs, particularly MMP-1 and MMP-7 (Elkins et al. 2002; Rowsell et al. 2002). A distinct feature of MMP-9 is Arg424, at the base of the S1′ pocket, that can adopt different conformations, allowing the accommodation of groups of varying size in substrates and inhibitors (Rowsell et al. 2002). The gelatinases also differ in the orientation of the second FnII insert in the catalytic domain; in MMP-2 it has substantial interactions with the rest of the domain in MMP-2, but not in MMP-9 (Elkins et al. 2002).
The MMPs are part of the larger metzincin metalloproteinase clan (Bode et al. 1996). The activities of MMPs and some other metzincins, specifically some disintegrin-metalloproteinases (ADAMs) and disintegrin-metalloproteinases with thrombospondin type I domains (ADAMTS), are tightly regulated by four endogenous inhibitors, the tissue inhibitors of metalloproteinases (TIMPs-1 to -4). The four mammalian TIMPs are generally broad-spectrum inhibitors of MMPs, with inhibition constants (Ki) in the low to subnanomolar range (Brew et al. 2000). An exception is the weak inhibition of certain MT-MMPs and MMP-19 by TIMP-1, in contrast to their strong inhibition by the other TIMPs (Will et al. 1996; Butler et al. 1997; Kolkenbrock et al. 1999; Shimada et al. 1999; Bigg et al. 2001). TIMPs have two domains, a larger N-terminal domain that carries the MMP-inhibitory activity, and a smaller C-domain that mediates other interactions, notably non-inhibitory interactions of TIMP-1 with pro-MMP-9 and of TIMP-2 with pro-MMP-2 (Brew et al. 2000). Truncated N-terminal domains (N-TIMPs) fold correctly and carry the full MMP-inhibitory activity of the parent protein. Their smaller size and fewer interactions make them amenable for solution structural studies and potentially more available in vivo.
Crystal structures have been determined for TIMP-1 complexed with the MMP-3 catalytic domain (CD) (Gomis-Ruth et al. 1997), N-TIMP-1 bound to the MMP-1 CD (Iyer et al. 2007), and for TIMP-2 bound to the CDs of MT1-MMP (Fernandez-Catalan et al. 1998) and MMP-13 (Maskos et al. 2007). These reveal similar inhibitory mechanisms in which a region surrounding the Cys1–Cys70 disulfide bond (TIMP-1 residue numbering) accounts for ∼75% of the contacts with the MMP. The conserved Cys1 of the TIMP coordinates the catalytic Zn2+ through bidentate interactions with its α-amino and carbonyl groups while the side chains of residues 2 and 4 interact with the S1′ and S3′ subsites, respectively; substitutions for residues 2 and 4 differentially affect the inhibitory activities of N-TIMPs for different metzincins (Butler et al. 1999; Meng et al. 1999; Lee et al. 2002a; Wei et al. 2003, 2005). The inhibitory domain has a five-stranded β-barrel fold with a Greek key topology (an OB-fold) and the connector between β-strands C and D also forms part of the MMP interaction site (▶). Also, the loop between β-strands A and B (the AB loop) makes varying contacts with the MMP in different complexes (Gomis-Ruth et al. 1997; Fernandez-Catalan et al. 1998; Iyer et al. 2007; Maskos et al. 2007). Previous studies have shown that TIMP specificity is also affected by mutations in the AB loop (Butler et al. 1999; Williamson et al. 2001) and substitutions for Ser68 and Thr98 (TIMP-1 sequence numbering; Lee et al. 2002a, 2003, 2004; Wei et al. 2003).
Figure 1.

Superimposed structures of N-TIMP-1 and N-TIMP-2. Ribbon representation of the N-terminal inhibitory domains of TIMP-1 colored orange (PDB code: 2JOT); (Iyer et al. 2007) and TIMP-2 colored green (PDB code: 1BQQ); (Fernandez-Catalan et al. 1998). The AB loop as well as the first six residues of the two proteins (shown as sticks in respective colors) have also been highlighted. The figure was generated using PyMOL (http://www.pymol.org).
Differences in contacts and chemistry within the interfaces of N-TIMP-1 complexes provide a basis for engineering inhibitory selectivity. Here, we describe the effects of changes in the N-terminal 6 residues and AB loop on binding to different MMPs. By combining mutations we have engineered a N-TIMP-1 variant that is highly selective for gelatinases, particularly MMP-9. A previously characterized Thr2 to Gly, mutation that largely abrogates the inhibitory activity for most MMPs (Meng et al. 1999; Wei et al. 2005), is found to be a good inhibitor of MMP-9 that is 2,500-fold more selective for MMP-9 over MMP-2.
Results
Role of AB loop in N-TIMP-1 inhibitory specificity
A comparison of structures of TIMP/MMP complexes indicates that there are major differences in the role of the TIMP AB loop in interface interactions. The longer AB loop in TIMP-2 makes extensive interactions with MT1-MMP and with MMP-13 (Fernandez-Catalan et al. 1998; Maskos et al. 2007), but the specific interactions differ in the two complexes. Also, the relatively short loop of TIMP-1 makes 17 contacts with MMP-3 but none with MMP-1 in their complex (Iyer et al. 2007). Mutational studies suggest that contacts with this loop are important for the high affinity binding of TIMP-2 to MT1-MMP, suggesting that differences in AB loop length might explain the weak inhibition of MT1-MMP by TIMP-1 (Williamson et al. 2001). However, TIMP-3, which has an AB loop similar in length to TIMP-1, is a good MT1-MMP inhibitor.
We investigated the role of the AB loop structure in selectivity of N-TIMP-1 by generating the N-TIMP-1(AB2) and N-TIMP-1(AB3) mutants (see ▶) in which the AB loop of N-TIMP-1 is replaced with those of TIMP-2 or TIMP-3, respectively. Both mutants have subnanomolar Ki values for MMP-1, indicating that the overall structure is intact, but are poor inhibitors of MT1-MMP, the AB2 mutant being similar in affinity to wild-type N-TIMP-1 while the AB3 mutant is a >60-fold weaker inhibitor (▶). The N-TIMP-1(AB2) mutant has essentially unchanged affinity for MMP-1 and MMP-2 but shows a 60-fold increase in the Ki for MMP-3 CD (▶). In contrast, N-TIMP-1(AB3) has an essentially unchanged Ki for MMP-3. This suggests that the AB2 loop disrupts the interaction with MMP-3, whereas the N-TIMP-1/MMP-1 interface can accommodate the extended loop. Also, in N-TIMP-1, the extended loop may not make the more extensive contacts seen in the MT1-MMP/TIMP-2 complex. The effect of changes in the AB loop provides an approach for designing N-TIMP-1 variants that are selective for certain MMPs.
Table 1.
Comparison of Ki values for wild-type N-TIMPs, N-TIMP-1 AB loop mutants, and combined mutants against different MMPs

Combination of AB2 with T2L/V4S or T2R mutations generates N-TIMP-1 variants that are selective for gelatinases
We previously identified a variant of N-TIMP-1 (T2L/V4S) that has enhanced selectivity for MMP-2 (Wei et al. 2003). Studies with additional MMPs show that this variant is also a potent inhibitor for matrilysin-1/MMP-7 and MMP-9 (▶). A combination of this mutation with the AB loop from TIMP-2 yielded a novel N-TIMP-1 variant that has Ki values in the subnanomolar range for both gelatinases and MMP-7, but lower affinity for MMP-1, MMP-3, and MT1-MMP (▶).
Investigation of the activity of a previously constructed mutant, T2R, with more MMPs showed that it is essentially inactive as an inhibitor of MMP-7, even at micromolar concentrations, but is a reasonably good inhibitor of MMP-9 with a Ki of 9 nM (▶); the bulky, cationic side chain of the arginine residue appears to disfavor N-TIMP-1 interactions with MMPs that have narrow or positively charged S1′ pockets (Meng et al. 1999). Modeled complexes of the T2R mutant with MMP-1 and -3 indicate that there are fewer contacts and H-bonds in the MMP-1 complex as well as structural rearrangements that may have unfavorable effects on the entropy of binding (Iyer et al. 2007). The weak inhibition of MMP-7 is not surprising since the S1′ pocket of MMP-7 is one of the narrowest among MMPs (Browner et al. 1995) whereas those of MMP-2 and MMP-9 are much wider and can accommodate larger side chains (Morgunova et al. 1999; Rowsell et al. 2002). Although the AB2 mutant has a slightly lower affinity than wild-type N-TIMP-1 for MMP-2, MMP-7, and MMP-9, when this mutation is combined with the T2R substitution, it enhances inhibition of these three MMPs. The resulting multisite mutant, T2R/AB2, is a gelatinase-specific inhibitor, with MMP-9/gelatinase B being favored over MMP-2/gelatinase A, and is at least two orders of magnitude less active for all other MMPs. (▶; ▶).
Figure 3.
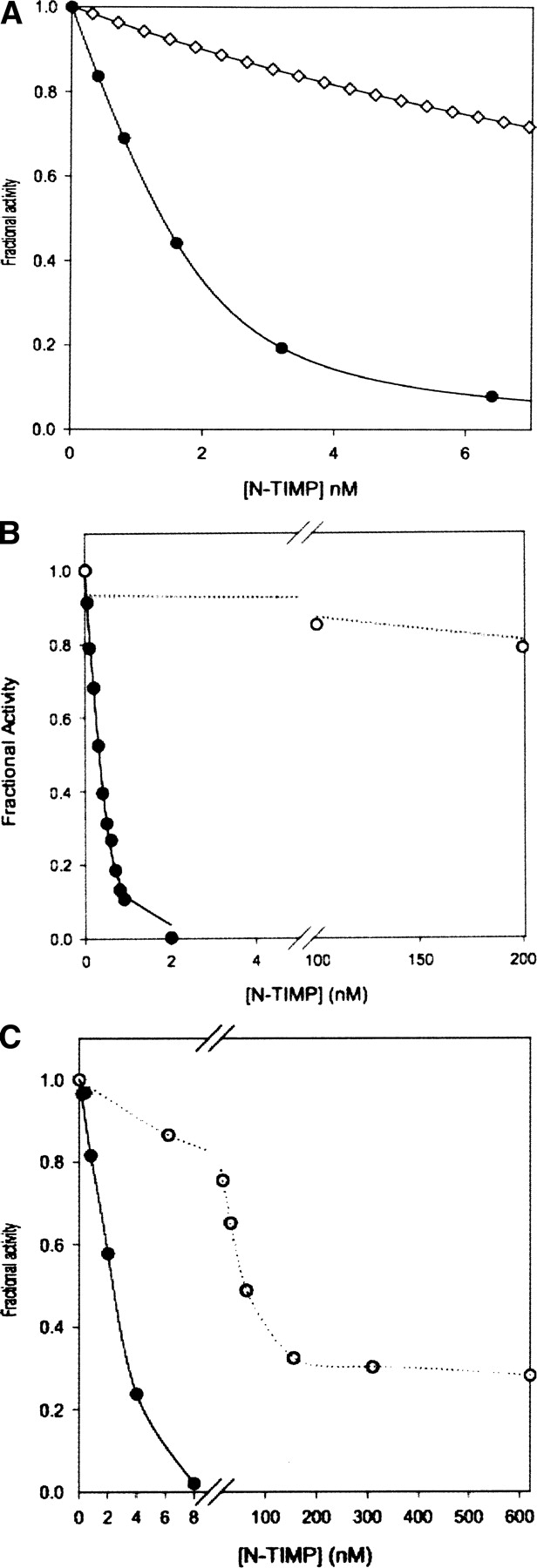
Inhibition profiles of selected N-TIMP-1 variants with different MMPs. (A) Inhibition of MMP-3(CD) by wild-type N-TIMP-1 (•) and N-TIMP-1(AB2) (⋄). (B) Inhibition of MMP-9 (•) and MMP-7 (○) by N-TIMP-1 T2R/AB2 mutant. (C) Inhibition of MT1-MMP (○) and MMP-1 (•) by N-TIMP-1 P5A mutant.
The T2G mutant is a good inhibitor of MMP-9
As discussed above, residue 2 of TIMP interacts with the S1′ pocket of MMPs (Gomis-Ruth et al. 1997; Fernandez-Catalan et al. 1998; Iyer et al. 2007; Maskos et al. 2007) and has a major influence on affinity for different MMPs. Substitution of Thr2 by glycine, which has no side chain, increases the Ki value of the N-TIMP-1 and -3 for MMP-1, MMP-2, MMP-3, and MT1-MMP by at least two to three orders of magnitude (▶; Meng et al. 1999; Wei et al. 2005). However, the same mutation has small effects on the inhibitory activity of N-TIMP-3 for ADAM17/TACE (Wei et al. 2005). Here, we find that this mutation in TIMP-1 has a much smaller ∼20-fold effect on the inhibition of MMP-7 and MMP-9 (▶). In contrast to N-TIMP-1 and other mutants described above, which have similar Ki values for MMP-2 and MMP-9 (▶), the T2G mutant has a >2500-fold higher affinity for MMP-9 relative to MMP-2, showing that TIMP-1 can be engineered to select between the two gelatinases. Similar results were obtained with N-TIMP-3 (data not shown), suggesting that the T2G mutation may have similar effects on activity in all TIMPs. Combination with the AB2 mutation did not improve the selectivity, since the AB2 loop generally increased the affinity for those enzymes that are poorly inhibited by the T2G mutant but not on the inhibition of MMP-7 or MMP-9 (▶). This suggests that looser interactions between the N-TIMP-1 mutant and the S1′ pocket of the protease can be partially compensated by stronger interactions with the extended AB loop, analogous to effects seen in other protease-protein inhibitor systems (Gillmor et al. 2000).
A comparison of the Ki values for mutants with the AB2 loop substitution plus other mutations with those calculated by adding together their effects on the free energy of binding (▶) shows that some combinations have synergistic rather than additive effects. Most notably, the AB2 loop mitigates the effect of the T2G and T2R mutation on binding to certain MMPs. In the case of the T2G mutant this compensation is particularly strong for MMP-1, -2, and -3 inhibition while, for the T2R mutant, MMP-3, MMP-7, and MMP-9 are most affected.
The hemopexin domain and the FnII inserts of MMP-9 do not contribute to the interaction with N-TIMP-1
To investigate the structural basis for relatively high affinity of the T2G mutant for MMP-9 we compared the inhibitory activity of this mutant toward full-length MMP, the isolated catalytic domain (MMP-9[CD]), and a truncated form of the catalytic domain with the FnII repeats deleted (MMP-9[CD-FN]). The inhibition of these forms of MMP-9 by wild-type N-TIMP-1 and the T2G and -1A mutants was also investigated. The latter mutation perturbs the interaction of Cys1 of TIMP with the catalytic zinc ion of the MMP and substantially reduces the affinity of TIMPs for different MMPs (Wingfield et al. 1999; Wei et al. 2005). Wild-type N-TIMP-1 and the mutants have similar Ki values for all three forms of MMP-9, suggesting that the C-terminal hemopexin domain of MMP-9 and the FnII inserts make no contribution to the interaction with N-TIMP-1 (▶).
Table 3.
Comparison of Ki(app) (nM) of N-TIMP-1 and two mutants (T2G and -1A) with different MMPs

This result indicates that the ability of the T2G mutant to select between the gelatinases does not relate to the difference in the position of their FnII inserts (Elkins et al. 2002). Deletion of this relatively large region (more than half of the catalytic domain) has little effect on the free energy of binding to wild-type N-TIMP-1, or either mutant (▶). Therefore, the much smaller effect of the T2G mutation on MMP-9 inhibition must reflect the interactions between the inhibitor and the main body of the catalytic domain.
The N-terminal alanine extension (-1A) produced a greater loss of inhibitory activity of N-TIMP-1 than the T2G mutation for all three forms of MMP-9, as well as MMP-7 (▶). This effect is similar to that observed previously for the corresponding mutation in N-TIMP-3 with different MMPs (Wei et al. 2005). Since both of these mutations are in the N-terminal region of N-TIMP-1, this observation suggests that the tighter binding of the N-TIMP-1(T2G) mutant to MMP-7 and MMP-9 may indicate that interactions with the S1′ specificity pocket are less important in stabilizing TIMP complexes with these two enzymes as compared with other MMPs.
Effects of substitutions adjacent to the Cys3–Cys99 disulfide bridge on inhibitory properties
TIMP-1 is unique in having proline at position 6 in its amino acid sequence. This produces a proline-rich region, V4PPHP8, that has a conformation different from the corresponding region of TIMP-2 (▶). Previous studies by Lee et al. (2003) indicate that the substitution of Leu for Thr98 alone greatly enhanced binding to MT1-MMP, producing a mutant with a Ki of about 11 nM. Thr98 is located above this section of the TIMP/MMP interface, adjacent to the Cys3–Cys99 disulfide bond. Additional substitutions for adjacent residues, Ala for Val4 and Val or Ala for Pro6, were found to further enhance binding to give a Ki of 1.7 nM. Under our experimental conditions, we find a more modest threefold improvement in Ki for the Thr98-to-Leu mutation, but our data support the general observation that substitution of more hydrophobic residues for Thr98 increase inhibitory activity toward MT1-MMP (▶). However, this is also accompanied by similar approximately threefold reductions in the Ki for MMP-3 and MMP-2. Combinations of V4A with P6A or P6V produced approximately additive effects on the free energy of binding, and the triple mutant, containing three mutations (V4A/P6V/T98L) that are favorable for MT1-MMP binding (▶), generated a variant with an ∼10-fold improvement in the Ki for this enzyme. However, this mutant remains a 20-fold weaker inhibitor for MT1-MMP than N-TIMP-2, indicating that the low affinity of TIMP-1 for MT1-MMP arises from multiple sequence changes in the interaction interface. As shown in ▶, while substitutions of Ala for Val4 or Val for Pro6 produce two- to threefold reductions in the Ki of N-TIMP-1 for MT1-MMP, the V4S and P6A mutations do not improve binding. As previously noted the V4A substitution also improves binding to MMP-3 (Wei et al. 2003). Our observations indicate that the V4S mutant is a less effective inhibitor of MT1-MMP than the wild-type inhibitor whereas Lee et al. (2003) find it to be slightly improved over wild type; this could possibly arise from differences in experimental conditions. We also characterized a P5A mutant that involves a residue that is conserved in all mammalian TIMPs. The inhibition data for this protein do not fit well to equations describing inhibition by an active site directed reversible enzyme inhibitor, particularly at higher concentrations of the mutant, and, instead, show that this mutant is a partial inhibitor of MT1-MMP with a maximum level of inhibition of ∼80%. Similar inhibition patterns were obtained by studies using different MT1-MMP preparations, and the results contrast with the data obtained for other MMPs (▶). These results suggest that this mutant is an exosite-type inhibitor that does not block the catalytic site but affects substrate binding.
Table 2.
Comparison of the effects of mutating residues 4–6 and T98, and combinations thereof on N-TIMP-1 affinity for MMPs-1, -2, -3, and MT1-MMP

Discussion
The four mammalian TIMP genes have originated by successive gene duplications and divergence in sequence and function. The weak inhibitory activity of TIMP-1 for MT1-MMP is an example of functional differentiation among the TIMPs. Although previous structural and mutational studies suggest a role for the AB loop of TIMP-2 in binding to MT1-MMP (Fernandez-Catalan et al. 1998; Williamson et al. 2001; Butler et al. 1999), the present study indicates that, when this loop is transferred to N-TIMP-1, it has no effect on affinity for MT1-MMP. The N-TIMP-1(AB2) mutant is of interest because of its selectively reduced affinity for MMP-3, which has provided a lead for engineering selective N-TIMP-1 variants that are weak inhibitors of MMP-3. Differences in the contributions by contacts between different regions of the binding site of TIMP-1 with various MMPs could explain the differing effects of the AB loop on Ki values (▶).
Previously, Lang et al. (2004) have discussed the basis of the weak inhibition of some MT-MMPs by TIMP-1. Their modeled complexes of TIMP-1 with MT1-MMP and MT3-MMP suggested possible clashes between Leu133 and Asn134 of the C-terminal domain of TIMP-1 with Asn234 and His235 of MT3-MMP and the corresponding region of MT1-MMP. However, this is inconsistent with the weak inhibition of MT1-MMP by both N-TIMP-1 and full-length TIMP-1, showing that the C-terminal domain is not a factor in MMP specificity. Lee et al. (2003) proposed that Thr98 is a key determinant of the weak inhibitory activity of N-TIMP-1 for MT1-MMP since substitution of Leu for Thr98 lowered the Ki for MT1-MMP from 178 to 11 nM. Our data confirm that the Thr98-to-Leu substitution improves MT1-MMP inhibition and indicates that the effect is only two- to threefold but also indicates that this mutation enhances binding to other MMPs, including MMP-3. The functional effects of the Thr98-to-Leu mutation are unlikely to arise from additional MMP interactions since the corresponding residue in TIMP-2 (Leu100) is >4 Å distant from MT1-MMP in their complex (Fernandez-Catalan et al. 1998). Previous calorimetric and NMR studies indicate that increased dynamics in the core of the N-TIMP-1 β-barrel contribute to the positive entropy change that drives its interaction with MMP-3 (Arumugam et al. 2003). Thr98 is perturbed in amide shift on binding MMP-3 (Arumugam et al. 2003) and it is conceivable that the Thr98-to-Leu mutation may promote the transmission of strain from the protein–protein interface to the N-TIMP-1 core, thereby raising the entropy of binding. Additional substitutions for Val4 and Pro6 enhance binding to MT1-MMP but even a triple mutant that also includes the Thr98-to-Leu substitution remains a much weaker inhibitor for MT1-MMP than N-TIMP-2, suggesting that the poor inhibition of MT1-MMP by TIMP-1 is a result of four or more substitutions, possibly linked to the structural differences between TIMP-1 and TIMP-2 in this region (▶).
Because gelatinases A and B have important roles in metastasis, tissue invasion, and angiogenesis in tumors, particularly breast cancer (Jinga et al. 2006), TIMP variants that selectively inhibit these enzymes are of interest for the development of treatments and for exploring the biological roles of MMPs. The catalytic domains of MMP-2 and MMP-9 have 64% sequence identity and similar substrate specificities. However, they also have distinct in vivo functions and unique substrates (McCawley and Matrisian 2001; Elkins et al. 2002) and contrasting physiological effects. Thus, platelet aggregation is stimulated by MMP-2 but inhibited by MMP-9 (Fernandez-Patron et al. 1999). Peptide substrates that have been identified by phage display are about 200-fold selective for MMP-2 over MMP-9, possibly reflecting differences in structure of their S2 pockets (Chen et al. 2002, 2003).
Previously we have shown that substitutions of amino acids with substitutions for residue 2 of N-TIMP-1 indicate that side chain size and charge affect the selectivity for MMPs with different depths and composition in their S1′ specificity pockets (Iyer et al. 2007). We find that the T2R and the T2G mutations have much less deleterious effects on binding to MMP-9 than to other MMPs. Interestingly, T2G is a weak inhibitor of MMP-2 but a good inhibitor of MMP-9, suggesting that interactions of MMP-9 with other regions the TIMP reactive site compensate for the loss of interactions between residue 2 and the S1′ pocket. A modeled structure of a complex of N-TIMP-1 (T2R) with MMP-9 suggests there are interactions of Glu67 and Val69 of the C–D connector of N-TIMP-1 with His411 and the phenyl ring of Phe107 of the MMP-9, respectively, that are absent from the TIMP-1/MMP-3 complex. Our results indicate that extended AB2 loop compensates for the loss of interactions in the T2G mutant and also enhances the binding of the T2R mutant with MMP-7 and MMP-9. The T2G and T2R(AB2) mutants developed in our study may be useful reagents for investigating the biological roles of the gelatinases and as possible leads for developing cancer therapies.
Materials and Methods
Materials
The plasmid pET-3a-timp-2 and the catalytic domain of MT1-MMP (MT1-MMP(CD)) were kindly provided by Dr. Hideaki Nagase (Kennedy Institute for Rheumatology, London). Macro-Prep Q-50 ion exchange resin and UNO Q1 columns were purchased from Bio-Rad, and DEAE-cellulose was from Sigma. Plasmid constructs were characterized by automated DNA sequencing (Davis Sequencing, LLC). All other materials were from the same sources as in previous studies (Wei et al. 2003, 2005).
Construction of wild-type and mutant N-TIMP expression systems
Using the pET-3a-timp-2 plasmid (containing the coding sequence for TIMP-2 between NdeI and BamHI sites) as template, T7 promoter primer, and the reverse primer 5′-AAAAAAAAGGATCC TTATCACTCGCAGCCCATCTGGTA-3′ (the BamHI site is in italics and stop codons are bold), we amplified the sequence encoding the N-TIMP-2 (N-timp-2) containing two stop codons in tandem following the GAG encoding E127 of TIMP-2. PCR reactions were carried out at 94°C for 30 sec, 60°C for 30 sec, and 72°C for 1 min, for 30 cycles after a 3-min hot start at 94°C. The PCR product was then cloned back into the pET-3a vector (Novagen) using the NdeI and BamHI sites.
N-TIMP-1 mutants with the N-TIMP-2 and TIMP-3 AB loops (N-TIMP-1[AB2] and N-TIMP-1[AB3]) were generated by overlap extension PCR (Ho et al. 1989) using the plasmid pET-3a (Novagen) containing the N-Timp-1 cDNA as template (see ▶). PCR reactions were carried out at 94°C for 1 min, 55°C for 1 min, and 72°C for 2 min, for 30 cycles after a 3-min hot start at 94°C. The PCR products were cloned back into pET-3a vector (Novagen) using NdeI and BamHI restriction sites.
Figure 2.

Comparison of regions of sequence from the four human TIMPs that were modified in the present study.
Multisite mutants of N-TIMP-1 were generated by megaprimer PCR as described previously (Huang et al. 1997; Meng et al. 1999) using N-TIMP-1(AB2) or wild-type N-TIMP-1 as template, and cloned into pET-3a using NdeI and BamHI sites. To simplify protein purification, some mutants of N-TIMP-1 (T2G, T2R, -1A, V4A, V4S, P5A, P6A, P6V, T98I, T98L, T98M, and combinations thereof) were expressed as His8-tagged fusion proteins. This was achieved by amplifying pET-3a-timp-1 in single PCR reactions with a forward primer introducing the corresponding mutation and a reverse primer introducing a NotI site, and cloning into pET-42b (Novagen).
Expression, purification, and folding of N-TIMP-1, N-TIMP-2, and mutants
N-TIMP-1, N-TIMP-2, and mutants were expressed in Escherichia coli BL21(DE3) cells (Novagen) as described for N-TIMP-1 (Huang et al. 1996). Cells were broken with a French press at 1000 psi; the inclusion bodies were collected and washed. The untagged N-TIMP-1 variants were purified and folded in vitro as described by Wei et al. (2003), whereas the His8-tagged proteins were purified and folded following the method previously described for N-TIMP-3 (Kashiwagi et al. 2001), except that no glycerol was added after folding, and the pH of the buffers used for ion exchange chromatography with CM-cellulose chromatography was 7.0 instead of 8.0.
N-TIMP-2 was extracted with 8 M urea and loaded at a flow rate of 0.5 mL/min onto a Macro-Prep Q-50 column (1.5 × 10 cm) pre-equilibrated with 20 mM Tris-HCl, pH 8.0, and 8 M urea. The column was eluted with a linear gradient of 0–0.5 N NaCl in equilibration buffer at a flow rate of 1.5 mL/min, and collected in 4.5 mL fractions. Fractions containing N-TIMP-2 were identified by SDS-PAGE, pooled, and folded in vitro as described for N-TIMP-1, except that the pH of the folding buffer was 8.0. The folded protein was applied to a column (1.5 × 10 cm) of DEAE-cellulose, equilibrated with 20 mM Tris-HCl, pH 8.0, and eluted with a gradient from 0 to 0.5 M NaCl in the same buffer at a flow rate of 1.5 mL/min. Further purification of N-TIMP-2 was carried out by anion exchange chromatography with a UNO Q1 column equilibrated with 20 mM Tris-HCl, pH 8.0, using a BioRad DuoFlow medium-pressure chromatography system; elution was performed with a linear salt gradient from 0 to 0.5 M NaCl.
Construction, expression, and folding of truncated forms of human gelatinase B (MMP-9)
The catalytic domain of human MMP-9 (residues 107–443) was amplified from the MGC clone 12,688 (ATCC; GenBank accession number: BF238288) using the primers: 5′-AAAAAAAACATATGTTCAAACCTTTGAGGGCGACCTC-3′ (forward), which incorporates a unique NdeI site and a translation initiating methionine, and 5′-AAAAAAAAGCGGCCGC CTAATAGAGGTGCCGGATGCCATTCAC-3′ (reverse), which incorporates a stop codon and a NotI site (restriction sites are in italics and the start and stop codons are bold). The PCR reactions were carried out as described previously. The product (∼1 kb) was digested with NotI and NdeI and cloned into pET-28b (Novagen). The protein encoded by this construct is designated MMP-9(CD).
The expression construct for MMP-9 catalytic domain with the FnII deleted (residues 216–390) was generated by overlap extension PCR (Ho et al. 1989). The final PCR product was subcloned into pET-28b using NdeI and NotI sites. This truncated enzyme protein is designated MMP-9(CD-FN).
MMP-9(CD) and MMP-9(CD-FN) were expressed in E. coli BL21 (DE3) cells. The proteins were extracted from inclusion bodies and partially purified using Macro-Prep Q-50 as described above for N-TIMP-2. Fractions containing MMP-9 were identified by SDS-PAGE and pooled.
The two forms of MMP-9 were folded in vitro, by 50-fold dilution with 20 mM Tris-HCl, 8 M urea, and dialyzing twice (24 h each) at 4°C against 20 mM, Tris-HCl, pH 8.0, 500 mM Arginine, 0.2 M NaCl, 5 mM CaCl2, and 50 μM ZnCl2, with (for MMP-9[CD]) or without (for MMP-9[CD-FN]) 5 mM β-mercaptoethanol and 1 mM 1-hydroxyethylsulfide. The folded protein was dialyzed against two changes of 20 mM Tris-HCl, pH 8.0, the precipitate was removed by filtration, and the MMP-9 further purified by chromatography with DEAE-cellulose as described above for N-TIMP-2. Fractions containing MMP-9 were identified by SDS-PAGE and pooled.
Cloning, expression, and folding of the catalytic domain of MT1-MMP
The coding sequence of the catalytic domain of human MMP-14 (104–308) was amplified from MGC clone 64,690 (ATCC; GenBank accession number: BC064803) using the primers: 5′-CGAAGGAAGCGCCATATGATCCAGGGTCTCAAATGGCAACATAAT-3′ (forward; incorporates a NdeI site and an initiating methionine) and 5′-GGTGTCAAAGTTGGATCC TCAGATGTTGGGCCCATAGGTGGGGTT-3′ (reverse; incorporates a stop codon and a BamH1 site). Restriction sites are in italics and start and stop codons are bold. The PCR reactions were carried out as described above. The product (∼0.7 kb) was digested with BamHI and NdeI and cloned into pET-MMP-14. The protein was expressed in E. coli BL21 (DE3) cells, and inclusion bodies were collected and extracted as described above. Initial purification was performed with a column of Q-Sepharose, pre-equilibrated with 20 mM Tris, 8 M urea, pH 8.0 buffer, and eluted with a gradient from 0 to 0.5 M NaCl. Fractions containing MMP-14 were identified by SDS-PAGE and pooled.
The catalytic domain of MMP-14 was folded in vitro by dialysis two times (24 h each) at 4°C against 20 mM, Tris-HCl, pH 8.0, 0.15 M NaCl, 5 mM CaCl2, and 50 μM ZnCl2 followed by desalting by dialysis against two changes of 20 mM Tris-HCl, pH 8.0. After removing the precipitate, the MMP-14 was purified using a Hi Prep DEAE-Sepharose column (GE Healthcare) pre-equilibrated with 20 mM Tris, pH 8.0 buffer, and eluted in a gradient of 0–0.5 M NaCl as described above. Active fractions were identified by assays and SDS-PAGE before pooling.
MMP inhibition kinetic studies
Inhibition kinetic studies for MMPs were carried out as described previously (Huang et al. 1996; Meng et al. 1999; Wei et al. 2003). Ki app values were calculated by fitting data to the following equations as appropriate:
(Equation 1) Tight binding inhibition (K < 100 nM):
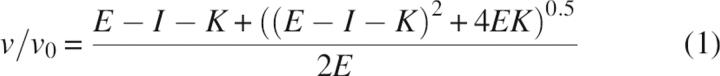 |
(Equation 2) Normal inhibition (K ≥ 100 nM):
where ν is the measured reaction velocity, ν0 is the uninhibited activity, E is enzyme concentration, I is inhibitor concentration, and K is the apparent inhibition constant (Ki (app)). As discussed previously (Huang et al. 1997), because the substrate concentration is very low relative to the Km, the apparent Ki is insignificantly different from the true Ki value.
Acknowledgments
We thank Shalini Iyer for preparing ▶. This work was supported by National Institutes of Health Grant AR40994.
Footnotes
Reprint requests to: Keith Brew, Department of Biomedical Sciences, Florida Atlantic University, 777 Glades Road, Boca Raton, FL 33431, USA; e-mail: kbrew@fau.edu; fax: (561) 297-2221.
Abbreviations: TIMP, tissue inhibitor of metalloproteinase; N-TIMP, N-terminal inhibitory domain of TIMP; MMP, matrix metalloproteinase; MT-MMP, membrane type-matrix metalloproteinase; FnII, fibronectin type II domain; ΔC, C-terminally truncated; CD, catalytic domain; Ki (app), apparent inhibition constant.
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.072978507.
References
- Arumugam S., Gao, G., Patton, B.L., Semenchenko, V., Brew, K., and Van Doren, S.R. 2003. Increased backbone mobility in β-barrel enhances entropy gain driving binding of N-TIMP-1 to MMP-3. J. Mol. Biol. 327 719–734. [DOI] [PubMed] [Google Scholar]
- Bigg H.F., Morrison, C.J., Butler, G.S., Bogoyevitch, M.A., Wang, Z., Soloway, P.D., and Overall, C.M. 2001. Tissue inhibitor of metalloproteinases-4 inhibits but does not support the activation of gelatinase A via efficient inhibition of membrane type 1-matrix metalloproteinase. Cancer Res. 61 3610–3618. [PubMed] [Google Scholar]
- Bode W., Grams, F., Reinemer, P., Gomis-Ruth, F.X., Baumann, U., McKay, D.B., and Stocker, W. 1996. The metzincin-superfamily of zinc-peptidases. Adv. Exp. Med. Biol. 389 1–11. [DOI] [PubMed] [Google Scholar]
- Brew K., Dinakarpandian, D., and Nagase, H. 2000. Tissue inhibitors of metalloproteinases (TIMPs): Evolution, structure and function. Biochim. Biophys. Acta 1477 267–283. [DOI] [PubMed] [Google Scholar]
- Brooks P.C., Stromblad, S., Sanders, L.C., von Schalscha, T.L., Aimes, R.T., Stetler-Stevenson, W.G., Quigley, J.P., and Cheresh, D.A. 1996. Localization of matrix metalloproteinase MMP-2 to the surface of invasive cells by interaction with integrin alpha v beta 3. Cell 85 683–693. [DOI] [PubMed] [Google Scholar]
- Brooks P.C., Silletti, S., von Schalscha, T.L., Friedlander, M., and Cheresh, D.A. 1998. Disruption of angiogenesis by PEX, a noncatalytic metalloproteinase fragment with integrin binding activity. Cell 92 391–400. [DOI] [PubMed] [Google Scholar]
- Browner M.F., Smith, W.W., and Castelhano, A.L. 1995. Matrilysin-inhibitor complexes: Common themes among metalloproteases. Biochemistry 34 6602–6610. [DOI] [PubMed] [Google Scholar]
- Butler G.S., Will, H., Atkinson, S.J., and Murphy, G. 1997. Membrane-type-2 matrix metalloproteinase can initiate the processing of progelatinase A and is regulated by the tissue inhibitors of metalloproteinases. Eur. J. Biochem. 244 653–657. [DOI] [PubMed] [Google Scholar]
- Butler G.S., Hutton, M., Wattam, B.A., Williamson, R.A., Knauper, V., Willenbrock, F., and Murphy, G. 1999. The specificity of TIMP-2 for matrix metalloproteinases can be modified by single amino acid mutations. J. Biol. Chem. 274 20391–20396. [DOI] [PubMed] [Google Scholar]
- Chen E.I., Kridel, S.J., Howard, E.W., Li, W., Godzik, A., and Smith, J.W. 2002. A unique substrate recognition profile for matrix metalloproteinase-2. J. Biol. Chem. 277 4485–4491. [DOI] [PubMed] [Google Scholar]
- Chen E.I., Li, W., Godzik, A., Howard, E.W., and Smith, J.W. 2003. A residue in the S2 subsite controls substrate selectivity of matrix metalloproteinase-2 and matrix metalloproteinase-9. J. Biol. Chem. 278 17158–17163. [DOI] [PubMed] [Google Scholar]
- Coussens L.M., Tinkle, C.L., Hanahan, D., and Werb, Z. 2000. MMP-9 supplied by bone marrow-derived cells contributes to skin carcinogenesis. Cell 103 481–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elkins P.A., Ho, Y.S., Smith, W.W., Janson, C.A., D'Alessio, K.J., McQueney, M.S., Cummings, M.D., and Romanic, A.M. 2002. Structure of the C-terminally truncated human ProMMP9, a gelatin-binding matrix metalloproteinase. Acta Crystallogr. D Biol. Crystallogr. 58 1182–1192. [DOI] [PubMed] [Google Scholar]
- Fernandez-Catalan C., Bode, W., Huber, R., Turk, D., Calvete, J.J., Lichte, A., Tschesche, H., and Maskos, K. 1998. Crystal structure of the complex formed by the membrane type 1-matrix metalloproteinase with the tissue inhibitor of metalloproteinases-2, the soluble progelatinase A receptor. EMBO J. 17 5238–5248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Patron C., Martinez-Cuesta, M.A., Salas, E., Sawicki, G., Wozniak, M., Radomski, M.W., and Davidge, S.T. 1999. Differential regulation of platelet aggregation by matrix metalloproteinases-9 and -2. Thromb. Haemost. 82 1730–1735. [PubMed] [Google Scholar]
- Gillmor S.A., Takeuchi, T., Yang, S.Q., Craik, C.S., and Fletterick, R.J. 2000. Compromise and accommodation in ecotin, a dimeric macromolecular inhibitor of serine proteases. J. Mol. Biol. 299 993–1003. [DOI] [PubMed] [Google Scholar]
- Gomis-Ruth F.X., Maskos, K., Betz, M., Bergner, A., Huber, R., Suzuki, K., Yoshida, N., Nagase, H., Brew, K., Bourenkov, G.P., et al. 1997. Mechanism of inhibition of the human matrix metalloproteinase stromelysin-1 by TIMP-1. Nature 389 77–81. [DOI] [PubMed] [Google Scholar]
- Ho S.N., Hunt, H.D., Horton, R.M., Pullen, J.K., and Pease, L.R. 1989. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene 77 51–59. [DOI] [PubMed] [Google Scholar]
- Huang W., Suzuki, K., Nagase, H., Arumugam, S., Van Doren, S.R., and Brew, K. 1996. Folding and characterization of the amino-terminal domain of human tissue inhibitor of metalloproteinases-1 (TIMP-1) expressed at high yield in E. coli . FEBS Lett. 384 155–161. [DOI] [PubMed] [Google Scholar]
- Huang W., Meng, Q., Suzuki, K., Nagase, H., and Brew, K. 1997. Mutational study of the amino-terminal domain of human tissue inhibitor of metalloproteinases 1 (TIMP-1) locates an inhibitory region for matrix metalloproteinases. J. Biol. Chem. 272 22086–22091. [DOI] [PubMed] [Google Scholar]
- Iyer S., Wei, S., Brew, K., and Acharya, K.R. 2007. Crystal structure of the catalytic domain of matrix metalloproteinase-1 in complex with the inhibitory domain of tissue inhibitor of metalloproteinase-1. J. Biol. Chem. 282 364–371. [DOI] [PubMed] [Google Scholar]
- Jinga D.C., Blidaru, A., Condrea, I., Ardeleanu, C., Dragomir, C., Szegli, G., Stefanescu, M., and Matache, C. 2006. MMP-9 and MMP-2 gelatinases and TIMP-1 and TIMP-2 inhibitors in breast cancer: Correlations with prognostic factors. J. Cell. Mol. Med. 10 499–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashiwagi M., Tortorella, M., Nagase, H., and Brew, K. 2001. TIMP-3 Is a potent inhibitor of ADAM-TS4 (aggrecanase 1) and ADAM-TS5 (aggrecanase 2). J. Biol. Chem. 276 12501–12504. [DOI] [PubMed] [Google Scholar]
- Kolkenbrock H., Essers, L., Ulbrich, N., and Will, H. 1999. Biochemical characterization of the catalytic domain of membrane-type 4 matrix metalloproteinase. Biol. Chem. 380 1103–1108. [DOI] [PubMed] [Google Scholar]
- Lang R., Braun, M., Sounni, N.E., Noel, A., Frankenne, F., Foidart, J.M., Bode, W., and Maskos, K. 2004. Crystal structure of the catalytic domain of MMP-16/MT3-MMP: Characterization of MT-MMP specific features. J. Mol. Biol. 336 213–225. [DOI] [PubMed] [Google Scholar]
- Lee M.H., Maskos, K., Knauper, V., Dodds, P., and Murphy, G. 2002a. Mapping and characterization of the functional epitopes of tissue inhibitor of metalloproteinases (TIMP)-3 using TIMP-1 as the scaffold: A new frontier in TIMP engineering. Protein Sci. 11 2493–2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M.H., Verma, V., Maskos, K., Nath, D., Knauper, V., Dodds, P., Amour, A., and Murphy, G. 2002b. Engineering N-terminal domain of tissue inhibitor of metalloproteinase (TIMP)-3 to be a better inhibitor against tumour necrosis factor-alpha-converting enzyme. Biochem. J. 364 227–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M.H., Rapti, M., and Murphy, G. 2003. Unveiling the surface epitopes that render tissue inhibitor of metalloproteinase-1 inactive against membrane type 1-matrix metalloproteinase. J. Biol. Chem. 278 40224–40230. [DOI] [PubMed] [Google Scholar]
- Lee M.H., Rapti, M., Knauper, V., and Murphy, G. 2004. Threonine 98, the pivotal residue of tissue inhibitor of metalloproteinases (TIMP)-1 in metalloproteinase recognition. J. Biol. Chem. 279 17562–17569. [DOI] [PubMed] [Google Scholar]
- Maskos K., Lang, R., Tschesche, H., and Bode, W. 2007. Flexibility and variability of TIMP binding: X-ray structure of the complex between collagenase-3/MMP-13 and TIMP-2. J. Mol. Biol. 366 1222–1231. [DOI] [PubMed] [Google Scholar]
- McCawley L.J. and Matrisian, L.M. 2001. Matrix metalloproteinases: They're not just for matrix anymore! Curr. Opin. Cell Biol. 13 534–540. [DOI] [PubMed] [Google Scholar]
- Meng Q., Malinovskii, V., Huang, W., Hu, Y., Chung, L., Nagase, H., Bode, W., Maskos, K., and Brew, K. 1999. Residue 2 of TIMP-1 is a major determinant of affinity and specificity for matrix metalloproteinases but effects of substitutions do not correlate with those of the corresponding P1′ residue of substrate. J. Biol. Chem. 274 10184–10189. [DOI] [PubMed] [Google Scholar]
- Morgunova E., Tuuttila, A., Bergmann, U., Isupov, M., Lindqvist, Y., Schneider, G., and Tryggvason, K. 1999. Structure of human pro-matrix metalloproteinase-2: Activation mechanism revealed. Science 284 1667–1670. [DOI] [PubMed] [Google Scholar]
- Page-McCaw A., Ewald, A.J., and Werb, Z. 2007. Matrix metalloproteinases and the regulation of tissue remodelling. Nat. Rev. Mol. Cell Biol. 7 221–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowsell S., Hawtin, P., Minshull, C.A., Jepson, H., Brockbank, S.M., Barratt, D.G., Slater, A.M., McPheat, W.L., Waterson, D., Henney, A.M., et al. 2002. Crystal structure of human MMP9 in complex with a reverse hydroxamate inhibitor. J. Mol. Biol. 319 173–181. [DOI] [PubMed] [Google Scholar]
- Shimada T., Nakamura, H., Ohuchi, E., Fujii, Y., Murakami, Y., Sato, H., Seiki, M., and Okada, Y. 1999. Characterization of a truncated recombinant form of human membrane type 3 matrix metalloproteinase. Eur. J. Biochem. 262 907–914. [DOI] [PubMed] [Google Scholar]
- Steffensen B., Wallon, U.M., and Overall, C.M. 1995. Extracellular matrix binding properties of recombinant fibronectin type II-like modules of human 72-kDa gelatinase/type IV collagenase. High affinity binding to native type I collagen but not native type IV collagen. J. Biol. Chem. 270 11555–11566. [DOI] [PubMed] [Google Scholar]
- Wei S., Chen, Y., Chung, L., Nagase, H., and Brew, K. 2003. Protein engineering of the tissue inhibitor of metalloproteinase 1 (TIMP-1) inhibitory domain. In search of selective matrix metalloproteinase inhibitors. J. Biol. Chem. 278 9831–9834. [DOI] [PubMed] [Google Scholar]
- Wei S., Kashiwagi, M., Kota, S., Xie, Z., Nagase, H., and Brew, K. 2005. Reactive site mutations in tissue inhibitor of metalloproteinase-3 disrupt inhibition of matrix metalloproteinases but not tumor necrosis factor-alpha-converting enzyme. J. Biol. Chem. 280 32877–32882. [DOI] [PubMed] [Google Scholar]
- Will H., Atkinson, S.J., Butler, G.S., Smith, B., and Murphy, G. 1996. Membrane-type-2 matrix metalloproteinase can initiate the processing of progelatinase A and is regulated by the tissue inhibitors of metalloproteinases. J. Biol. Chem. 271 17119–17123. [DOI] [PubMed] [Google Scholar]
- Williamson R.A., Hutton, M., Vogt, G., Rapti, M., Knauper, V., Carr, M.D., and Murphy, G. 2001. Tyrosine 36 plays a critical role in the interaction of the AB loop of tissue inhibitor of metalloproteinases-2 with matrix metalloproteinase-14. J. Biol. Chem. 276 32966–32970. [DOI] [PubMed] [Google Scholar]
- Wingfield P.T., Sax, J.K., Stahl, S.J., Kaufman, J., Palmer, I., Chung, V., Corcoran, M.L., Kleiner, D.E., and Stetler-Stevenson, W.G. 1999. Biophysical and functional characterization of full-length, recombinant human tissue inhibitor of metalloproteinases-2 (TIMP-2) produced in Escherichia coli. Comparison of wild type and amino-terminal alanine appended variant with implications for the mechanism of TIMP functions. J. Biol. Chem. 274 21362–21368. [DOI] [PubMed] [Google Scholar]