

Abstract
Diversity analysis of 71 published dengue virus gene sequences revealed several strains that appeared to be mosaics comprising gene regions with conflicting evolutionary histories. Subsequent maximum likelihood breakpoint estimation identified seven recombinants, including members of three of the four dengue virus serotypes, with breakpoints in the premembrane/membrane gene, the envelope gene, and at the junction of the envelope and first nonstructural genes. Many of the individual recombinants contain sequence representing separate genetic subtypes. The results were highly statistically significant and were confirmed by phylogenetic analysis of the regions of interest. These findings indicate that recombination may play a very significant role in shaping genetic diversity in dengue virus and, as such, have important implications for its biology and its control.
The last 25 years have seen dengue fever and dengue hemorrhagic fever (DHF) emerge as the most important vector-borne viral diseases of human beings (1, 2). Transmitted primarily in a human-mosquito cycle, dengue viruses now cause an estimated 100 million cases of serious febrile illness annually (1). In addition, every year several hundred thousand patients are hospitalized with DHF, an immunopathologic disease most often occurring in persons who experience sequential dengue virus infections (1, 3). Without appropriate supportive therapy, DHF may be fatal in approximately 5% of cases (1).
Dengue virus, a single-stranded, positive-sense RNA virus, belongs to the family Flaviviridae and is closely related to the etiologic agent of yellow fever, which also may cause hemorrhagic disease. Its genome of approximately 11 kb encodes three structural genes and seven nonstructural genes in one ORF. Its principal vector, Aedes aegypti, a domestic, daytime-biting mosquito that thrives in tropical and subtropical cities, has enjoyed spectacular success in recent years as increases in human population size, poverty, and mobility have led to its current pan-tropical distribution. The impressive spread of Ae. aegypti has been matched by the ever-increasing distributions of the four genetically distinct serotypes of dengue virus (DEN-1–DEN-4), which now cocirculate in much of Asia, Africa, and the Americas and put some 2.5 billion people at risk of infection (1).
Within each of the four dengue virus serotypes, phylogenetic studies have identified genetic subtypes that differ in nucleotide sequence by up to 12% in the envelope (E) gene, which determines most antigenic characteristics of the virus (4). DEN-1 virus comprises five known subtypes (I–V, ref. 5) and DEN-2 virus comprises six, although DEN-2 virus subtype III has been further divided into sublineages IIIa and IIIb (4). DEN-3 and DEN-4 viruses currently are classed into four and two different subtypes, respectively (6, 7). The complexity of the geographic distribution of dengue virus revealed by recent phylogenetic analyses—with closely related strains often present in distant parts of the world and multiple serotypes and subtypes commonly cocirculating (4–7)—has confirmed its efficient dispersal.
One undoubted contributing factor to the genetic diversity of dengue virus is its relatively high mutation rate, a characteristic of many RNA viruses that lack the proofreading enzymes used by DNA-based organisms to enhance the fidelity of genome replication. Another, largely ignored but potentially important, means by which genetic diversity could be generated in dengue virus is recombination. For recombination to take place different viruses need to coinfect the same individual human or mosquito host and then replicate in the same cell. Simultaneous infection of human hosts with different serotypes of dengue virus has, in fact, been carefully documented (8), and simultaneous presence of more than one species of Flavivirus is known to occur in the vector Ae. aegypti (8). However, although the possibility of genetic exchange between divergent dengue virus strains—and the serious consequences this has for the generation of new types of dengue virus—have been considered previously (2, 9), nearly all of the work to date on the biology, evolution, and control of dengue virus has rested on the implicit assumption that recombination plays little or no role in producing the genetic differences between strains. Here we report the results of a phylogenetic analysis of dengue virus sequences that reveals widespread intra-serotype recombination among them.
Recombination involving divergent strains can be detected when phylogenetic analysis reveals different evolutionary histories for different regions of a single genome as, for example, in HIV (10) and Neisseria bacteria (11, 12). Recently, in a paper that outlines a likelihood method for detecting crossover breakpoints in putative recombinants, we reported just such phylogenetic evidence suggesting that two South American strains of DEN-1 virus may be descendants of a single recombinant ancestor produced by genetic exchange between viruses belonging to two different subtypes (13). In the present study we used a sliding window analysis to identify possible recombinants among published sequences of dengue virus, and then applied the phylogenetic methods described in ref. 13 to define their presumed recombination breakpoints and to test the significance of these results. Strong phylogenetic evidence indicated that a surprisingly large proportion of sequenced dengue virus strains were recombinant.
MATERIALS AND METHODS
Sequence Data.
To conduct a systematic search for recombination within each serotype, we collected published dengue viruses from GenBank for which extensive sequence data exist. Because the chance of finding a recombination crossover point, and the ability to characterize it, grow with increasing sequence length, for each serotype we constructed a nucleotide sequence alignment by using the longest contiguous stretch of the viral genome for which several individual sequences were available: for DEN-1 virus, this resulted in a 2,325-nt alignment of the capsid (C), prM/M (premembrane/membrane), and E genes of six isolates (nucleotides 1–2325 counting from the 5′ end of the coding region); for DEN-2 virus, a 2,541-nt alignment of the E and NS1 (first nonstructural) genes of 24 isolates (nucleotides 841-3381); for DEN-3 virus, a 1,983-nt alignment of the prM/M and E genes of 22 isolates (nucleotides 343-2325); and for DEN-4 virus, a 1,485-nt alignment of the E gene for 19 isolates (nucleotides 841-2325). Alignments also were made of the homologous sequences from all four serotypes. All sequences were aligned manually and sites with gaps were removed in all cases. All of the alignments are available from the authors on request.
Diversity Plots.
To test the hypothesis that some dengue virus strains might be the result of recombination between parent strains belonging to different serotypes, or to different intra-serotype subtypes, we first examined the pairwise patterns of divergence between sequences in each alignment. By using the program divert (available at http://193.50.234.246/∼beaudoin/anrs/Diversity.html) the percentage pairwise divergence between query sequences and comparison sequences was determined by sliding a window of 150 bp along the alignments in 3-bp increments. The resulting diversity profiles then were used to identify sequences that appeared to have conflicting profiles in different genomic regions. Sequences most closely resembling viruses of a particular subtype in one region, but similar to representatives of a divergent lineage or subtype in another were identified as putative recombinants and subjected to further analyses (see ref. 14 for a similar method applied to HIV).
Breakpoint Analysis.
Once putative recombinants and their putative “parents” (the sequences in the data set that most closely matched the putative recombinant in a part of its sequence) were identified, their optimal recombination breakpoints were determined by using a maximum likelihood method (13). Briefly, for every possible breakpoint, or pair of breakpoints, the sequence alignment was divided into two independent regions for which the branch lengths of a tree of the putative recombinant and its two parental sequences were optimized. The two likelihoods obtained by using the separate regions then were combined to give a likelihood score for that breakpoint position and the breakpoint position that yielded the highest likelihood was identified. This “recombination model” likelihood then was compared, by using a likelihood ratio test, to the likelihood obtained from the same data under a model that permitted no recombination.
To assess whether the recombination model gave a significantly better fit to the data than the null hypothesis of no recombination, the likelihood ratio obtained by using the real data was evaluated for significance against a null distribution of likelihood ratios produced by using Monte Carlo simulation of sequences generated without recombination. In each instance, sequences were simulated 200 times by using the maximum likelihood model parameters and sequence lengths from the real data. The simulated sequences then were subjected to the same breakpoint analyses as the real data to produce the distribution of likelihood ratios expected if no recombination had occurred among them.
Phylogenetic Trees and Bootstrap Support.
We used phylogenetic trees to better evaluate the evidence for conflicting evolutionary histories of different sequence regions identified by the previous analyses. As far as possible, these trees included representatives of the full range of the genetic diversity within each serotype. In each case, separate maximum likelihood trees were constructed for each putative recombinant region, and the phylogenetic position of the putative recombinant was compared between them. Phylogenetic discrepancies then were assessed by using the percentage of bootstrap replicates supporting the conflicting phylogenetic positions. Bootstrapping was conducted both with maximum likelihood (100 replicates) and neighbor joining (1,000 replicates) trees. Because the maximum likelihood bootstrap analyses consistently resulted in equal or lower bootstrap support for the conflicting phylogenetic positions of putative recombinants than those recovered under neighbor joining, only these (more conservative) results are presented. All trees (including those used for breakpoint analyses) were produced with the 4.0d64 test version of paup* kindly provided by David L. Swofford (Smithsonian Institution, Washington, DC) using the HKY85 model of nucleotide substitution, with transition/transversion ratios and shape parameters of the gamma distribution of among-site rate heterogeneity estimated during tree reconstruction.
RESULTS
Diversity Plots.
Diversity plots for most sequence comparisons revealed a pattern of more-or-less consistent divergence between pairs of dengue virus sequences across the whole length of sequence surveyed. No inter-serotype recombinants were detected, a finding that was not surprising given the often extensive sequence divergence between serotypes and because each isolate analyzed was amplified and sequenced by using primers specific for just one of the four serotypes. Several strains, however, did exhibit strikingly discordant regions, showing close affinity to very divergent viruses over different parts of their sequences. In some of these cases, it was clear that different parts of individual sequences closely resembled viruses from distinct subtypes within the same serotype. Such discordant relationships between regions within the same sequence strongly suggested the existence of intra-serotype mosaic genomes produced by recombination between divergent parent lineages.
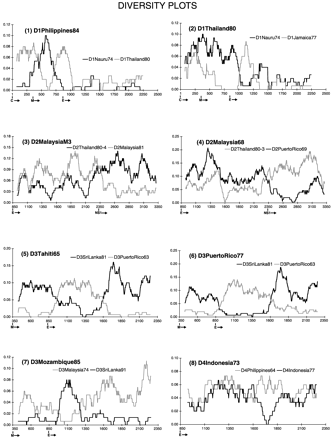
Fig. 1 shows the diversity profiles of one putative recombinant from each serotype. (The others, not shown in Fig. 1, are listed with their parents in Table 1 and are available as supplemental data on the PNAS web site, www.pnas.org.) Strain D1Philippines84 (Fig. 1a) was clearly most similar to strain D1Nauru74 along most of the sequence analyzed, but for a portion of its premembrane/M gene diverged from D1Nauru74 and became more similar to an otherwise divergent virus, D1Thailand80. D1Thailand80, in turn (Table 1), contained a short portion of sequence near the beginning of its E gene that was similar to D1Nauru74 and quite different from D1Jamaica77, the sequence it most closely resembled across the remainder of its sequence. Based on a phylogeny of DEN-1 viruses inferred from a 240-bp fragment spanning the E/NS1 junction (5), all of these viruses have been classed as DEN-1 virus subtype IV. However, it is clear from analyzing a larger portion of their sequences that a good deal more diversity exists among them than was revealed by looking at this short region alone.
Figure 1.

Diversity plots for putative recombinants and their parents. The vertical axis is the percent difference (Hamming distance) between the query sequence and each parent plotted at the midpoint of each window. The horizontal axis is nucleotide number counting from the 5′ end of the translated sequence (i.e., the start of the C gene). The beginning of each gene is indicated below this axis. Window size and increment were 150 and 3 bp, respectively, in each case. The query sequence is written in bold above the plot, and the gray and black lines represent the results of pairwise comparisons between it and its two parents as identified for each plot. The plots indicate different patterns of relationship for different sequence regions within the same virus, with parts being alternately similar to one, then the other, parent.
Table 1.
Recombinant strains, their parents, and their breakpoint analysis results
| Sequence, “strain” | GenBank accession no. (reference) | Region | Gene | “Parents” | Subtype | P-value |
|---|---|---|---|---|---|---|
| D1Philippines84 | D00503 | 1-372 | C+M | D1Nauru74 | IV | |
| “162” | (15) | 373-690 | M | D1Thailand80 | IV | P < 0.005 |
| 691-2325 | M+E | D1Nauru74 | IV | |||
| D1Thailand80 | D00502 | 1-1053 | C+M+E | D1Jamaica77 | IV | |
| “AHF82-80” | (15) | 1054-1230 | E | D1Nauru74 | IV | P < 0.005 |
| 1231-2325 | E | D1Jamaica77 | IV | |||
| FrenchGuiana | Not available | 1-1146 | C+M+E | D1Jamaica77 | IV | |
| “FGA/89” | (13, 16) | 1147-2325 | E | D1Singapore90 | I | P < 0.005 |
| Brazil | S64849 | 1-1152 | C+M+E | D1Jamaica77 | IV | |
| “BR/90” | (13, 16) | 1153-2325 | E | D1Singapore90 | I | P < 0.005 |
| D2MalaysiaM3 | X15214/X17340 | 841-2322 | E | D2Thailand80-4 | IIIb | |
| “M3” | (17, 18) | 2323-3381 | E+NS1 | D2MalaysiaM1 | IIIa | P < 0.005 |
| D2Malaysia68 | U89517 | 841-2289 | E | D2Thailand80-3 | IIIa | |
| “P7-863” | (19) | 2290-3381 | E+NS1 | D2PuertoRico69 | V | P < 0.005 |
| D3Tahiti65 | L11619 | 343-942 | M+E | D3PuertoRico63 | IV | |
| “2167” | (6) | 943-1623 | E | D3SriLanka81 | III | P < 0.005 |
| 1624-2325 | E | D3PuertoRico63 | IV | |||
| D3PuertoRico77 | L11434 | 343-921 | M+E | D3PuertoRico63 | IV | |
| “1340” | (6) | 922-1623 | E | D3SriLanka81 | III | P < 0.005 |
| 1624-2325 | E | D3PuertoRico63 | IV | |||
| D3Mozambique85 | L11430 | 343-1101 | M+E | D3SriLanka91 | III | |
| “1558” | (6) | 1012-1212 | E | D3Malaysia74 | I | P < 0.005 |
| 1213-2325 | E | D3SriLanka91 | III | |||
| D4Indonesia73 | U18428 | 841-1653 | E | D4Philippines64 | I | |
| “30153” | (7) | 1654-1821 | E | D4Indonesia77 | II | P < 0.105 |
| 1822-2325 | E | D4Philippines64 | I |
Two DEN-2 virus strains also had profiles suggestive of genetic exchange. D2MalaysiaM3 (Table 1) most closely resembled a DEN-2 subtype IIIb virus (D2Thailand 80–4) over its E gene sequence, but was much closer in its NS1 gene to another Malaysian virus (D2Malaysia81) that belongs to subtype IIIa. D2Malaysia68 (Fig. 1b), closest to a subtype IIIa virus (D2Thailand80–3) over most of its E gene, was much more similar in its NS1 sequence to D2PuertoRico69, a representative of the distant subtype V DEN-2 virus strains.
Similarly, three DEN-3 virus sequences clearly showed patterns suggestive of recombination. D3Mozambique85 (Table 1) appeared to be a normal subtype III DEN-3 virus over most of its premembrane/M and E sequence, based on a close affinity with D3SriLanka91. However, the existence of a part of its E gene sequence that dramatically diverged from the subtype III viruses and became very similar to a subtype I virus (D3Malaysia74), suggested it was not a simple subtype III dengue virus. D3Tahiti65 (Table 1) and D3PuertoRico77 (Fig. 1c) flip-flopped, in a similar fashion, from subtype IV (closest relative D3PuertoRico63) to subtype III (closest relative D3SriLanka81) then back to subtype IV over the region analyzed. Finally, one DEN-4 virus strain, D4Indonesia73, while quite divergent from the DEN-4 subtype II viruses along most of its E gene, appeared to be nearly identical to them in one region (Fig. 1d).
Breakpoint Analysis.
Table 1 summarizes the results of the breakpoint analyses. For each putative recombinant, the maximum likelihood estimated breakpoints are listed along with the genes or partial genes comprising each region and the parent and subtype affiliated with that region. The two recently characterized DEN-1 virus sequences (BR/90 and FGA/89, ref. 13) are included for completeness. In all cases the maximum likelihood breakpoint estimates were in close agreement with the crossover points depicted in the corresponding diversity plots. Moreover, the likelihood ratios for all but one of the putative recombinants were greater than any of the 200 likelihood ratios produced by using Monte Carlo simulation to test each result, with likelihood ratio test P values < 0.005. In other words, the results of the simulation tests indicated that the discordant relationships for different regions of each putative recombinant (except D4Indonesia73, which we will return to later) were highly unlikely to be the result of chance.
The breakpoint analyses also strengthened our suspicion that the epidemiologically linked (6) pair of DEN-3 virus sequences (D3PuertoRico77 and D3Tahiti65) with similar diversity profiles might represent descendants of a single ancestral recombination event. Of the two breakpoints inferred for each of the two DEN-3 virus sequences (Table 1), one was identical (after nucleotide 1623) and the other was similar (after nucleotide 942 in D3Tahiti65, and after nucleotide 921 in D3PuertoRico77). Assuming that breakpoints are randomly distributed across the genome of real recombinants, the probability that independent recombination events would produce such similar breakpoints is very small (less than 10−4). Thus, as with the pair of South American DEN-1 viruses, the breakpoint analyses suggested that these two putative recombinants are more likely descendants of one recombinant ancestor than independently derived.
In the case of the DEN-4 virus D4Indonesia73, although breakpoint analysis identified the expected optimal recombination region, statistical support for recombination was not significant (P < 0.105). However, a few observations lead us to speculate that D4Indonesia73 may be a genuine recombinant, and not simply the product of extreme rate variation in different parts of its sequence. First, no other DEN-4 virus sequence comparisons give any evidence that the regions identified for D4Indonesia73 evolve at the anomalous rates needed to explain its diversity profile (data not shown). Second, although it has been classed as a (DEN-4) subtype II virus, D4Indonesia73 is the most divergent member of this subtype and is antigenically distinct (7). Finally, phylogenetic evidence based on trees reconstructed by using the two regions defined by our breakpoint analysis supports robust topological differences in the position of D4Indonesia73 in the DEN-4 virus phylogeny (Fig. 2d). Hence, the majority of its sequence may represent an as-yet-uncharacterized lineage of DEN-4 virus that differs consistently across its E gene sequence from subtype I and II strains. Current data, however, are insufficient to clearly demonstrate recombination in this case.
Figure 2.

Phylogenetic analysis of putative recombinant genomes. Maximum likelihood trees were constructed by using the portions of the sequence alignments corresponding to the recombinant regions identified for each putative recombinant. The recombinant sequence (and abbreviation) is written in bold at the top of each pair of trees and highlighted with a box on the trees. The putative parents in each case were: (a) D1Nauru74 (N74) and D1Thailand80 (T80); (b) D2Thailand80–3 (T80–3) and D2PuertoRico69 (PR69); (c) D3PuertoRico63 (PR63) and D3SriLanka81 (SL81); and (d) D4Indonesia77 (I77) and D4Philippines64 (P64). The sequence region used for each tree is written above it and corresponds with Table 1 and Fig. 1. The subtypes within each serotype are identified with shading and are labeled with roman numerals. All branch lengths are drawn to scale. The trees (and their associated maximum likelihood bootstrap values) strongly support different phylogenetic positions for the recombinants within different parts of their genomes, indicating their mosaic nature.
Phylogenetic Trees and Bootstrap Support.
Phylogenetic trees constructed by using the different regions identified by breakpoint analyses for the putative recombinants strongly supported the hypothesis of recombination. The four sets of phylogenetic trees corresponding to Fig. 1 a-d are shown in Fig. 2. (The others are not included but show the same kind of phylogenetic conflict, supported by high bootstrap values. They are available as supplemental data on the PNAS web site, www.pnas.org.) The different topological positions of the putative recombinants for different parts of their genomes, and the robustness of these differences demonstrated by high bootstrap values indicated that regions with very different evolutionary histories were present within individual strains. Both D1Philippines84 (Fig. 2a) and D1Thailand80 (not shown) contain short sequence regions similar to otherwise divergent subtype IV viruses. One DEN-2 virus strain, D2MalaysiaM3 (not shown), currently classed as a subtype IIIb virus (based on an analysis of E gene sequences), was a subtype IIIa virus with respect to its NS1 gene. Interestingly, based on this portion of its sequence, its two closest relatives were two other (subtype IIIa) sequences also from Malaysia. Another Malaysian DEN-2 virus sequence, D2Malaysia68 (Fig. 2b), contained both some subtype IIIa sequence and some very divergent subtype V sequence.
The DEN-3 virus responsible for the Mozambique epidemic in 1985 (D3Mozambique85) is known through epidemiological (20) and genetic evidence (6) to be related to viruses from India and Sri Lanka and has been classed with them in DEN-3 virus subtype III (6). Our phylogenetic analysis showed that, for a portion of its E gene, this relationship did not hold: this African strain also contained sequence from subtype I viruses, being most similar to another Asian strain isolated in Malaysia in 1974 (Table 1).
Even stronger phylogenetic evidence for recombination came from the two other DEN-3 virus strains. D3Tahiti65 and D3PuertoRico77 have been classed in DEN-3 virus subtype IV (6). However, phylogenetic trees confirmed that these two strains were not straightforward subtype IV viruses at all. Both, in fact, possessed similar, approximately 700-bp regions in their E genes that clearly belonged to the subtype III DEN-3 viruses (Table 1, Fig. 2c). Subtype IV viruses, notably, have been known to cause outbreaks of only classical dengue fever, but never DHF, leading to suggestions that they may possess distinct biological properties (6). Attempts to inspect the E genes of these two “subtype IV” DEN-3 viruses for the genetic basis of these properties would be much improved by the knowledge that almost half of their E gene sequence came from another subtype.
Given that several published dengue virus sequences comprise regions with different evolutionary histories, it is important, of course, to assess alternatives to the hypothesis that they resulted from recombination in natural virus populations. If they are not the result of natural genetic exchange then either (i) they are real recombinants but this process took place in the laboratory rather than in nature, during, for instance, cell culture replication or PCR, or (ii) the “mosaic” genomes identified never actually existed and are the product of mistakenly combining regions of two or more real genomes that were, for example, unwittingly sequenced together.
For every one of the putative recombinants (with the possible exception of D2MalaysiaM3, whose estimated breakpoint was near the junction of separately sequenced genes) the second option is unlikely: sequencing multiple strains simultaneously by the methods used would produce multiple peaks at single nucleotide sites on the electropherogram rather than a mosaic genome. Furthermore, some of the putative recombinants were plaque-purified before sequencing, effectively ruling out the presence of more than one virus strain during sequencing.
The other option, that the mosaic sequences are real, but laboratory-derived, implies the presence (through contamination or multiple infection) of divergent strains that have exchanged gene sequence in the laboratory. One opportunity for this process to occur would be during PCR. Yet although we would expect only a vanishingly small proportion of chimeric sequences produced by template switching during PCR to retain the same sequence length and nucleotide arrangement as natural dengue viruses, every one of the putative recombinants we describe appears to be the product of homologous recombination.
Homologous recombination during virus replication in cell culture, on the other hand, is more plausible because cell culture recombination would rely on the same selective process as “wild” recombination to weed out less viable, nonhomologous recombinants. However, it is difficult to see how even successful cell culture recombination could explain the identical mosaic patterns found within two separate pairs of putative recombinants. In DEN-1 virus and DEN-3 virus we found independent examples of pairs of sequences with identical or nearly identical inferred breakpoints and parents. Significantly, (i) they did not appear to be cobbled together from portions of other known sequences; and (ii) while showing near-identical mosaic patterns, the viruses did differ from each other slightly over their entire aligned sequence in precisely the manner expected for related viruses that have diverged through independent evolution: D3Tahiti65 and D3PuertoRico77, for example, differ at 2.0% of their subtype IV sites and at 2.4% of their subtype III sites. In short, they appear to possess the signature of “real” evolutionary change. The members of each pair are geographically and temporally related to their separate parental lineages, and to each other, in ways that make very good sense in light of a single ancestral recombination event linking them. It would be asking a great deal of laboratory artifacts to explain so well such apparently well-ordered evolutionary relationships.
DISCUSSION
The purpose of this study was to use the wealth of sequence data available for dengue virus to test a crucial assumption about the nature of its genetic diversity, namely, that recombination plays no significant role in its generation. To do this, we compared published sequences within each serotype to search for recombinant strains. Our results provide strong evidence not only that recombination occurs in natural populations of dengue virus but also that it may play a very important part in generating new, biologically successful strains. Of the 71 strains surveyed in this study we have found at least seven to be recombinant, suggesting either that genetic exchange is quite common among dengue viruses, that recombinant viruses are particularly successful at leaving descendants, or both. As such, we think the assumption of “clonality” in dengue virus is no longer tenable and that the role recombination plays in dengue virus evolution and biology demands immediate and serious consideration.
At the simplest level, we need to analyze new dengue virus sequences carefully to determine whether they are recombinant and to prevent misclassification of strains that may be used in future inference. However, to best identify recombinants it is necessary to compare them against a panel of well-characterized, full-length representative sequences. The apparent inadequacy of short sequence fragments to provide a complete picture of recombinant dengue virus phylogenetic relationships further demonstrates the value of full-length sequencing for dengue virus research. It is worth noting that a previous investigation using split decomposition analysis failed to detect evidence for recombination in three of the dengue virus serotypes (13). Although for DEN-2 virus the greater sequence length analyzed in the present study accounts for this difference of results, for the other serotypes the methods used in the present study were clearly more effective.
More challenging than simply documenting recombination will be the effort to answer some of the many questions its presence raises. First, what is the molecular basis of recombination in dengue virus? The high frequency of intra-serotype recombinants suggests that a sizeable proportion of individual hosts (either human or mosquito) can be simultaneously infected by divergent strains of dengue virus, and that these viruses can recombine in vivo to produce successful hybrids. However, we do not yet know the circumstances under which coinfection can occur, the proportion of coinfected hosts that experience simultaneous infection of individual cells, or the probability of recombination events given these prerequisites. There is an immediate need not only for laboratory confirmation of intra-serotype recombination but also for experimental studies of recombination between different serotypes of dengue virus.
Second, what is the biological significance of recombination in dengue virus and for RNA viruses in general? The relative importance of recombination versus the accumulation of point mutations for evolutionary change is still an open, and fundamental, question. As methods of sequence analysis are developed that are increasingly able to distinguish recombination from other evolutionary processes we expect genetic exchange to be found more often among RNA viruses. Whatever its past role in dengue virus, recombination seems poised to become ever more important with the increased prevalence and hyper-endemicity of dengue. The high proportion of recombinants in the database, and the two pairs of related recombinants that show how chimeric viruses can spread over impressive spans of both time and space, hint at important biological properties of hybrid strains. More pragmatically, recombination hinders studies of whether certain viral strains are more often associated with DHF than others because a single gene region no longer can be assumed to represent a marker for the entire genome (21).
Third, what is the significance of recombination for the ongoing (and as yet unsuccessful) effort to produce a safe and effective vaccine against dengue infection? Clearly, the production of new varieties via genetic exchange does not make the challenge of dengue virus immunization any easier. However, whether or not recombinants will prove to be a serious impediment to the success of vaccines will depend on the types of vaccines used. The unique obstacle to dengue virus immunization, that prior infection by one serotype seems to enhance subsequent infection by other serotypes (3), has prompted the development of a “four-in-one” dengue virus candidate vaccine that combines live, attenuated strains of all four serotypes (22). Although no inter-serotype dengue virus recombinants have yet been identified, our results suggest that dengue virus is recombinogenic, at least within serotypes. As such, we think it would be wise at least to consider the possibility of recombination between these vaccine strains and the unique epidemiological consequences this could have if it resulted in a viable escape strain (2). A key question in this context is what level of sequence divergence prevents recombination from taking place? Disease-associated recombinants resulting from genetic exchange between live, attenuated strains in a multivalent vaccine would not be unprecedented. A high proportion of patients with vaccine-associated paralytic poliomyelitis exhibit vaccine-derived inter-serotype recombinant strains that have lost the attenuated phenotype (23).
The fact that we do not yet know whether this scenario is a real danger seems, to us, good reason for urgent investigation into the nature of recombination in dengue virus. Evidence of apparently widespread genetic exchange between divergent strains adds a new layer of complexity to both the biological and medical understanding of dengue. If genetic exchange in dengue virus affects genetic diversity as much as our results suggest, it is clear that any effort to discern its biological properties, or to immunize against it, must be informed by an understanding of its natural history that encompasses recombination.
Supplementary Material
Acknowledgments
We thank Paulo Zanotto and Paul Harvey for invaluable discussions and comments and two anonymous reviewers for helpful advice. This work was supported by grants from The Royal Society, The Wellcome Trust, and The Rhodes Trust.
ABBREVIATIONS
- DHF
dengue hemorrhagic fever
- E
envelope
- NS
nonstructural
- C
capsid
- M
membrane
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
References
- 1.Gubler D J. Clin Microbiol Rev. 1998;11:480–496. doi: 10.1128/cmr.11.3.480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Monath T P. Proc Natl Acad Sci USA. 1994;91:2395–2400. doi: 10.1073/pnas.91.7.2395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Halstead S B. Science. 1988;239:476–481. doi: 10.1126/science.3277268. [DOI] [PubMed] [Google Scholar]
- 4.Lewis J G, Lanciotti R S, Chang G-J, Kinney R M, Trent D W. Virology. 1993;197:216–224. doi: 10.1006/viro.1993.1582. [DOI] [PubMed] [Google Scholar]
- 5.Rico-Hesse R. Virology. 1990;174:479–493. doi: 10.1016/0042-6822(90)90102-w. [DOI] [PubMed] [Google Scholar]
- 6.Lanciotti R S, Lewis J G, Gubler D J, Trent D W. J Gen Virol. 1994;75:65–75. doi: 10.1099/0022-1317-75-1-65. [DOI] [PubMed] [Google Scholar]
- 7.Lanciotti R S, Gubler D J, Trent D W. J Gen Virol. 1997;78:2279–2286. doi: 10.1099/0022-1317-78-9-2279. [DOI] [PubMed] [Google Scholar]
- 8.Gubler D J. Am J Trop Med Hyg. 1985;34:170–173. doi: 10.4269/ajtmh.1985.34.170. [DOI] [PubMed] [Google Scholar]
- 9.Kuno G. In: Dengue and Dengue Hemorrhagic Fever. Gubler D J, Kuno G, editors. Wallingford, U.K.: CAB International; 1997. pp. 61–88. [Google Scholar]
- 10.Robertson D L, Sharp P M, McCuthan F E, Hahn B H. Nature (London) 1995;374:124–126. doi: 10.1038/374124b0. [DOI] [PubMed] [Google Scholar]
- 11.Spratt B G, Bowler L D, Zhang Q-Y, Zhou J, Maynard Smith J. J Mol Evol. 1992;34:115–125. doi: 10.1007/BF00182388. [DOI] [PubMed] [Google Scholar]
- 12.Maynard Smith J. J Mol Evol. 1992;34:126–129. doi: 10.1007/BF00182389. [DOI] [PubMed] [Google Scholar]
- 13.Holmes E C, Worobey M, Rambaut A. Mol Biol Evol. 1999;16:405–409. doi: 10.1093/oxfordjournals.molbev.a026121. [DOI] [PubMed] [Google Scholar]
- 14.Gao F, Robertson D L, Carruthers C D, Morrison S G, Jian B X, Chen Y L, Barré-Sinoussi F, Girard M, Srinivasan A, Abimiku A G, et al. J Virol. 1998;72:5680–5698. doi: 10.1128/jvi.72.7.5680-5698.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chu M C, O’Rourke E J, Trent D W. J Gen Virol. 1989;70:1701–1712. doi: 10.1099/0022-1317-70-7-1701. [DOI] [PubMed] [Google Scholar]
- 16.Desprès P, Frenkiel M-P, Deubel V. Virology. 1993;196:209–219. doi: 10.1006/viro.1993.1469. [DOI] [PubMed] [Google Scholar]
- 17.Samuel S, Koh C L, Blok J, Pang T, Lam S K. Nucleic Acids Res. 1989;17:8887. doi: 10.1093/nar/17.21.8887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fong M Y, Koh C L, Samuel S, Pang T, Lam S K. Nucleic Acids Res. 1990;18:1642. doi: 10.1093/nar/18.6.1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fong M Y, Koh C L, Lam S K. Res Virol. 1999;149:457–464. doi: 10.1016/s0923-2516(99)80014-4. [DOI] [PubMed] [Google Scholar]
- 20.Gubler D J, Sather G E, Kuno G, Cabral J R. Am J Trop Med Hyg. 1986;35:1280–1284. doi: 10.4269/ajtmh.1986.35.1280. [DOI] [PubMed] [Google Scholar]
- 21.Rico-Hesse R, Harrison L M, Nisalak A, Vaughn D W, Kalayanarooj S, Green S, Rothman A L, Ennis F A. Am J Trop Med Hyg. 1998;58:96–101. doi: 10.4269/ajtmh.1998.58.96. [DOI] [PubMed] [Google Scholar]
- 22.Bhamarapravati N, Yoksan S. In: Dengue and Dengue Hemorrhagic Fever. Gubler D J, Kuno G, editors. Wallingford, U.K.: CAB International; 1997. pp. 367–377. [Google Scholar]
- 23.Georgescu M-M, Delpeyroux F, Tardy-Panit M, Balanant J, Combiescu M, Combiescu M M, Guillot S, Crainic R. J Virol. 1994;68:8089–8101. doi: 10.1128/jvi.68.12.8089-8101.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}